

Abstract
A previously introduced POSSIM (POlarizable Simulations with Second order Interaction Model) force field has been extended to include parameters for alanine peptides and protein backbones. New features were introduced into the fitting protocol, as compared to the previous generation of the polarizable force field for proteins. A reduced amount of quantum mechanical data was employed in fitting the electrostatic parameters. Transferability of the electrostatics between our recently developed NMA model and the protein backbone was confirmed. Binding energy and geometry for complexes of alanine dipeptide with a water molecule were estimated and found in a good agreement with high-level quantum mechanical results (for example, the intermolecular distances agreeing within ca. 0.06Å). Following the previously devised procedure, we calculated average errors in alanine di- and tetra-peptide conformational energies and backbone angles and found the agreement to be adequate (for example, the alanine tetrapeptide extended-globular conformational energy gap was calculated to be 3.09 kcal/mol quantim mechanically and 3.14 kcal/mol with the POSSIM force field). However, we have now also included simulation of a simple alpha-helix in both gas-phase and water as the ultimate test of the backbone conformational behavior. The resulting alanine and protein backbone force field is currently being employed in further development of the POSSIM fast polarizable force field for proteins.
Keywords: polarizable force fields, Monte Carlo simulations, torsional parameters, alanine peptides
I. Introduction
While quantum mechanical calculations offer valuable data in a variety of biological and biomedical calculations, applications of empirical force fields remain the only way of approaching the majority of problems of interest. On one hand, they require less computer resources. On the other hand, the issue of choosing the best level of quantum theory is still a non-trivial one, and the level of quantum mechanical accuracy in a specific application is far from being guaranteed.
When empirical force fields are employed, accurate assessment of energy often requires explicit treatment of the electrostatic polarization.1 The properties which depend on it include dimerization energies and acidity constants of small molecules, energies of protein-ligand interactions, protein pKa values, or even the very thermodynamic stability of complexes in solutions. For example, we have demonstrated that that pKa values for acidic and basic residues of the OMTKY3 can be reproduced within 0.6 and 0.7 pH units of the experimental data with a polarizable force field. The corresponding errors with the non-polarizable OPLS were 3.3 and 2.2 pH units.2 Formation of sugar-protein complexes represents yet another example when polarization is critical for predicting a thermodynamically stable structure.3 It is generally acknowledged that polarization is an important component in many computational studies of proteins and protein-ligand complexes, although it is sometimes included in surrogate forms, such as, for example, conformation-specific protein charges.4
There are two main issues related to the empirical polarizable force field development. The first one is in the functional form of the electrostatic polarization. Using fluctuating charges saves time and is computationally efficient in simulating uniform systems such as pure liquid water.5 However, it causes problems when out-of-plane polarization response is required or when a bifurcated hydrogen bond is formed. Therefore, the inducible dipoles approach is more adequate when arbitrary systems have to be simulated with a high degree of accuracy. On the other hand, it is known that the inducible dipole technique slows down polariable calculations very significantly. In order to reduce the severity of this problem, we are applying the second-order approximation in treatment of the electrostatic polarization. It has been demonstrated to increase the speed by ca. an order of magnitude without sacrificing the accuracy.6 Moreover, this approximation makes the so called polarization catastrophe (the resonance-like infinite growth of the induced dipole moment values) impossible. Our previous paper described development of the POSSIM (POlarizable Simulations with Second order Interaction Model) software and force field parameters for a series of small molecules, including water and NMA. In this work, we describe creation of alanine and protein backbone parameter sets in the POSSIM framework.
The second issue is choosing the source of fitting data for a polarizable force field. High-level quantum mechanical results are very attractive in this respect,7,8 but experimental data can be more robust. We follow the middle-of-the-road path by relying on experimental data whenever possible and making heavy use of quantum mechanical calculations when needed. One important issue is the standard procedure of producing torsional parameters for peptides by fitting to conformational energies of di- and tetrapeptides.8 We include it in our work and are describing an improved procedure for creation of the torsional parameters in the Methods section below. At the same time, the quantum mechanical conformers employed in such calculations are created by gas-phase quantum mechanical optimizations and they often belong to parts of the conformational space which are not found in experimental protein structures. Therefore, we have included an additional conformational test in the alanine and backbone parameter fitting. It is known that the tridecaalanine peptide (ala-13) forms a stable–α helix.9 Therefore, we studied the stability of our POSSIM ala-13 α–helix and compared it to that of the OPLS-AA8 for benchmarking. We have also discovered that the quality of the force field in reproducing the quantum mechanical di- and tetrapeptide conformational energies has a relatively weak effect on the stability of the tridecaalanine peptide in water.
Overall, the following has been derived, developed or otherwise calculated in this work: (i) the torsional parameters for the alanine residues and protein backbones have been produced; (ii) binding energies of a water molecule with the alanine dipeptide as calculated with the POSSIM and OPLS-AA force fields have been compared with the quantum mechanical data to confirm transferability of the non-bonded parameters and to justify using the latter from the POSSIM NMA model in protein studies; (iii) the resulting parameters were employed in gas-phase and aqueous solution simulations of an α-helix to validate the resulting POSSIM parameters as acceptable in protein and peptide simulations. Moreover, the additional optimizations in the step (i) above and the steps (ii) and (iii) altogether represent a novel development in our methodology of protein force field production, as compared to that used to create the previous version of the force field.
The rest of the paper is organized as follows: Given in Section II is description of the methodology involved. Section III contains results and discussion. Finally, conclusions are presented.
II. Methods
A. Force Field
The total energy Etot is a sum of the electrostatic interactions Eelectrostatic, the van-der-Waals energy EvdW, harmonic bond stretching and angle bending Estretch and Ebend and the torsional term Etorsion:
| (1) |
Electrostatic Energy
The electrostatic polarization energy as calculated with inducible point dipoles μ is:
| (2) |
where E0 is the electrostatic field in the absence of the dipoles.
| (3) |
where α are scalar polarizabilities and Tij is the dipole-dipole interaction tensor. The self-consistent Equation 3 is usually solved iteratively. Let us explicitly write down the first two iterations:
| (4a) |
| (4b) |
We are using the second-order expression in Equation (4b). It has been previously shown to yield a ca. order of magnitude increase of the computational speed with no loss of accuracy.6 The electrostatic energy also includes the pairwise-additive contribution from interactions of permanent charges:
| (5) |
The factor fij is set to zero for 1,2- and 1,3-pairs (atoms which belong to the same valence bond or angle), to 0.5 for 1,4-interactions (atoms in the same dihedral angle) and to 1.0 otherwise.
To avoid unphysical increase of the electrostatic interactions at short distances, each atom type has a cutoff parameter Rcut. When the overall distance Rij is smaller than the sum of these parameters for the atoms i and j, Rij is replaced by an effective smooth function
| (6) |
The following important points about the second-order approximation in equation 4b should be made. First of all, we do not fit parameters using the full-scale polarization solution to Equation 3 to later employ Equation 4b as an approximate technique during the simulations. For our practical purposes, Equation 4b is, in fact, the representation of the many-body interactions. It does differ from the true physical point-dipole approximation, and thus we always carefully monitor whether any errors are introduced by not computing inducible dipoles with the complete iterative procedure. So far, simulations of gas-phase dimers, quantum mechanical electrostatic three-body energies, pure liquids, solutions and peptides have given us no indication that the second-order approximation leads to any deficient physical results, and we have always been able to produce fitting to quantum mechanical and experimental data which was as good as for the full-scale polarization.6,10 Moreover, application of the second-order approximation given by Equation 4b turns the expression for the inducible dipoles into an analytical one, thus eliminating the possibility of the polarization catastrophe. This can also become a very useful feature in future developments, e. g. in creating a continuum dielectric model, as convergence issues are known to be of importance for continuum solvation techniques.
The Rest of the Force Field
We are using the standard Lennard-Jones formalism for the van-der-Waals energy:
| (7) |
Geometric combining rules are applied (εij = (εi · εj)1/2, σij= (σi · σj)1/2). Bond stretching and angle bending are computed with the usual harmonic formalism, and the torsional term is calculated as:
| (8) |
The fixed-charges OPLS-AA force field used for benchmarking is functionally the same, except that it lacks the polarization part of the electrostatic energy.
B. Parameterization of the Force Field
Whenever possible, the force field parameters for the alanine peptides were adopted directly from the previously created NMA parameter values.10 The only completely new parameters were those for the backbone torsions. This is different from the previous version of the PFF for proteins in which electrostatic parameters for the alanine (and thus for the backbone) were also refitted.7 Therefore, we believe that the present work demonstrates a greater degree of utilizing parameter transferability.
Fitting of torsional parameters for the protein backbone φ and ψ angles (Figure 1) cannot be done separately from each other as the torsions are coupled.
Figure 1.

Protein backbone angles φ and ψ shown in the alanine dipeptide molecule.
The initial part of our torsional fitting was the same as used before.7,8 (i) The fitting was done to an ab initio data obtained previously8 at the LMP2/cc-pVTZ(-f)//HF-6-31G** level with Jaguar software suite.11 (ii) The choice of the fitting subspace is illustrated on Figure 2. Out of the six alanine dipeptide local minima previously used,7,8 only two are shown for the sake of clarity.
Figure 2.

Torsional fitting subspace, for the alanine dipeptide φ/ψ potential energy surface. Such crosses were centered at each of the six minima and each arm contained four fitting points (here some crosses and points are omitted for the sake of clarity).
(iii) We used the following non-Boltzmann weighting scheme for the error at the fitting points:
| (9) |
Here Gi is the magnitude of the torsional surface gradient at the point i, and Wi is the weight. This way more importance was given to the points with low gradients (near the minima).
In the presented work, we used the procedure described above only to produce the initial guess for the torsional parameters to be employed in Equation 8. After that, the following approach was taken. The errors in the conformational energies were combined with the errors in the conformational angles φ and ψ to produce the error function as shown in Equation 10:
| (10) |
Here and Ei are the quantum mechanical and empirical conformational energies for all the conformers i, and the second sum contains the values of the backbone angles φ and ψ. The error function was minimized as a function of the torsional parameters in Equation 8.
C. Calculating Dimerization Energies for the Alanine Dipeptide Complexes with Water
In order to test the transferability of the NMA nonbonded parameters employed for our alanine and protein backbone model, we calculated energies of interaction of the alanine-dipeptide with a water molecule. Structures and energies obtained for these systems with the POSSIM program were compared to the quantum mechanical results obtained with Jaguar.11 For hydrogen bonds, a good level of accuracy can be achieved via MP2 calculations extrapolated to the basis set limit, where the contribution of higher level excitations (e.g. CCSD (T)) has been shown to be negligible (except for some cases, such as pi stacking of aromatic rings, where the MP2 level has been shown to not be sufficient).
Briefly, dimer geometries were obtained by LMP2 optimizations with a cc-pVTZ(-f) basis set. The final quantum mechanical dimer binding energy Ebind, as used in this work, is a linear combination of the LMP2 binding energy for a smaller cc-pVTZ(-f) basis set (Eccpvtz) and the LMP2 binding energy with a larger cc-pVQZ(-g) basis set (Eccpvq).15 This method has been previously demonstrated to produce a high-quality fitting and benchmarking data for force field development.7,8
D. Gas-Phase and Liquid-State Simulations of the Tridecaalanine peptide
In order to give our alanine and backbone model a final test, we carried out simulations of a tridecaalanine (ala-13) peptide both in gas-phase and in aqueous solution at 25°C and 1 atm. The initial structure was set at the α-helix conformation, with φ = 296° and ψ = 319°, and the simulations proceeded with all the degrees of freedom completely unconstrained. It is known experimentally that an α-helix represents a stable conformation of alanine peptides, including ala-13, both in gas-phase and in aqueous solution.9 We intended to show that our POSSIM force field for the alanine and backbone protein systems performs reasonably well under these conditions and is thus sufficiently robust to be successfully employed in protein and protein-ligand studies.
Gas-phase and hydrated simulations consisted of at least 18 × 106 and 25 × 106 Monte Carlo configurations, respectfully, to ensure convergence. A 7 Å dipole-dipole cutoff was used. An 8 Å cutoff was employed for the intermolecular interactions in solution (including both the solute-solvent and solvent-solvent interactions). The standard correction procedure to account for the Lennard-Jones interactions beyond the cutoff was used. The electrostatic interactions were quadratically feathered over the last 0.5 Å before the cutoff distance. A rectangular box with periodic boundary conditions was used. The box contained 948 water molecules. The initial box setup was done to have 10 Å of water on each side of the hydrated ala-13 molecule. After that, the isobaric-isothermal (NPT) ensemble was used, with Metropolis Monte Carlo technique. In the case of the OPLS simulations, a three-site model was used with TIP3P13 non-bonded parameters and flexible bond lengths and bond angles. A flexible three-site POSSIM water model10 was employed in the polarizable runs.
All the calculations which did not involve quantum mechanics (i. e., geometry optimizations and Monte Carlo runs) were performed with our previously introduced POSSIM software suite.10 Whenever possible, comparison with the fixed-charges OPLS-AA force field was done, and the OPLS-AA results were also calculated with the POSSIM program.
III. Results and Discussion
A. Alanine Dipeptide and Tetrapeptide Conformational Energies and Angles
We have followed the previously established procedure of calculating the alanine di- and tetrapeptide conformational energies and φ and ψ values as the initial assessment of the quality of the parameters for the alanine and protein backbones. The same set of the conformers that was employed in the previous studies was used.7,8 The production of the torsional parameters proceeded as described in the Methods section. First, weighted fitting to rotamer energies was carried out. The resulting parameters are shown in Table 1 (torsional parameters which are not listed were the same as in the NMA model10). We denote this set of parameters as tors.1, as opposed to the final set tors.final. Given in Table 2 are conformational energies and φ and ψ values, as computed with the quantum mechanics, POSSIM and OPLS. In addition to the six conformers used in previous studies, we have also added PII and αR which are more relevant in aqueous solution.14 Quantum mechanical optimizations were done at the LMP2/cc-pVTZ(-f) level. In both OPLS and POSSIM calculations, conformers β2, αL, PII and αR had the backbone dihedral angles fixed at the quantum mechanical values. It is known that molecular mechanics usually does not reproduce these conformers well. Overall, the performance of both POSSIM and OPLS is satisfactory. The POSSIM results have a slightly lower error in the conformational energies, while the OPLS results are closer to the quantum mechanics in terms of the geometries.
Table 1.
Backbone torsional parameters, set tors.1. The coefficients are given in kcal/mol.
| Parameter | V1 | V2 | V3 |
|---|---|---|---|
| C-N-αC-C, φ | 0.667 | −0.012 | −4.003 |
| N-αC-C-N, ψ | −2.011 | 2.528 | −4.829 |
| C-N-αC-βC, φ′ | −2.165 | 0.024 | 4.221 |
| βC-αC-C-N, ψ′ | 0.594 | −0.386 | 4.378 |
Table 2.
Conformational energies in kcal/mol and angles in degrees for alanine dipeptide. POSSIM refers to the polarizable force field with the tors.1 version of the torsional parameters. Angles φ and ψ for conformers β2, αL, PII, αR were fixed at their QM values. QM energy minimizations were unconstrained except for PII.
| Conformer | Energy | φ | ψ | ||||||
|---|---|---|---|---|---|---|---|---|---|
| QM | OPLS | POSSIM | QM | OPLS | POSSIM | QM | OPLS | POSSIM | |
| C7eq | 0.00 | 0.00 | 0.00 | −81.4 | −79.5 | −83.8 | 85.6 | 61.8 | 53.2 |
| C5 | 1.00 | 0.91 | 0.78 | −160.5 | −149.8 | −151.3 | 165.9 | 159.9 | 150.9 |
| C7az | 2.71 | 2.40 | 2.85 | 70.3 | 77.5 | 76.5 | −76.8 | −46.6 | −50.3 |
| β2 | 2.56 | 2.82 | 2.57 | −105.1 | −105.1 | −105.1 | 10.6 | 10.6 | 10.6 |
| αL | 4.21 | 5.96 | 5.41 | 68.3 | 68.3 | 68.3 | 22.4 | 22.4 | 22.4 |
| α′ | 5.47 | 5.96 | 5.53 | −162.0 | −156.5 | −149.5 | −33.2 | −48.5 | −100.3 |
| PII | 2.78 | 2.18 | 3.96 | −85.0 | −85.0 | −85.0 | 160.0 | 160 | 160.0 |
| αR | 2.71 | 2.39 | 1.95 | −83.7 | −83.7 | −83.7 | −3.9 | -3.9 | −3.9 |
|
| |||||||||
| error | – | 0.73 | 0.67 | – | 3.2 | 3.8 | – | 9.4 | 17.6 |
We have also calculated relative energies of the extended and globular conformations of the alanine tetrapeptide (shown on Figures 3 and 4, respectively). We determined the quantum mechanical energy difference for these conformers to be 3.09 kcal/mol, the globular form being the global energy minimum. At the same time, this quantity is known to have a relatively large range of calculated quantum mechanical energies. For example, Reference 15 lists the globular – extended energy gap for the alanine tetrapeptide to be between 2.88 and 4.99 kcal/mol. The POSSIM result with the tors.1 torsional parameters set was 2.53 kcal/mol, and the OPLS result was 3.51 kcal/mol.7,8
Figure 3.

LMP2/cc-pVTZ(-f) geometry of the extended alanine tetrapeptide conformation.
Figure 4.

LMP2/cc-pVTZ(-f) geometry of the globular alanine tetrapeptide conformation.
We then further refined the backbone torsional parameters as described in the Methods section. The resulting values of the torsional Fourier coefficients and the conformational energies and angles are given in Tables 3 and 4, respectively. This set of the torsional parameters is termed tors.final, and this is the final set for the POSSIM protein backbone φ and ψ. The average dipeptide conformational energy error is now slightly higher at 0.97 kcal/mol, but the average errors in the backbone angles φ and ψ are reduced to 1.6° and 12.9°, respectively. Moreover, the globular – extended energy gap in the tetrapeptide is 3.14 kcal/mol, in a better agreement with the quantum mechanical results (3.09 kcal/mol with our calculations and 2.88–4.99 kcal/mol from the data Reference 15). The value of the ψ for the C7eq conformer is lower now, but this part of the conformational space is not relevant in practical protein applications. The overall average error in both backbone angles was reduced.
Table 3.
Backbone torsional parameters, set tors.final. The coefficients are given in kcal/mol.
| Parameter | V1 | V2 | V3 |
|---|---|---|---|
| C-N-αC-C, φ | 2.000 | −0.500 | −3.772 |
| N-αC-C-N, ψ | −2.837 | 3.942 | −3.328 |
| C-N-αC-βC, φ′ | −2.718 | 1.757 | 5.202 |
| βC-αC-C-N, ψ′ | 0.372 | −0.915 | 3.321 |
Table 4.
Conformational energies in kcal/mol and angles in degrees for alanine dipeptide. POSSIM refers to the polarizable force field with the tors.final version of the torsional parameters. Angles φ and ψ for conformers β2, αL, PII, αR were fixed at their QM values. QM energy minimizations were unconstrained except for PII.
| Conformer | Energy | φ | ψ | ||||||
|---|---|---|---|---|---|---|---|---|---|
| QM | OPLS | POSSIM | QM | OPLS | POSSIM | QM | OPLS | POSSIM | |
| C7eq | 0.00 | 0.00 | 0.00 | −81.4 | −79.5 | −77.2 | 85.6 | 61.8 | 34.4 |
| C5 | 1.00 | 0.91 | 1.37 | −160.5 | −149.8 | −160.3 | 165.9 | 159.9 | 159.2 |
| C7az | 2.71 | 2.40 | 2.17 | 70.3 | 77.5 | 78.1 | −76.8 | −46.6 | −36.2 |
| β2 | 2.56 | 2.82 | 2.77 | −105.1 | −105.1 | −105.1 | 10.6 | 10.6 | 10.6 |
| αL | 4.21 | 5.96 | 5.79 | 68.3 | 68.3 | 68.3 | 22.4 | 22.4 | 22.4 |
| α′ | 5.47 | 5.96 | 5.98 | −162.0 | −156.5 | −162.9 | −33.2 | −48.5 | −38.0 |
| PII | 2.78 | 2.18 | 3.52 | −85.0 | −85.0 | −85.0 | 160.0 | 160 | 160.0 |
| αR | 2.71 | 2.39 | 0.99 | −83.7 | −83.7 | −83.7 | −3.9 | −3.9 | −3.9 |
|
| |||||||||
| error | – | 0.73 | 0.97 | – | 3.2 | 1.6 | – | 9.4 | 12.9 |
B. Alanine Dipeptide – Water Dimerization Energies and Distances
There are four possible water hydrogen bonding sites in the alanine dipeptide – two NH hydrogens and two carbonyl oxygen atoms. However, our quantum mechanical energy minimizations have demonstrated that water molecules prefer to make two hydrogen bonds at the same time, one with the H and one with the O atom. Therefore, there are only two water-alanine dipeptide heterodimer structures, as shown on Figures 4 and 5.
Figure 4.
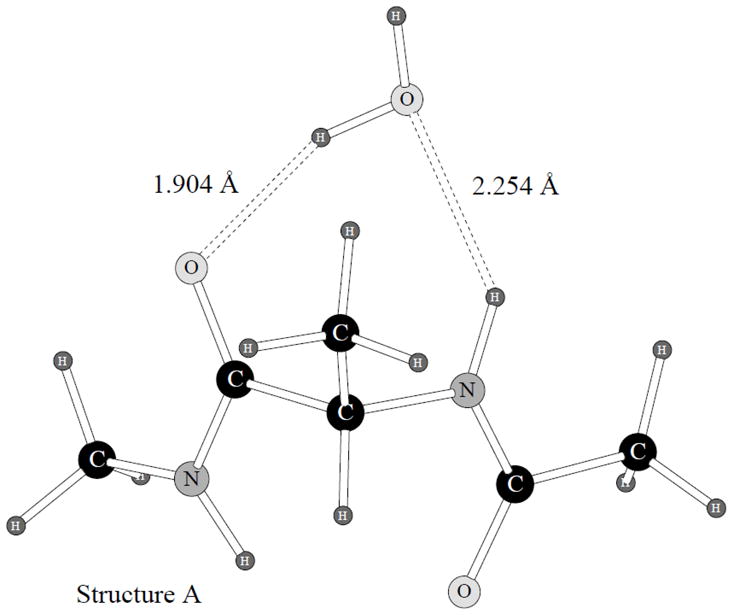
Alanine dipeptide hydrogen bonded complex with a water molecule, structure A.
Figure 5.

Alanine dipeptide hydrogen bonded complex with a water molecule, structure B.
The quantum mechanical structures were used as the initial guesses for the POSSIM optimizations. Both POSSIM and OPLS-AA were utilized. We compared the binding energies, as well as the geometries of the complexes. Both hydrogen bonding distances (O…H-N) and H…O=C) and the φ and ψ angles of the alanine dipeptide backbone were used for the comparison. The results of these calculations are presented in Table 5. The quantum mechanical energy of the dimerization is reproduced slightly better with the OPLS, the average error being 0.89 kcal/mol vs. 1.12 kcal/mol with POSSIM. The latter tends to underestimate the magnitude of the binding energy. This is not unexpected. The non-bonded parameters for the alanine dipeptide have been adopted from NMA fitting.
Table 5.
Results of simulating alanine dipeptide complexes with water. Energies are in kcal/mol, distances in Å, angles in degrees.
| property | Structure A | Structure B | ||||
|---|---|---|---|---|---|---|
| QM | OPLS | POSSIM | QM | OPLS | POSSIM | |
| Binding energy | −9.80 | −9.48 | −7.95 | −9.73 | −11.19 | −9.34 |
|
| ||||||
| R(O…O) | 2.83 | 2.75 | 2.84 | 2.83 | 2.72 | 2.73 |
| R(O…N) | 3.10 | 2.89 | 3.09 | 3.05 | 2.86 | 2.95 |
|
| ||||||
| φ, dimer | −83.4 | −87.3 | −80.2 | −84.3 | −89.4 | −86.9 |
| ψ, dimer | 90.3 | 114.6 | 83.7 | 131.2 | 113.3 | 122.2 |
|
| ||||||
| φ, monomer | −79.7 | −79.5 | −77.2 | −79.7 | −79.5 | −77.2 |
| ψ, monomer | 88.1 | 61.8 | 34.4 | 88.1 | 61.8 | 34.4 |
And the same tendency was also present in the NMA case, with the POSSIM underestimating the NMA-water binding energy by an average of 0.89 kcal/mol.10 The overall performance of the NMA parameters was very good. This included reproducing liquid NMA heat of vaporization and density. Which lead us to the conclusion that our quantum mechanical NMA-water binding energies are probably somewhat overestimated. Therefore, a similar trend in the alanine dipeptide complex formation with water could have been expected and is not at all an indication of problems with the protein POSSIM force field. Moreover, it can be easily seen from the data in Table 5 that the POSSIM performed noticeably better than the OPLS in reproducing the hydrogen bond lengths, which are probably given much more accurate than the energies by the quantum mechanics. The average errors in these lengths are 0.15 and 0.06Å with the OPLS and POSSIM calculations, respectively.
It is also worth noting that the values of the φ and ψ backbone angles in this complex, as computed with the POSSIM, are much closer to the resulting quantum mechanical values of these angles than their OPLS counterparts, with the average error of only 5.3° vs. 12.8°. This is so even though the POSSIM gives the lowest-energy monomer conformer (C7eq) ψ angle of only 34.4° vs. the quantum mechanical 88.1° and the OPLS 61.8°. We believe that this fact confirms that (i) the conformational energy surface is rather flat at that region, and so the precise location of the minimum is not entirely crucial; (ii) the POSSIM force field is robust and adequate in reproducing important binding geometries.
We have further investigated the alanine dipeptide – water binding properties by running calculations, in which the values of φ and ψ were kept the same as in the fully optimized quantum mechanical dimers in all the cases (quantum mechanical, OPLS and POSSIM monomers and also the OPLS and POSSIM dimers). The results are presented in Table 6. The structure B dimerization energy as computed with the POSSIM is slightly greater than the quantum mechanical one in this case (−12.2 vs. −11.7 kca/mol), otherwise the trends are the same as in the fully relaxed geometry optimizations. The average errors in the dimerization energies with the POSSIM and OPLS are 0.76 and 0.39 kcal/mol, respectively. The POSSIM and OPLS errors in the hydrogen bonding distances are 0.09 and 0.05Å. Interestingly, the improvement in geometry achieved by fixing the backbone angles is greater with the OPLS than it is with the POSSIM. Once again, we believe this indicates that, even though the C7eq conformational geometry is better reproduced with the OPLS, the more important binding properties are better assessed with the POSSIM force field.
Table 6.
Results of simulating alanine dipeptide complexes with water. φ and ψ of both dimers and monomers are fixed in the QM dimer positions. Energies are in kcal/mol, distances in Å, angles in degrees.
| property | Structure A | Structure B | ||||
|---|---|---|---|---|---|---|
| QM | OPLS | POSSIM | QM | OPLS | POSSIM | |
| Binding energy | −10.71 | −10.04 | −9.75 | −11.68 | −11.79 | −12.24 |
|
| ||||||
| R(O…O) | 2.83 | 2.81 | 2.82 | 2.83 | 2.75 | 2.74 |
| R(O…N) | 3.10 | 2.94 | 3.06 | 3.05 | 2.94 | 2.98 |
|
| ||||||
| φ, dimer | −83.4 | −83.4 | −83.4 | −84.3 | −84.3 | −84.3 |
| ψ, dimer | 90.3 | 90.3 | 90.3 | 131.2 | 131.2 | 131.2 |
|
| ||||||
| φ, monomer | −83.4 | −83.4 | −83.4 | −84.3 | −84.3 | −84.3 |
| ψ, monomer | 90.3 | 90.3 | 90.3 | 131.2 | 131.2 | 131.2 |
C. Gas-Phase and Hydrated Simulations of the Tridecaalanine Peptide (Ala-13)
We have carried out Monte Carlo simulations of the ala-13 in order to test the robustness of the POSSIM force field by assessing stability of this experimentally known α-helical peptide. While quantum mechanical gas-phase alanine dipeptide conformational energies and geometries are important in fitting, these simulations provided a direct comparison with the available experimental observations. In particular, we were assessing the general stability of the helix and the average values of the backbone φ and ψ angles. Figures 6 and 7 show graphs of the average values of these angles as a function of the simulation length (in millions of Monte Carlo configurations) for the OPLS force field, as well as with POSSIM, using both the tors.1 and tors.final torsional parameters. Each angle value represents averaging over the last 200,000 configurations before the indicated simulation length.
Figure 6.

Average φ angles in α-helix gas-phase simulations vs. the simulation length.
Figure 7.

Average ψ angles in α-helix gas-phase simulations vs. the simulation length.
The experimental values of the backbone φ and ψ in an α-helix are 296° and 319°, respectively, with a 7° uncertainty.16 In finding the average values of the backbone angles, we disregarded one residue on each end of the helix.
Two conclusions can be made from the presented results. First, the final version of the POSSIM, as well as the OPLS force field, yield better agreement with the experimental data than the POSSIM version with the tors.1 parameters. Second, the φ values are more stable than those of the angle ψ with all the force fields tested.
But one should keep in mind that the experimental data represent crystallographic results, and thus the thermal motion allowed in the Monte Carlo calculations can cause oscillations beyond the ±7° experimental lines. Overall, we can conclude that the gas-phase simulations confirm that the newly developed POSSIM force field is stable and robust. They reproduce the experimentally observed α-helix gas-phase stability. The stability of the simulated helixes can also be evaluated by studying the final structure of the system shown on Figures 8–10. One can see that, while the OPLS and POSSIM with tors.final produce a stable α-helix, the POSSIM/tors.1 helix denaturates. At the same time, the average φ and ψ angles in the tors.1 version of POSSIM are not extremely far from the experimental data, therefore the helicity of the structure is at least partially conserved.
Figure 8.

Structure of the ala-13 a-helix simulated with OPLS in gas-phase, after 19 × 106 Monte Carlo configurations.
Figure 10.

Structure of the ala-13 a-helix simulated with POSSIM, version tors.final in gas-phase, after 19 × 106 Monte Carlo configurations.
Average values of the φ and ψ angles as a function of the simulation length for the tridecaalanine (ala-13) peptide in water are shown on Figures 11 and 12. In this case, as can be expected, the stability of the both angles is greater, and the deviations are smaller. It should be noted that the angle φ tends to be too low compared to the experimental crystallographic values, while the angle ψ is somewhat too high, thus their sum stays roughly at the same spot as the experimental one (255° or −105°), and the α-helicity of the structure for all the force fields employed is good.
Figure 11.

Average φ angles in α-helix simulations in aqueous solution vs. the simulation length, in millions of Monte Carlo configurations.
Figure 12.

Average ψ angles in α-helix simulations is aqueous solution vs. the simulation length, in millions of Monte Carlo configurations.
Structures of the ala-13 peptide after 25 × 106 Monte Carlo configurations in water are given on Figures 13–15. Water molecules are not removed for clarity. It can be seen from the figures, in combination with the graphs and the table for the liquid-state simulations, that in this case (hydrated ala-13) all three force fields (OPLS and the two versions of POSSIM) perform adequately, and no denaturation of the tridecaalanine α-helix is observed.
Figure 13.

Structure of the ala-13 a-helix simulated with OPLS, in aqueous solution, after 25 × 106 Monte Carlo configurations. Water molecules are not shown for the sake of clarity.
Figure 15.

Structure of the ala-13 a-helix simulated with POSSIM, version tors.final, in aqueous solution, after 25 × 106 Monte Carlo configurations. Water molecules are removed for clarity.
IV. Conclusions
We have presented results of developing a fast polarizable POSSIM force field for alanine and protein backbones. The quantum mechanical data set used for fitting was streamlined and simplified as compared to the previous version of the complete polarizable force field for proteins, and a high degree of transferability of the potential energy parameters has been demonstrated.
We have included a previously unused step of calculating dipeptide dimerization energies with a water molecule as an additional proof of validity of the technique and the resulting force field. The POSSIM force field performs well in this test.
The torsional fitting procedure has been augmented by a new step, a direct optimization-type fitting of the torsional parameters to the quantum mechanical conformational energies and structures.
At the same time, we believe that quantum mechanical dipeptide conformers in themselves are not a sufficient tool in validation of a force field. One of the reasons for this assumption is that most of these conformers belong to parts of the total conformational space which are rarely found in experimentally known proteins. Therefore, we have included an additional step to further test the robustness of the POSSIM force field. We have simulated the tridecaalanine peptide (ala-13) in both gas-phase and aqueous solution with the Monte Carlo technique. This peptide is experimentally known to form an α-helix under these conditions. The POSSIM ala-13 (and the OPLS used for benchmarking) was found to maintain a stable α-helical conformation as well.
We conclude that the resulting polarizable POSSIM force field is adequately accurate and we will use this model for the alanine and protein backbones as the basis for further development of a complete polarizable POSSIM force field for proteins.
Supplementary Material
Figure 9.

Structure of the ala-13 a-helix simulated with POSSIM, version tors.1, in gas-phase, after 19 × 106 Monte Carlo configurations.
Figure 14.

Structure of the ala-13 a-helix simulated with POSSIM, version tors.1, in aqueous solution, after 25 × 106 Monte Carlo configurations. Water molecules are not shown for the sake of clarity.
Acknowledgments
This project was supported by Grant Number R01GM074624 from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. The authors express gratitude to Schrödinger, LLC for the Jaguar and Impact software.
Footnotes
Supporting Information Available
Tabulated values of φ and ψ angles of the α-helix in gas-phase and solution as a function of simulation length. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(1) See, for example, Caldwell JW, Kollman PA. J Am Chem Soc. 1995;117:4177–4178.Cieplak P, Caldwell J, Kollman P. J Comp Chem. 2001;22:1048–1057.Kaminski GA. J Phys Chem B. 2005;119:5884–5890. doi: 10.1021/jp050156r.Jiao D, Zhang JJ, Duke RE, Li GH, Schneiders MJ, Ren PY. J Comp Chem. 2009;30:1701–1711. doi: 10.1002/jcc.21268.Hernandez G, Anderson JS, LeMaster DM. Biochemistry. 2009;48:6482–6494. doi: 10.1021/bi900526z.Wang XY, Zhang JZH. Chem Phys Lett. 2011;501:508–512.
- 2.(a) MacDermaid CM, Kaminski GA. J Phys Chem B. 2007;111:9036–9044. doi: 10.1021/jp071284d. [DOI] [PubMed] [Google Scholar]; (b) Click TH, Kaminski GA. J Phys Chem B. 2009;113:7844–7850. doi: 10.1021/jp809412e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veluraja K, Margulis CJ. J Biomol Struct & Dynamics. 2005;23:101–111. doi: 10.1080/07391102.2005.10507051. [DOI] [PubMed] [Google Scholar]
- 4.(a) Ji C, Mei Y, Zhang JZH. Biophys J. 2008;95:1080–1088. doi: 10.1529/biophysj.108.131110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ji CG, Zhang JZH. J Phys Chem B. 2009;113:16059–16064. doi: 10.1021/jp907999e. [DOI] [PubMed] [Google Scholar]
- 5.For representative publications see: Rick SW, Stuart SJ, Berne BJ. J Chem Phys. 1994;101:6141–6156.Liu YP, Kim K, Berne BJ, Friesner RA, Rick SW. J Chem Phys. 1998;108:4739–4755.Ramon JMH, Rios MA. Chem Phys. 1999;250:155–169.Gonzalez MA, Enciso E, Bermejo FJ, Bee M. J Chem Phys. 1999;110:8045–8059.Soetens JC, Jansen G, Millot C. Mol Phys. 1999;96:1003–1012.Dang LX. J Chem Phys. 2000;113:266–273.Chen B, Xing JH, Siepmann JI. J Phys Chem B. 2000;104:2391–2401.Jedlovszky P, Vallauri R. J Chem Phys. 2001;115:3750–3762.Ribeiro MCC. Phys Rev B. 2001;6309:4205.Rinker S, Gunsteren WF. J Chem Phys. 2011;134:084110. doi: 10.1063/1.3553378.Jiang W, Hardy DJ, Phillips JC, MacKerrel AD, Schulten K, Roux B. J Phys Chem Lett. 2011;2:87–92. doi: 10.1021/jz101461d.
- 6.Kaminski GA, Zhou R, Friesner RA. J Comp Chem. 2003;24:267–276. doi: 10.1002/jcc.10170. [DOI] [PubMed] [Google Scholar]
- 7.Kaminski GA, Stern HA, Berne BJ, Friesner RA, Cao YX, Murphy RB, Zhou R, Halgren T. J Comput Chem. 2002;23:1515–1531. doi: 10.1002/jcc.10125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 9.(a) Marqusee S, Robbins VH, Baldwin RL. Proc Natl Acad Sci USA. 1989;86:5286–5290. doi: 10.1073/pnas.86.14.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Scholtz JM, York EJ, Steward JM, Baldwin RL. J Am Chem Soc. 1991;113:5102–5104. [Google Scholar]; (c) Scholtz JM, Baldwin RL. Annu Rev Biophys Biomol Struct. 1992;21:95–119. doi: 10.1146/annurev.bb.21.060192.000523. [DOI] [PubMed] [Google Scholar]; (d) Kinnear BS, Kaleta DT, Kohtani M, Hudgins RR, Jarrold MF. J Am Chem Soc. 2000;122:9243–9256. [Google Scholar]; (e) Wei Y, Nader W, Hansmann UHE. J Chem Phys. 2007;126:204307. doi: 10.1063/1.2734967. [DOI] [PubMed] [Google Scholar]
- 10.Kaminski GA, Ponomarev SY, Liu AB. J Chem Theory Comput. 2009;5:2935–2943. doi: 10.1021/ct900409p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Jaguar v3.5. Schrödinger, Inc; Portland, OR: 1998. [Google Scholar]; (b) Jaguar v4.2. Schrödinger, Inc; Portland, OR: 2000. [Google Scholar]
- 12.Kaminski GA, Maple JR, Murphy RB, Braden D, Friesner RA. J Chem Theory Comput. 2005;1:248–254. doi: 10.1021/ct049880o. [DOI] [PubMed] [Google Scholar]
- 13.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 14.Takekiyo T, Imai T, Kato M, Taniguchi Y. Biopolymers. 2004;73:283–290. doi: 10.1002/bip.10548. [DOI] [PubMed] [Google Scholar]
- 15.Distasio RA, Steele RP, Rhee YM, Shao Y, Head-Gordon M. J Comput Chem. 2007;28:839–856. doi: 10.1002/jcc.20604. [DOI] [PubMed] [Google Scholar]
- 16.Berndt Kurt D. [accessed on December 10, 2010];Protein Secondary Structure. http://www.cryst.bbk.ac.uk/PPS2/course/section8/ss-960531_6.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.