

Abstract
In a previous 52-week trial, treatment with alglucosidase alfa markedly improved cardiomyopathy, ventilatory function, and overall survival among 18 children <7 months old with infantile-onset Pompe disease. Sixteen of the 18 patients enrolled in an extension study, where they continued to receive alglucosidase alfa at either 20 mg/kg biweekly (n=8) or 40 mg/kg biweekly (n=8), for up to a total of 3 years. These children continued to exhibit the benefits of alglucosidase alfa at the age of 36 months. Cox regression analyses showed that over the entire study period, alglucosidase alfa treatment reduced the risk of death by 95%, reduced the risk of invasive ventilation or death by 91%, and reduced the risk of any type of ventilation or death by 87%, as compared to an untreated historical control group. Cardiomyopathy continued to improve and 11 patients learned and sustained substantial motor skills. No significant differences in either safety or efficacy parameters were observed between the 20 mg/kg and 40 mg/kg biweekly doses. Overall, long-term alglucosidase alfa treatment markedly extended survival as well as ventilation-free survival, and improved cardiomyopathy.
Pompe disease is characterized by a deficiency of acid α-glucosidase (GAA). The GAA enzyme degrades lysosomal glycogen, and insufficient GAA activity causes glycogen to accumulate in various tissues. Accumulation of lysosomal glycogen in cardiac muscle and skeletal muscle causes progressive cardiomyopathy and generalized muscle weakness and hypotonia, resulting in severely delayed motor development and cardio-respiratory failure (1–3).
The presentation and course of Pompe disease can vary widely. Patients may exhibit signs and symptoms as early as prenatally or in the first few days of life or as late as the sixth decade. The most severe and rapidly progressive form is designated as infantile-onset Pompe disease, in which patients generally develop significant clinical manifestations within the first months of life. If left untreated, most children with infantile-onset Pompe disease succumb to cardiac and/or respiratory failure before the age of 1 year (1,4–6).
Enzyme replacement therapy with recombinant human alglucosidase alfa (rhGAA, MyozymeR) was approved for treating patients with Pompe disease in 2006. Previous studies demonstrated that enzyme replacement therapy changes the natural history of Pompe disease in infants and children (7–12). The largest of these studies evaluated the effects of alglucosidase alfa in 18 young infants with severe infantile-onset Pompe disease; these patients exhibited cardiomyopathy and profound deficiency of GAA activity at age <7 months (12). Fifty-two weeks of treatment with alglucosidase alfa markedly improved survival, respiratory function, cardiomyopathy and, among a subset of patients, motor function, as compared to an untreated historical control group (12). This report describes the long-term effects of continued alglucosidase alfa treatment (up to 3 years) in the same cohort. These data represent a substantial addition to the literature, as long-term enzyme replacement outcome data were previously available for only 4 cases of infantile-onset Pompe disease (9).
METHODS
Study design and treatment
The design of the 52-week open-label study has been described in detail elsewhere (12). Briefly, patients must have had documented symptoms of infantile-onset Pompe disease and been <7 months old at enrollment. Patients with respiratory insufficiency, heart failure or any prior replacement therapy with GAA were excluded. A closely matched untreated historical control group of 61 infants who had infantile-onset Pompe disease was used as a comparator population (12).
Parents or guardians gave written informed consent for patients’ participation and consent could be withdrawn at any time. Local Institutional Review Boards or Independent Ethics Committees approved protocols and consent forms at each of the primary and extension study sites.
Alglucosidase alfa was supplied by Genzyme Corporation (Cambridge, MA). Eligible patients were randomized in a 1:1 ratio to receive intravenous infusions of alglucosidase alfa at either 20 mg/kg or 40 mg/kg every other week (12). Fifty-two weeks after the last patient was randomized to treatment, patients were eligible to participate in an extension study, where they continued to receive alglucosidase alfa at the same dose to which they were originally assigned. Patients were treated in 52-week modules until the study was terminated June 15, 2006, shortly after Myozyme® was approved for commercial use. Because patients were enrolled over a period of 1 year and survived to different ages, the duration of data collection varied among patients, ranging from 60 weeks to 150 weeks.
Clinical Assessments Of Safety
Safety data were analyzed for the duration of treatment. Patients were observed and vital signs were monitored during each infusion and for 2 hr afterward. Safety assessments included evaluating blood and urine chemistry, vital signs, 12-lead electrocardiograms, and the incidence and nature of adverse events, including infusion associated reactions (IARs). The development of anti-rhGAA IgG antibodies was assessed in serum samples taken prior to each infusion every 4 weeks from baseline through week 24 and subsequently at 12-week intervals, as described previously (11).
Clinical Assessments Of Efficacy
Survival and ventilator use (invasive and non-invasive) were analyzed at ages 24 and 36 months; all other efficacy data were analyzed with respect to changes from baseline to the final assessment. Left ventricular mass (LVM) and other cardiac parameters were evaluated by echocardiogram at 12-week intervals. Echocardiograms were centrally read by a pediatric cardiologist who was blinded to dose, patient, and time point. Motor development was centrally evaluated using the Alberta Infant Motor Scale (AIMS) (13) until patients reached the maximum total AIMS score to allow for continuous monitoring on this scale.
Anti-rhGAA antibody and Inhibitory Antibody testing
The presence of anti-rhGAA IgG antibodies was assessed using enzyme-linked immunosorbent assays and confirmed using radioimmunoprecipitation, as described previously (11). Serum samples from seropositive patients were retrospectively analyzed for inhibition of rhGAA enzymatic activity and uptake in vitro (14).
Statistical Methods
The Kaplan-Meier method was used to calculate the proportion of patients with an event (i.e., death or ventilator use) (15). The Cox regression analysis was used to compare the risk of an event (i.e., death or ventilator use) in alglucosidase alfa-treated patients to the untreated historical control group (16). Statistical analyses were performed using the SASR statistical software system (version 8).
Eighteen infants participated in the initial open-label trial (12). As shown in Figure 1, 16 surviving patients enrolled in the extension study. One patient, a male in the 20 mg/kg dose group, died at the age of 19.8 months, after receiving alglucosidase alfa for 61 weeks, but before the extension study began. A female patient in the 40 mg/kg dose group completed the initial open-label study, but did not enroll in the extension study after becoming ventilated; the patient continued therapy in an international Expanded Access Program until her death at age 32 months. Although these 2 patients did not enroll in the extension study, available data from baseline to their last assessment are included in these analyses, for completeness.
Figure 1.
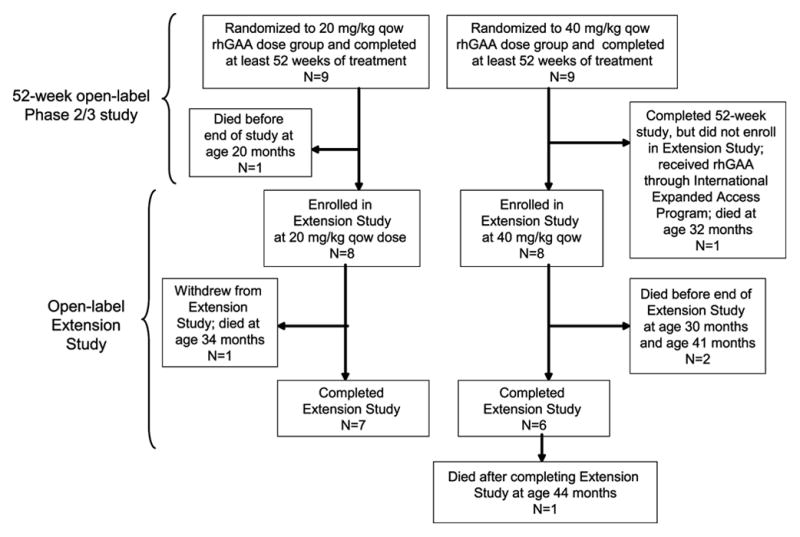
Disposition of patients in 20 mg/kg qow and 40 mg/kg qow groups.
Results
The duration of alglucosidase alfa treatment ranged from 1.1 years to 3.0 years, with a median duration of treatment of 2.3 years. All 18 patients who participated in the initial open-label study survived to the age of 18 months (12). As shown in Figure 1, 5 of the 18 patients died before the end of the extension study. Patient C discontinued from the initial study, but was receiving alglucosidase alfa through an international Expanded Access Program at the time of death. Patient D died during the initial study, and Patients A and Q died during the extension study. Patient L withdrew from the extension study after a decline in clinical status, at the request of family. The survival analyses do not include the death of a sixth patient, Patient P, who was reported to have died approximately 1 year after completing the extension study.
Survival status for individual patients is shown in Table 1. Seventeen of the 18 patients reached the age of 24 months, for a survival rate of 94.4% 95% CI: 83.9% to 100%). The survival rate at age 36 months was 72% (95% CI: 47.9% to 96.0%), including seven of the 18 patients who reached age 36 months by the end of the study. Four patients died before age 36 months, including Patient D at 19.8 months, Patient C at 31.9 months, Patient L at 34.3 months, and Patient Q at 30.1 months. The other 7 patients were right-censored from this analysis because they had not reached age 36 months by the end of the study, although they were alive at that time, at ages ranging from 27.1 to 35.7 months. In contrast, only 1 of 61 patients in the untreated historical control group survived to the ages of 24 and 36 months (1.9%; 95% CI: 0% to 5.5%). Survival rate by age estimated by the Kaplan-Meier method is shown in Figure 2A.
Table 1.
Survival, Ventilation Status, and Motor Function in Individual Patients
| Patient ID | Dose | Survival Status | Age at End of Study (or death) in months | Ventilation Status | Motor Function at Final AIMS Assessment
|
||
|---|---|---|---|---|---|---|---|
| Age (months) | Study Week | Motor Function Status | |||||
| A | 40 | Died | 40.7 | Invasive | 35.7 | Week 130 | Minimal gains |
| B | 20 | Alive | 41.5 | No vent. | 29.5 | Week 104 | Walking |
| C *, § | 40 | Died | 31.9 | Invasive | 26.4 | Week 90 | Minimal gains |
| D† | 20 | Died | 19.8 | Invasive | 17.8 | Week 52 | Minimal gains |
| E | 20 | Alive | 39.4 | Invasive | 37.6 | Week 142 | Minimal gains |
| F | 40 | Alive | 39.4 | No vent. | 21.8 | Week 64 | Walking |
| G | 40 | Alive | 35.7 | No vent. | 16.2 | Week 52 | Walking |
| H | 20 | Alive | 36.7 | No vent. | 35.7 | Week 130 | Walking |
| I | 20 | Alive | 36.1 | No vent. | 35.7 | Week 130 | Walking |
| J | 40 | Alive | 38.1 | No vent. | 36.7 | Week 130 | Sitting |
| K | 20 | Alive | 34.1 | No vent. | 33.0 | Week 116 | Sitting |
| L § | 40 | Died | 34.3 | Invasive | 25.1 | Week 104 | Minimal gains |
| M § | 20 | Alive | 30.0 | Invasive | 33.0 | Week 116 | Sitting |
| N | 40 | Alive | 28.8 | No vent. | 16.0 | Week 34 | Walking |
| O | 20 | Alive | 32.8 | No vent. | 32.7 | Week 116 | Walking |
| P ‡ | 40 | Alive | 32.0 | Invasive | 30.6 | Week 104 | Minimal Gains |
| Q | 40 | Died | 30.1 | Invasive | 24.2 | Week 78 | Sitting |
| R | 20 | Alive | 27.1 | Invasive | 25.6 | Week 104 | Minimal Gains |
Patient C completed the initial study, but did not enroll in the extension study. This patient continued to receive alglucosidase alfa through an international expanded access program.
Patient D died during the initial 52-week study.
Patients C, L, and M each exhibited some level of walking skills at earlier assessments. Each of these patients experienced declines in respiratory function that led to invasive ventilation and the subsequent loss of walking skills.
Patient P was alive at the end of the extension study, but died in May 2007, approximately 1 year after completing the study, at age 44 months.
Vent. = ventilation
Figure 2.

Kaplan-Meier analyses of survival, survival free of invasive ventilation, and survival free of any ventilation. In each panel, thick solid lines show the Kaplan-Meier estimates for the treated patient group; thin solid lines show those for the historical control group, with 95% confidence intervals given by the corresponding dashed lines. Solid circles indicate right-censored observations. (A) Kaplan-Meier estimate of time from date of birth to death. Seven patients were right-censored from this analysis because they had not reached age 36 months by the end of the study, although they remained alive at that time. (B) Kaplan-Meier estimate of time from birth to invasive ventilator use or death. Four patients were right-censored from this analysis because they had not reached age 36 months by the end of the study, although they remained free of invasive ventilation that time. (C) Kaplan-Meier estimate of time from birth to any ventilator use or death. Four patients were right-censored from this analysis because they had not reached age 36 months by the end of the study, although they remained free of any ventilation that time. *Asterisk indicates that 1 patient from the historical control group remained alive at 36 months of age; this patient died at age 44 months.
Ventilation status for individual patients at the end of the study or at the time of their death is shown in Table 1. Three of the 18 patients (Patients A, P, and R) required invasive ventilator support at age 18 months (12). Six of the 18 patients (the 3 noted above plus Patients C, E, and L) required invasive ventilator support at age 24 months or at the time of death. A total of 9 patients (the 6 noted above plus Patients D, M, and Q) required invasive ventilator support at age 36 months, at the time of death, or at the end of the study. The age at which patients became dependent on invasive ventilator support ranged from 9.1 months to 29.6 months. Overall, 12 patients remained free of invasive ventilation at the age of 24 months, and 9 patients remained free of invasive ventilation at age 36 months, including 4 patients who were right-censored from this analysis because they had not reached age 36 months by the end of the study, though they remained free of invasive ventilatory support at that time. The Kaplan-Meier invasive ventilation-free survival rate (the primary efficacy endpoint) was 66.7% (95% CI: 44.9% to 88.4%) at age 24 months and 49.4% (95% CI: 26.0% to 72.8%) at age 36 months. This is far greater than the overall survival rate of untreated patients (with or without ventilation), as shown in Figure 2B.
The proportion of patients who were alive and free of any ventilator support (invasive or non-invasive) was evaluated as a secondary endpoint. No additional patients required non-invasive ventilatory support at the ages of 24 months or 36 months or at the time of death. Thus, the Kaplan-Meier ventilation-free survival rate was the same as the invasive ventilation-free survival rate: 66.7% (95% CI: 44.9% to 88.4%) at age 24 months and 49.4% (95% CI: 26.0% to 72.8%) at age 36 months; again, this is much higher than the overall survival rate of untreated patients (with or without ventilation), as shown in Figure 2C.
Cox regression analyses demonstrated that alglucosidase alfa reduced the risk of death by 95% (hazard ratio of 0.05, 95% CI: 0.02 to 0.14); the risk of invasive ventilation or death by 91% (hazard ratio of 0.09, 95% CI: 0.04 to 0.22); and the risk of any type of ventilation or death by 87% (hazard ratio of 0.13 95% CI: 0.06 to 0.29). No difference in the effects of alglucosidase alfa on survival or ventilator-free survival was observed between the 2 dose groups.
Cardiomyopathy was defined as an LVM value >2 standard deviations from the normal mean. Mean LVM Z-scores progressively decreased from 7.1 to 3.3 over the first 52 weeks of alglucosidase alfa treatment (12). During the extension study, mean LVM Z-scores continued to gradually decrease. At the end of the extension study, mean LVM Z-scores remained stable at slightly above the upper limit of the normal range (2.0), as shown in Figure 3. LVM Z-scores for individual patients are shown in Table 2. Among individual patients, 7 had LVM Z-scores within the normal range at the time of the final assessment (Patients B, G, H, I, J, N, and O) and all but 1 of the patients (Patient L) showed reductions in LVM of at least 1 Z-score from the first assessment to the time of the final assessment (Table 2). Patient L had exhibited more robust improvement at 52 weeks (12), but worsening cardiomyopathy thereafter. This patient eventually withdrew from the study after progression to invasive ventilation. No difference in the effects of alglucosidase alfa on cardiomyopathy was observed between the 2 dose groups.
Figure 3.
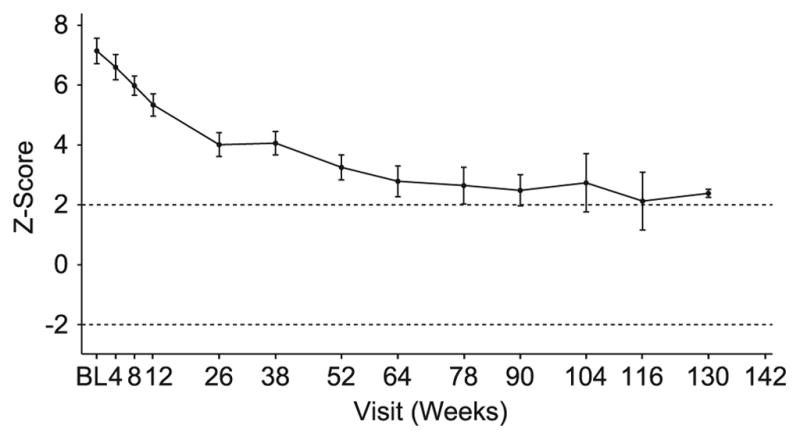
Mean left ventricular mass Z-scores of treated patients from baseline to week 52; vertical bars represent standard error.
Table 2.
Effect of Alglucosidase Alfa Treatment on Left Ventricular Mass in Individual Patients
| Patient ID | Dose | LVM Z-score at Baseline* | LVM Z-score at Final Assessment | Change in LVM Z-score from Baseline | Study Week of Final LVM Assessment |
|---|---|---|---|---|---|
| A | 40 | 8.39 | 2.06 | −6.33 | Week 124 |
| B | 20 | 3.03* | 1.93 | −1.10 | Week 98 |
| C † | 40 | 7.04* | 2.94 | −4.10 | Week 26 |
| D † | 20 | 9.27 | 4.36 | −4.91 | Week 52 |
| E | 20 | 6.73 | 2.52 | −4.21 | Week 130 |
| F | 40 | 8.99 | 2.25 | −6.74 | Week 130 |
| G | 40 | 8.00 | 1.81 | −6.19 | Week 116 |
| H | 20 | 7.88 | 0.47 | −7.41 | Week 116 |
| I | 20 | 6.84 | 1.46 | −5.38 | Week 64 |
| J | 40 | 6.60 | 0.76 | −5.84 | Week 116 |
| K | 20 | 7.77 | 2.19 | −5.58 | Week 104 |
| L | 40 | 6.50 | 5.84 | −0.66 | Week 116 |
| M | 20 | 6.33 | 2.91 | −3.42 | Week 84 |
| N | 40 | 2.62 | 1.09 | −1.53 | Week 104 |
| O | 20 | 5.43 | 0.76 | −4.67 | Week 104 |
| P | 40 | 7.57 | 4.57 | −3.00 | Week 90 |
| Q | 40 | 7.55* | 2.47 | −5.08 | Week 90 |
| R | 20 | 8.23 | 7.02 | −1.21 | Week 104 |
Baseline LVM values were unavailable for Patients B, C, and Q. For these patients, the first available LVM values are shown (from Study Week 4 or Week 8).
Patients C and D did not enroll in the extension study (see Table 1).
Shaded rows indicate patients who had died prior to database lock (November 30, 2006), as shown in Table 1.
LVM Z-score values in bold are within normal limits (LVM values within 2 standard deviations from the normal mean (i.e., Z-score -2 to +2) [12].
LVM = left ventricular mass
As shown in Table 1, 7 of the 18 patients (Patients B, F, G, H, I, N, and O) gained motor skills and were walking by the time of their final AIMS assessment. An additional 4 patients (Patients J, K, M, and Q) gained motor skills and were sitting independently at that time. The remaining 7 patients (Patients A, C, D, E, L, P, and R) made minimal motor gains, or were unable to achieve sustained motor gains and had very limited gross motor skills. Each of these 7 patients had become dependent on invasive ventilation at the end of the extension study or at the time of their deaths. All but 1 of the 7 patients who had learned to walk by Study Week 52 (12) retained this skill by the end of the study. Patient C, who was walking at Study Week 52 (12), became dependent on invasive ventilation at age 24 months (Study Week 75) and lost the ability to walk. Patients L and M had each gained standing skills by week 52 (12), which were subsequently lost. No difference in motor function was observed between patients in the 2 dose groups.
Eleven of the 18 patients experienced 224 IARs, defined as any treatment-related AE that occurred during an infusion or within 2 hr after the infusion. All IARs were mild or moderate in intensity; none were severe. The most common IARs were urticaria (47 events), fever (27 events), and decreased oxygen saturation (24 events). IARs were typically managed by slowing or interrupting infusions and all 11 patients who experienced IARs recovered without sequelae.
Overall, more IARs were reported for patients in the 40 mg/kg dose group. Five patients in the 20 mg/kg dose group experienced 47 events (21% of all IARs) and 6 patients in the 40 mg/kg dose group experienced 177 events (79% of all IARs). Most of these events (65%) were experienced by 2 patients, both in the 40 mg/kg dose group: Patient C (43 events) and Patient Q (102 events), as shown in Table 3. These patients experienced recurrent episodes of urticaria, flushing, fever, agitation, rash, tachypnoea, cough, and vomiting at multiple infusions. The events were managed by slowing the infusion rate of alglucosidase alfa or temporarily stopping the infusions and administering antihistamines, antipyretics, and/or corticosteroids. As shown in Table 3, both of these patients also exhibited sustained high anti-rhGAA IgG titer levels.
Table 3.
Summary of Immunological Characteristics and Infusion-Associated Reactions
| Patient ID | Dose Group | Number of Infusions Received | Maximum anti- rhGAA IgG Titer Level Reported | Anti-rhGAA IgG Titer Level at Final Study Visit | Number of IARs | CRIM Status | Inhibitory Antibody Activity >20% in Serum |
|---|---|---|---|---|---|---|---|
| A | 40 | 76 | 204,800 | 51,200 | 4 | + | No |
| B | 20 | 74 | 12,800 | 200 | 2 | + | No |
| C * | 40 | 41 | 409,600 | 409,600 | 43 | − | Yes |
| D * | 20 | 31 | 6,400 | 200 | 4 | + | No |
| E | 20 | 71 | 1,600 | 200 | 5 | + | No |
| F | 40 | 69 | 3,200 | 400 | 0 | + | No |
| G | 40 | 67 | 400 | 100 | 0 | + | No |
| H | 20 | 65 | 800 | 400 | 0 | + | No |
| I | 20 | 65 | 800 | 400 | 14 | + | No |
| J | 40 | 63 | 25,600 | 1600 | 4 | + | No |
| K | 20 | 58 | N/A | N/A | 0 | + | No |
| L | 40 | 58 | 3,276,800 | 1,638,400 | 14 | − | Yes |
| M | 20 | 62 | 12,800 | 3,200 | 0 | + | No |
| N | 40 | 59 | N/A | N/A | 0 | + | No |
| O | 20 | 53 | 400 | 100 | 0 | + | No |
| P | 40 | 53 | 409,600 | 102,400 | 10 | − | Yes |
| Q | 40 | 53 | 409,600 | 51,200 | 102 | + | No |
| R | 20 | 53 | 102,400 | 25,600 † | 22 | − | No |
Patients C and D did not enroll in the extension study (see Table 1).
No IgG titer level was available for Patient R at the final study visit; the value shown reflects titer levels measured at Week 38.
N/A, not applicable; Patients K and N remained seronegative at all assessments.
Note that the number of IARs represents individual AEs and may include the same term multiple times. Patients C and Q experienced recurrent IARs at multiple infusions.
Shaded rows indicate patients who had died prior to database lock (November 30, 2006), as shown in Table 1.
Four of the 18 patients (Patients C, L, P, and R) did not exhibit any detectable endogenous full-length or partial GAA protein product, designated as cross-reacting immunologic material (CRIM) (12). Serum samples from 3 of the 4 CRIM-negative patients (Patients C, L, and P) exhibited in vitro inhibitory antibody activity, as determined by analysis of enzyme activity or rhGAA uptake assays. Overall, patients in the 40 mg/kg dose group tended to have higher anti-rhGAA IgG titers (Table 3). However, due to the small number of CRIM-negative patients in this study and the fact that 3 of the 4 CRIM-negative patients (Patients C, L, and P) were randomized to the 40 mg/kg dose group, statistical analyses regarding the relationship between dose, immune response, and IARs were not feasible.
Discussion
This study includes the largest number of patients and represents one of the longest trials of rhGAA enzyme replacement therapy to date for the most rapidly progressing form of infantile-onset Pompe disease. In the absence of treatment, the life expectancy of children with classical infantile-onset Pompe disease is usually less than 1 year, with death typically caused by cardiac and/or respiratory failure (1,5,6). A report based on the first 52 weeks of treatment in this cohort demonstrated that alglucosidase alfa treatment significantly improved survival, respiratory function, cardiomyopathy and, in a subset of patients, motor function, up to the age of 18 months (12). This report describes the status of these patients through up to 3 years of alglucosidase alfa treatment. Seventeen of the 18 patients survived to age 24 months, whereas only 1 of 61 untreated patients in the historical control group survived to this age. Over the course of the study, alglucosidase alfa treatment markedly improved overall survival, as well as ventilator-free and invasive ventilator-free survival. Cox regression analyses demonstrated that alglucosidase alfa reduced the risk of death by 95%, the risk of invasive ventilation or death by 91%, and the risk of any type of ventilation or death by 87%. These were conservative comparisons, as patients’ ventilator-free survival (invasive or any ventilation) was compared to the overall survival (without regard to ventilation status) in the historical untreated cohort.
Deficiency of GAA leads to extensive accumulation of glycogen in cardiac muscle. Untreated patients exhibit cardiomyopathy and frequently succumb to cardiorespiratory failure (1,5,6). In contrast, 17 of the 18 treated patients in this study exhibited substantial reductions in LVM from baseline to the final assessment and 7 patients had LVM values within normal limits at that time. Three of the 4 CRIM-negative patients exhibited less improvement in LVM (LVM Z-score >4 at their final assessment), whereas only 1 of the 14 CRIM-positive patients had LVM Z-scores >4 at their final assessment. Overall, long-term alglucosidase alfa treatment results in sustained improvement in cardiomyopathy. This likely plays an important role in the extended survival observed in response to alglucosidase alfa treatment, regardless of CRIM or ventilatory status.
Accumulation of glycogen in skeletal muscle is associated with severe muscle weakness and hypotonia in patients with Pompe disease (1). In infants, this generally prevents the acquisition of key gross motor skills normally exhibited by children, such as rolling, independent sitting, and walking. Eleven patients exhibited a steady acquisition of motor skills, including 7 who sustained the ability to walk and 4 who were sitting independently at their final assessment. This represents a major departure from the natural progression of infantile-onset Pompe disease, where children seldom acquire significant gross motor skills. However, 7 children had not acquired substantial gross motor skills at the end of the study or lost significant motor skills acquired during treatment, such as ambulation. Each of these 7 patients also required invasive ventilation at the end of the study or at the time of their deaths, suggesting that disease progression affects both respiratory and skeletal muscle function. Interestingly, each of the 7 patients who had LVM Z-scores within the normal range at the time of the final assessment also exhibited sustained gains in gross motor function over the course of the study; 4 were walking and 3 had learned to sit independently.
No differences were observed between the 2 dose groups in terms of the effects of long-term alglucosidase alfa treatment on the proportion of patients with prolonged survival, ventilation-free survival, or degree of improvement in cardiomyopathy or motor function.
Therapeutic recombinant proteins frequently induce an immune response, the extent of which varies between individuals. Patients in the 40 mg/kg dose group tended to develop higher anti-rhGAA IgG titers than patients in the 20 mg /kg dose group. However, 3 of the 4 CRIM-negative patients, all of whom exhibited high anti-rhGAA IgG titers, were randomized to the 40 mg/kg dose group. Given the small number of patients in this study and the various confounding factors involved, it is not clear whether a higher dose is more immunogenic. However, this and previous published reports contribute to a growing body of evidence suggesting that CRIM-negative patients, who produce little or no endogenous GAA, develop a stronger immunologic response against alglucosidase alfa than CRIM-positive patients, who produce some form of endogenous GAA protein (7,11,12,17,18). Three of the 4 CRIM-negative patients also exhibited in vitro inhibitory antibody activity, which could impact the long-term efficacy of alglucosidase alfa. Indeed, each of these 3 patients showed signs of clinical decline during treatment, became dependent on invasive ventilation, and eventually died; Patient C died at age 31.9 months, Patient L died at 34.3 months, and Patient P died after completing the study, at age 44.4 months. Thus, there is a relationship between CRIM status, high antibody titers, inhibitory antibody activity, and long-term clinical outcome in Pompe disease.
In various clinical trials and expanded access programs, approximately 14% of patients treated with alglucosidase alfa (38 of 280 patients) developed allergic reactions involving at least 2 of the following 3 body systems: cutaneous, respiratory or cardiovascular (19). The 2 patients who experienced the highest number of IARs in this study (Patients C and Q) also exhibited signs and/or symptoms suggestive of allergic reactions. Patient C, who was CRIM-negative, experienced urticaria, cough, and serious rales and Patient Q, who was CRIM-positive, experienced recurrent events of hypotension, rash, cough, and urticaria on multiple occasions. Both patients were receiving the 40 mg/kg dose of alglucosidase alfa and both patients had high antibody titers. It is possible that some IARs experienced by these 2 patients could be IgG-mediated reactions, and may have occurred due to development of IgG antibodies that bind to complement and release specific mediators from complement activation. IARs were generally dose-related or infusion rate-related. Each patient tested negative for IgE and positive for complement on multiple occasions, with serum tryptase levels that were slightly elevated (Patient C) or within the normal range (Patient Q) (data not shown). Further studies of more patients with long-term data will be necessary to delineate possible relationships between dose, CRIM status, antibody titers, and IARs.
Taken together, these findings show that the beneficial effects of alglucosidase alfa in children with infantile-onset Pompe disease are sustained after prolonged treatment. All 18 patients who enrolled in the initial study survived significantly longer and with fewer ventilation events than untreated historical control patients and improvements in cardiomyopathy were generally sustained over the course of the extension study. However, morbidity and mortality remain substantial, with a 28% mortality rate and a 51% invasive ventilation rate at age 36 months. The effect of treatment on gross motor function was somewhat variable. Eleven patients exhibited clinically meaningful gains and 7 patients either did not acquire or did not sustain substantial gains in motor function. Various factors, including invasive ventilation, CRIM status, and patients’ immunological response to alglucosidase alfa may have contributed to the poorer motor skills exhibited by these children. Interestingly, each of the 7 patients who were walking at the end of the study exhibited little or no cardiomyopathy and relatively low IgG antibody titers at the final visit.
Increasing evidence suggests that CRIM-negative patients are at considerable risk of developing a high, sustained immunological response that may reduce the clinical benefit of alglucosidase alfa and worsen the long-term outcome in this subpopulation of patients (11,12,18). However, the favorable risk-to-benefit ratio suggests that infants with rapidly progressive Pompe disease should be treated with alglucosidase alfa. Immune modulation strategies to control the development of antibodies during long-term administration of alglucosidase alfa could be a viable option in high risk, CRIM-negative patients (18,20).
The young ages at which these patients began receiving alglucosidase alfa and the onset of therapy at early stages of disease progression (i.e. prior to ventilation or cardiac failure) may have contributed to the robust improvements in survival, cardiac, and motor function observed. Findings from this and other published reports with shorter follow-up periods demonstrate that alglucosidase alfa is an effective treatment for most patients with infantile-onset Pompe disease. However, while this treatment provides dramatic improvement, it is clearly not a cure; further investigation into the profound individual variability in response to treatment is needed.
Acknowledgments
This study was supported by Genzyme Corporation and grant number RR024128 from the Duke Clinical Research Unit Program, National Center for Research Resources, National Institutes of Health.
We would like to thank the GCRC staff at Duke University for the excellent care provided to patients at the Duke site. The authors are especially grateful to the patients, who participated in this clinical study, and their families.
ABBREVIATIONS
- AIMS
Alberta Infant Motor Scale
- CRIM
cross-reacting immunologic material
- GAA
acid α-glucosidase
- IAR
infusion-associated reactions
- LVM
left ventricular mass
- rhGAA
recombinant human acid α-glucosidase
Footnotes
Publisher's Disclaimer: Pediatric Research Articles Ahead of Print contains articles in unedited manuscript form that have been peer-reviewed and accepted for publication. As a service to our readers, we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting and review of the resulting proof before it is published in its final definitive form. Please note that during the production process errors may be discovered, which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hirschhorn R, Reuser AJ. Glycogen storage disease type II: acid α-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8. McGraw Hill; New York: 2001. pp. 3389–3420. [Google Scholar]
- 2.Koeberl DD, Kishnani PS, Chen YT. Glycogen storage disease types I and II: treatment updates. J Inherit Metab Dis. 2007;30:159–164. doi: 10.1007/s10545-007-0519-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kishnani PS, Howell RR. Pompe disease in infants and children. J Pediatr. 2004;144:S35–S43. doi: 10.1016/j.jpeds.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 4.Chen YT, Amalfitano A. Towards a molecular therapy for glycogen storage disease type II (Pompe disease) Mol Med Today. 2000;6:245–251. doi: 10.1016/s1357-4310(00)01694-4. [DOI] [PubMed] [Google Scholar]
- 5.van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, Smit GP, Poll-The BT, Bakker HD, Loonen MC, de Klerk JB, Reuser AJ, van der Ploeg AT. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- 6.Kishnani PS, Hwu WL, Mandel H, Nicolino M, Yong F, Corzo D Infantile-Onset Pompe Disease Natural History Study Group. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–676. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 7.Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA, 3rd, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT. Recombinant human acid α-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–138. doi: 10.109700125817-200103000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Van den Hout JM, Reuser AJ, de Klerk JB, Arts WF, Smeitink JA, Van der Ploeg AT. Enzyme therapy for Pompe disease with recombinant human alpha-glucosidase from rabbit milk. J Inherit Metab Dis. 2001;24:266–274. doi: 10.1023/a:1010383421286. [DOI] [PubMed] [Google Scholar]
- 9.Van den Hout JM, Kamphoven JH, Winkel LP, Arts WF, De Klerk JB, Loonen MC, Vulto AG, Cromme-Dijkhuis A, Weisglas-Kuperus N, Hop W, Van Hirtum H, Van Diggelen OP, Boer M, Kroos MA, Van Doorn PA, Van der Voort E, Sibbles B, Van Corven EJ, Brakenhoff JP, Van Hove J, Smeitink JA, de Jong G, Reuser AJ, Van der Ploeg AT. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. 2004;113:e448–e457. doi: 10.1542/peds.113.5.e448. [DOI] [PubMed] [Google Scholar]
- 10.Klinge L, Straub V, Neudorf U, Schaper J, Bosbach T, Görlinger K, Wallot M, Richards S, Voit T. Safety and efficacy of recombinant acid alpha-glucosidase (rhGAA) in patients with classical infantile Pompe disease: results of a phase II clinical trial. Neuromuscul Disord. 2005;15:24–31. doi: 10.1016/j.nmd.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Kishnani PS, Nicolino M, Voit T, Rogers RC, Tsai AC, Waterson J, Herman GE, Amalfitano A, Thurberg BL, Richards S, Davison M, Corzo D, Chen YT. Results from a phase II trial of Chinese hamster ovary cell-derived recombinant human acid α-glucosidase in infantile-onset Pompe disease. J Pediatr. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, Leslie N, Levine J, Spencer C, McDonald M, Li J, Dumontier J, Halberthal M, Chien YH, Hopkin R, Vijayaraghavan S, Gruskin D, Bartholomew D, van der Ploeg A, Clancy JP, Parini R, Morin G, Beck M, De la Gastine GS, Jokic M, Thurberg B, Richards S, Bali D, Davison M, Worden MA, Chen YT, Wraith JE. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease [published correction appears in Neurology 2008; 71:1748] Neurology. 2007;68:99–109. doi: 10.1212/01.wnl.0000251268.41188.04. [DOI] [PubMed] [Google Scholar]
- 13.Piper MC, Darrah J. Motor assessment of the developing infant. Philadelphia: W.B. Saunders; 1994. [Google Scholar]
- 14.Nicolino M, Byrne B, Wraith JE, Leslie N, Mandel H, Freyer DR, Arnold GL, Pivnick EK, Ottinger CJ, Robinson PH, Loo J-C, Smitka M, Jardine P, Tatò L, Chabrol B, McCandless S, Kimura S, Mehta L, Bali D, Skrinar A, Morgan C, Rangachari L, Corzo D, Kishnani PS. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med. 2009;11:210–219. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. JAm Stat Soc. 1958;53:457–481. [Google Scholar]
- 16.Cox DR. Regression models and life tables (with discussion) J Roy Statist Soc Ser B Methodological. 1972;34:187–220. [Google Scholar]
- 17.Mandel H, Gruber M, Goldsher D, Chistyakov A, Kaplan B, Zaaroor M, Hafner H. Longer survival by enzyme replacement therapy unmasks the underrecognition of otoneurologic involvement in infantile-onset Pompe disease. Clin Ther. 2007;29:S109–S110. [Google Scholar]
- 18.Sun B, Bird A, Young SP, Kishnani PS, Chen YT, Koeberl DD, Slonim AE, Bulone L, Ritz S, Goldberg T, Chen A, Martiniuk F. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am J Hum Genet. 2007;81:1042–1049. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Myozyme [package insert] Genzyme Corporation; Cambridge, MA: 2006. [Google Scholar]
- 20.Mendelsohn NJ, Messinger YH, Rosenberg AS, Kishnani PS. Elimination of antibodies to recombinant enzyme in Pompe's disease. N Engl J Med. 2009;360:194–195. doi: 10.1056/NEJMc0806809. [DOI] [PubMed] [Google Scholar]