

Abstract
Escherichia coli DNA polymerase III is a highly processive replicase due to the presence of the β clamp protein that tethers DNA polymerases to DNA. The β clamp is a head-to-tail ring-shaped homodimer, in which each protomer contains three structurally similar domains. Although multiple studies have probed the functions of the β clamp, a detailed understanding of the conformational dynamics of the β clamp in solution is lacking. Here we used hydrogen exchange mass spectrometry to characterize the conformation and dynamics of the intact dimer β clamp and a variant form (I272A/L273A) with diminished ability to dimerize in solution. Our data indicate that the β clamp is not a static closed ring but rather is dynamic in solution. The three domains showed different dynamics though they share a highly similar tertiary structure. Domain I, which controls the opening of the clamp by dissociating from Domain III, contained several highly flexible peptides that underwent partial cooperative unfolding (EX1 kinetics) with a half-life ~4 h. The comparison between the β monomer variant and the wild-type β clamp showed that the β monomer was more dynamic. In the monomer, partial unfolding was much faster and additional regions of Domain III also underwent partial unfolding with a half-life ~1 h. Our results suggest that the δ subunit of the clamp loader may function as a “ring holder” to stabilize the transient opening of the β clamp, rather than as a “ring opener”.
Keywords: processivity clamp, hydrogen exchange, mass spectrometry, EX1 kinetics, PCNA, clamp loader
Chromosomal DNA synthesis in Escherichia coli (E. coli) is primarily carried out by DNA polymerase III (pol III) holoenzyme that consists of ten different proteins (1–7). The catalytic core, including α, ε, and θ subunits, is responsible for DNA synthesis and proofreading. With only the catalytic core, replication is slow (~20 nt/s) and processivity is weak because the polymerase core dissociates from DNA templates frequently (8). For efficient replication, the core polymerase requires a processivity factor, called β clamp in E. coli, which works as a sliding platform and tethers the DNA polymerase to the template during replication. In the presence of the β clamp, replication is very efficient (~750 nt/s) and processive (>50 kb) (9). The clamp loader complex (subunits γ, δ, δ’, τ, χ, and ψ) of pol III loads the β clamp onto primed DNA (6, 10–11). Therefore, the β clamp plays an important role in supporting pol III as a highly efficient and processive DNA synthesis machine (12).
The architecture and mechanism of processivity clamps are well-conserved throughout evolution (5, 13–16). Clamp proteins (β clamp, bacteriophage T4 gp45, and eukaryotic PCNA, for example) form planar ring structures with six similarly folded domains, though there is no detectable sequence similarity among the clamps (14). The sizes of the central channel in the clamps are also similar with sufficient width to accommodate double-stranded DNA (17). The study presented here focuses on the E. coli β clamp protein.
The crystal structure of β clamp was reported by Kong et al. (18) at 2.5-Å resolution and by Oakley et al. (19) at 1.85-Å resolution with similar results. As reported, the β clamp is a ring-shaped homodimer that is composed of two monomers in a head-to-tail arrangement to generate two identical interfaces (Figure 1, PDB code: 1MMI (19)). Each monomer contains three domains which share little primary structure conservation, but good tertiary structure conservation (Figure S1 of the Supporting Information). In each domain, two four-stranded antiparallel β sheets compose the outer layer of the β clamp, which provide a scaffold to support the two α helices in the inner layer. A hydrophobic cleft located between Domain II and Domain III is the surface with which β interacts with different DNA polymerases, such as the α and δ subunits of pol III, pol II, Y family polymerases pol IV and pol V, and with UmuD (20–31). The outer diameter of the ring is ~80 Å and the inner diameter is ~35 Å (Figure 1) (18–19), which easily accommodates double-stranded DNA. The β clamp has been co-crystallized with primed DNA, revealing that the β clamp directly binds DNA via interactions between the positively charged residues on the inside of the clamp and the negatively charged DNA backbone. The duplex DNA was found in the central channel of the clamp tilted by 22° from the C2 rotation axis of β clamp (17). This finding also suggested a possible mechanism of DNA polymerase switching mediated by the β clamp, as the tilt of DNA in the β clamp could facilitate specific interactions with different polymerases bound to either monomer of the dimeric clamp (17).
Figure 1.
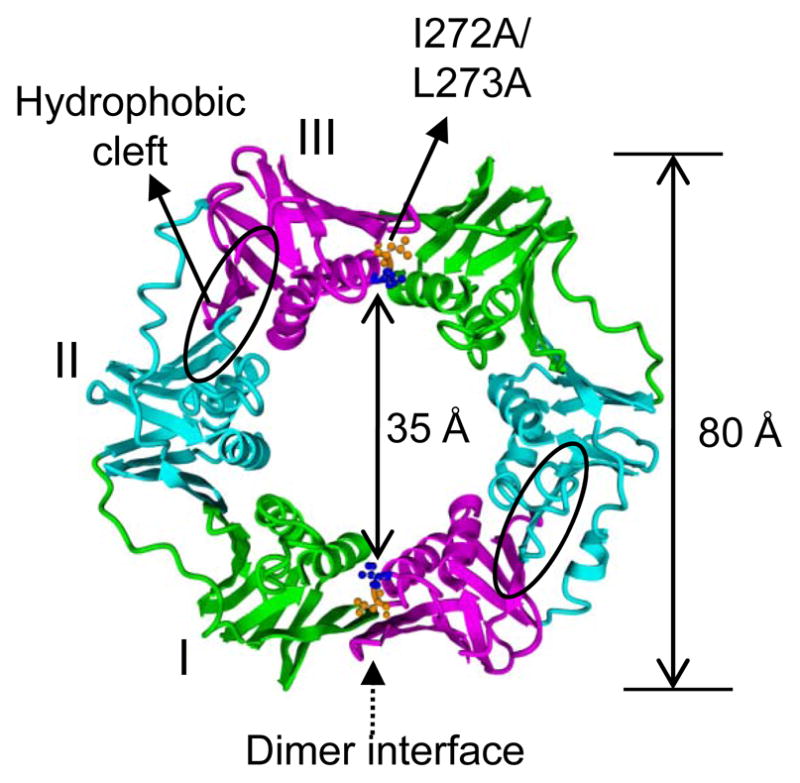
Ribbon representation of the crystal structure of the β clamp (PDB code: 1MMI) (19). The overall structure shows it is a ring-shaped homodimer, approximate diameter 80 Å, with a 35-Å central hole (18–19). The three domains of each monomer are labeled with different colors (Domain I in green, Domain II in blue and Domain III in magenta). The hydrophobic cleft located between Domains II and III is circled. Two residues, Ile272 and Leu273, in the dimer interface are highlighted in ball and stick (Ile272 is shown in blue and Leu273 in orange). The double mutation I272A/L273A severely disrupts the β dimer interface and results in a monomeric protein.
Although structural and biochemical studies have revealed a great deal about the mechanism of β clamp opening and loading onto DNA, a detailed analysis of the conformation and dynamics of the β clamp in solution is missing. We investigated the conformational dynamics of the β clamp in solution and compared that to the dynamics of a monomeric variant (32) by using hydrogen exchange mass spectrometry (HXMS). Due to the relatively short half-life of the β clamp on DNA (~2 h) (33), we focus here on the free β proteins in solution. HXMS is a powerful technique to monitor conformational changes in proteins (34–36). Based on the fact that the deuterium exchange rates of backbone amide hydrogens are highly dependent on the solvent accessibility of amide hydrogens and their participation in hydrogen-bonding networks, this methodology has the advantage of allowing analysis of the conformation and dynamics of the protein backbone caused by protein motions.
In this study, we found that the β clamp is not a static closed ring in solution. Domain I displayed partial local unfolding and was observed to be much more dynamic than the other domains. Therefore, the dimer interface of the β clamp may open spontaneously by Domain I dissociating from Domain III of its partner protomer. In comparison, the monomeric form of the β clamp displayed significant differences in exchange and was far more dynamic than the dimeric β clamp. Overall our results provide significant advancements in the understanding of dynamics within the β clamp and offer more clues about the opening mechanism.
MATERIALS AND METHODS
Protein cloning, expression and purification
The dnaN gene encoding the β clamp was cloned from a plasmid encoding His-HMK- β (37) into expression plasmid pET11T using NdeI and BamHI restriction sites (38). A double mutation (I272A, L273A), which caused the β clamp to be monomeric (32), was made by using QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) using the following forward and reverse oligonucleotide primers: 5′-GCTCGCGCGGCGGCTGCCTCTAACGAGAAATTC and 5′-GAATTTCTCGTTAGAGGCAGCCGCCGCGCGAGC. E. coli strain BL21 (DE3) pLysS was transformed with pET11T-β expressing WT β or the monomer variant.
Transformed BL21 (DE3) cells expressing either WT or monomeric β clamp were plated on LB agar plates that contained 100 μg/mL ampicillin and incubated at 37 °C overnight. An individual colony was selected for a starter culture (50 mL LB with 100 μg/mL ampicillin) and grown in a shaker (200 rpm) at 37 °C for ~12 h. A 1.0-L culture for WT β clamp and a 2.0-L culture for the monomer variant were seeded with the starter culture and grown until the optical density (600 nm) reached 0.8. Protein expression was induced by adding isopropyl-β-D-thiogalactoside (IPTG) to 1 mM at 30 °C. After 4-h induction, cells were harvested by centrifugation for 10 min at 6750 x g and 4 °C, and resuspended in 0.85% sodium chloride. After resuspension, cells were frozen and stored at −80 °C or were lysed immediately. SDS-PAGE was used to confirm overexpression of the β proteins.
For purification, frozen cells were thawed on ice at 4 °C and fresh phenylmethanesulfonyl fluoride (PMSF) to 10 μg/mL and protease inhibitor cocktail (Mini Complete, Roche) were added. All subsequent steps were performed at 4 °C. Sonication was used to lyse cells in the presence of lysozyme and DNase. After lysis, cell debris was pelleted by centrifugation (14000 x g) at 4 °C for 1 h. The lysate was loaded onto a DEAE weak anion exchange column (for the WT β clamp, HiTrap™ IEX FF Column, 2 × 5 mL, GE healthcare was used; for the β monomer, HiPrep 16/10 DEAE FF column, 20 mL, GE healthcare was used). Protein was eluted with a gradient of 0–0.5 M NaCl in Buffer A (20 mM Hepes, 0.1 mM EDTA and 3 mM DTT, pH 7.5). The fractions containing the protein of interest were pooled and diluted 1:1 (v/v) with Buffer A containing 1 M ammonium sulfate. The solution was loaded onto a Phenyl Sepharose column (HiPrep™ Phenyl FF column, 2 × 5 mL, GE Healthcare) and eluted with a 1.0 - 0 M gradient of ammonium sulfate in Buffer A. The fractions with the protein of interest were pooled and concentrated to less than ~2 mL using Vivaspin 6 centrifugal concentrator (5000 Da MWCO, Sartorius Stedim Biotech) and were loaded onto a size exclusion column (Superdex™ 75, 26 mm × 70 cm, GE Healthcare) that was equilibrated with 20 mM Hepes, 50 mM NaCl, 0.1 mM EDTA and 1 mM TCEP. Fractions containing protein were collected. The final purity and mass of all proteins were verified by electrospray mass spectrometry (LCT premier, Waters) and each theoretical mass matched the measured mass to within 0.5 Da (Figure S2 of the Supporting Information).
Deuterium exchange and MS analysis
Deuterium labeling
Purified β proteins were labeled by dilution of protein stock solutions (~50 μM in Buffer A) 17-fold (v/v) with D2O buffer (20 mM Hepes, 50 mM NaCl, 0.1 mM EDTA, 1 mM TCEP, pD 7.5), 25 ºC. At several time points (ranging from 10 s to 4 h), ~200 pmol (4.7 μL) of protein were removed from the exchange reaction and the labeling was quenched by addition of 75 μL phosphoric acid buffer (150 mM H3PO4-NaH2PO4, 6 M guanidine HCl, pH 2.1) to reduce the pH to 2.6. The quenched protein solution was diluted 1:1 using pre-chilled H2O with 0.05% formic acid to reduce the concentration of GdHCl to 1.5 M before injection into the LC system.
Intact protein analysis
The deuterated samples were immediately injected onto a 2 × 20 mm refillable guard column (Alltech, Deerfield, IL) that contained POROS 20 R2 (Applied Biosystems, Foster City, CA). Before elution with acetonitrile, protein on the column was rapidly desalted with several injections of 250 μL of H2O with 0.05% formic acid (pH ~2.5). Proteins were eluted and directed into a Waters LCT-Premier mass spectrometer with a 15–70% gradient of acetonitrile (containing 0.05% formic acid, pH ~2.5) at a flow rate of 50 μL/min. During sample analysis, the mobile phases, the injector, sample loop, column and all transfer lines were placed in an ice bath to minimize deuterium back-exchange (39). The instrument was calibrated by infusing 500 fmol/mL myoglobin (5 μL/min) at the end of each run. The relative deuterium level was determined by subtracting the mass of the undeuterated protein from the mass of the deuterated protein at each labeling time point. No adjustment was made for deuterium back-exchange during analysis, and therefore all results are reported as the relative deuterium level (40). In this experimental system, the average back-exchange was ~20% as determined by an analysis of completely deuterated control peptide.
Peptide analysis
To provide more spatial resolution, identical deuterated samples were prepared as described above and subjected to protolytic digestion before mass analysis (39). For in-solution digestion, approximately 20 pmol of the quenched protein solution was incubated with 5-fold excess (by weight) porcine pepsin on ice for 5 minutes. Pepsin-digested samples were injected into a Waters UPLC system designed for HXMS (41) and trapped on a VanGuard Pre-Column (2.1 mm × 5 mm, ACQUITY UPLC BEH C18, 1.7 μm) for 4 min. Then the trap was placed in-line with an ACQUITY UPLC BEH C18 1.7-μm 1.0 mm × 100 mm column (Waters Corp.) and an 8 - 40% gradient of acetonitrile over 8 min at a flow rate of 40 μL/min was used to separate the peptides. Formic acid (0.05%) was added to both mobile phases to maintain pH 2.5. Peptides were directed into a QTOF-Premier mass spectrometer (Waters Corp.) with electrospray ionization and lock-mass correction (Glu-fribrinogen peptide was used as an internal standard). Mass spectra were acquired over the m/z range 100 - 1700. Peptic peptides were identified using a combination of exact mass and MSE, aided by Waters IdentityE software (42). The spectra of deuterated peptides of WT β clamp and β monomer at different time points (including time 0 which was the undeuterated control) were extracted from the chromatograms by Waters HDX browser software. For peptides that displayed EX2 kinetics (a single binomial isotopic distribution), the peak width was measured at full-width at half-maximum (FWHM) by HX-express software (43). For peptides that displayed EX1 kinetics (a bimodal isotopic distribution), the two isotope distributions were analyzed with Gaussian-fitting using PeakFit software. The centroid mass of each EX1 distribution and the width of EX1 distributions were manually measured. The average relative deuterium incorporation was calculated by subtracting the centroid mass of the isotopic distribution of undeuterated peptide from the centroid mass of the isotopic distribution of deuterium labeled peptides. The resulting relative deuterium incorporations were plotted versus the labeling time.
RESULTS
In order to determine the dynamics of the β clamp and its monomeric variant in solution, HXMS was used to measure deuterium incorporation at the level of both the intact protein and peptic peptides. In solution HXMS, only deuterium incorporation at backbone amide positions is monitored because side-chain deuterium undergoes rapid back-exchange during chromatography analysis (39). Backbone amide hydrogen exchange is a good indicator of the dynamic properties of a protein (34). Fast amide hydrogen exchange implies high solvent exposure around the amide or lack of hydrogen bonding, while a slow amide hydrogen exchange implies a relatively low solvent exposure or that the amide hydrogen is involved in hydrogen bonding. Protected amide hydrogens cannot exchange until molecular motions, ranging from local fluctuations in conformation to global unfolding, expose the protected hydrogens to deuterium solvent and break hydrogen bonds (44). Thus, the exchange rates of amide hydrogens provide information about protein motions.
HXMS analysis of intact β proteins
We first compared the deuterium exchange into the WT dimeric β clamp with exchange into a variant form (I272A/L273A) that is a monomer (32). The purity and correct mass of each protein were verified by electrospray mass spectrometry (Figure S2 of the Supporting Information). Intact WT β clamp and β monomer were labeled with deuterium for various periods of time ranging from 10 s to 4 h. For both β proteins, HXMS measurements were obtained under identical experimental conditions, which allowed for comparison of deuterium exchange without back-exchange corrections (40). The average mass increase from triplicate experiments in each protein was plotted (Figure 2). In each protomer of the β clamp, there are 366 amino acids and 20 proline residues, which lack amide hydrogens. Therefore, there are 345 backbone amide hydrogens available for exchange calculated by subtracting the number of prolines and one for the N-terminus from the total number of residues (40). Within the first 10 min of labeling, the β clamp and its monomer variant showed similar deuterium exchange and appeared to be fairly well protected overall as evidenced by the very modest 19% deuterium incorporation (64 out of 345 exchangeable residues after 10 seconds of labeling, without taking into account back-exchange, Figure 2). These data indicate that the WT β clamp is resistant to deuteration, consistent with a well-folded, stable protein. Upon longer labeling time, the masses of both WT β clamp and β monomer increased due to molecular motions, as the deuterium incorporation increased to ~45% in the β clamp after 4 h of labeling.
Figure 2.

Relative deuterium uptake as a function of time for intact β clamp (red) and β monomeric variant (blue). The level of deuterium exchange was monitored at 10 s and 1, 10, 120, and 240 min for each protein. The experiment was repeated three times and the data shown here are the average of the three replicates. The number of maximum exchangeable backbone amide hydrogens is 345, the maximum y-axis value in the graph.
After 10 min of labeling, the β monomer began to show more deuteration than WT β clamp and the level of deuterium incorporation reached 57% after 4 h of labeling. The maximum difference at the 4 h time point between the WT β clamp and β monomer was approximately 40 deuterons. These data indicate that the β monomer underwent more dynamic motion compared with WT β clamp, as the WT β clamp was significantly more resistant to deuteration than the monomer. We next used pepsin digestion (39) to obtain more detailed information about the location of the differences.
Localizing the conformational differences between the β clamp and β monomer
By digesting deuterated protein into peptides after the labeling reaction has been quenched, the location of differences in deuteration can be determined. The deuteration reactions described for the intact proteins were repeated and the samples digested with pepsin prior to mass analysis. Different time points (10 s, 1 m, 10 m, 2 h, and 6 h) were chosen empirically based on analysis of the intact protein to best represent the dynamic range of the exchange reactions for WT and monomeric β clamp. The maximum labeling time (6 h) is longer than the intact protein analysis which was 4 h. To increase the pepsin digestion efficiency, guanidine hydrochloride was pre-mixed with the quenching buffer (45), and labeled proteins were digested in solution on ice. Digestion of the β proteins generated a large number of peptides. Figure S3 of the Supporting Information shows the peptides that were obtained in at least three experiments for both the WT β clamp and the monomer variant; the sequence coverage was ~98% for both forms of the β clamp. All peptic peptides produced during the digestion were identified by MSE and validated by manual inspection of the MS/MS spectra. The exchange level as a function of time was determined for each peptide under identical experimental conditions for both proteins. The peptide analysis of WT β clamp was repeated five times and the analysis of β monomer was repeated three times and provided great reproducibility; therefore, the observed differences were reliable indicators of the exchange differences but only one set of data is shown.
In the comparison of exchange into the WT and monomeric form of β, evidence was found for exchange by both EX2 and EX1 mechanisms. The majority of proteins display EX2 kinetics in HXMS where there is a steady, gradual increase of the isotopic distribution for a deuterated peptide up the m/z scale during the labeling time. Spectra other than the simple binomial isotope patterns are indicative of partial cooperative unfolding in solution (so-called EX1 kinetic signatures) (46–48). EX1 kinetics occurs when the deuterium exchange rate for a segment of the protein is faster than the protein refolding rate and multiple amide hydrogen atoms in a specific region can be exchanged simultaneously. This type of exchange gives rise to two distinct mass envelopes. Sixteen peptic peptides in one protomer of the WT β clamp and 21 peptides of the β monomer were found to exhibit spectra with bimodal isotope distributions, i.e., EX1 kinetics. Processing data with EX1 kinetics can sometimes be challenging. One simple solution is to find the centroid mass of the entire bimodal distribution. Each component of the bimodal distribution can also be processed separately. In the analysis of the data, we have used both approaches. For the peptic peptides that displayed EX1 kinetics, both the centroid mass and the width of each EX1 distribution were measured manually (in total, 37 EX1 peptides for both WT β and β monomer were found in each run, and six time points for each peptide were analyzed for peak width, totaling 222 isotopic distributions interrogated). We will first discuss the results of simply finding the centroid mass of all isotopic distributions, EX2 or EX1 in order to make a general comparison between the WT β clamp and the monomer. In a subsequent section, we present a more thorough processing of the EX1 data which reveals additional features of the proteins.
To give a general overview of the differences between the WT β clamp and β monomer, the average centroid of the isotope peaks for each time point was measured, the relative percent deuteration was calculated, and the results represented on the crystal structure of the β clamp (Figure 3). The comparison of the three domains in each protein indicated that Domain I of both WT β and β monomer underwent more deuterium exchange compared with Domain II and Domain III especially at the earlier time points (see the 10-sec time point), indicating the more dynamic nature of Domain I. At the earlier time points (10 sec and 1 min), the differences in deuterium uptake of WT β clamp and the monomer were modest, while the differences between them increased over time. Although the three domains share nearly identical 3D structure (Figure S1B, of Supporting Information), the dynamic behavior of each domain was very different. With increasing labeling time, the deuterium incorporation of WT β gradually increased due to molecular motions. Domain I maintained a higher level of deuterium uptake compared with other domains throughout the timecourse, which indicates that Domain I may play a specific role in β clamp dimer interface opening. Furthermore, the comparison of the three domains between dimer and monomer indicated that Domain III at the dimer interface of WT β clamp displayed less deuterium exchange compared with Domain III of the β monomer, which would not be involved in dimer interface interactions. Overall, the β monomer was more deuterated, especially after 10 minutes of deuteration, which is consistent with the intact protein analysis.
Figure 3.

Average deuterium incorporation into regions of the WT β clamp and β monomer as a function of time. Deuterium incorporation data for the two proteins are mapped onto the crystal structure of β clamp (PDB code: 1MMI) (19). Monomeric β is shown as one protomer of the β dimer in 1MMI. For each time point, WT β clamp is shown on the right and the β clamp monomer is shown on the left. For the peptides that display EX1 kinetics, the center of the two distributions was measured and used to determine relative deuteration (see main text); for the peptides displaying EX2 kinetics, the center of the single distribution was used. Color coding is shown at the right bottom of the figure. Regions shown in gray represent residues where deuterium levels were not determined.
EX1 kinetics or EX1/EX2 mixture identified in both β proteins
EX1 kinetics is a relatively rare phenomenon in stable, folded proteins in physiological conditions (48–50); however, when observed it provides important clues concerning protein motion, especially in native conditions. In EX1 kinetics, multiple amide hydrogens exchange simultaneously during cooperative unfolding events. The mass spectra of proteins undergoing EX1 are distinctive (51) and generally the peaks become broader (48), especially near the half-life (t1/2) of the unfolding event. In the characteristic bimodal pattern,the lower isotope distribution represents the molecules that have not yet unfolded and exchanged and the higher mass distribution represents the molecules that have undergone the correlated unfolding and exchange event.
Multiple peptides in both the WT β clamp and the β clamp monomer displayed EX1 kinetics (examples shown in Figures 4 and 5). The data reveal the existence of conformational heterogeneity in solution with markedly different levels of deuteration in the conformational forms under native conditions. It should be noted that although the vast excess of deuterium present makes the labeling reaction essentially unidirectional, the unfolding process itself, like most all other similar EX1 processes detected by HX MS (48), is reversible. In the WT β clamp (Figure 4A), the peptides with EX1 kinetics span most regions of Domain I, but only a few regions in Domain II and III. The regions in Domains II and III with EX1 kinetics are those regions nearest Domain I and so their EX1 kinetics may be influenced by the dynamics of Domain I. As shown in Figure 3, these results suggest that although the three domains of β clamp have almost identical tertiary structure, their solution dynamics are remarkably different. Domain I motions were quite different from those in the other domains. In the β monomer, EX1 kinetics was much more widespread and was observed in all three of the domains (Figure 4B). This observation is consistent with a protein that is much more dynamic and flexible in solution compared with the WT β clamp. Without formation of the dimer interface to support the structure, the monomer is much more dynamic.
Figure 4.

Residues showing EX1 kinetics in WT and monomeric β. All peptides that showed EX1 kinetics are highlighted on the crystal structure (PDB code: 1MMI (19)) of β clamp: in red for the WT β clamp (A) and in blue for the β clamp monomer (B). Deuterium uptake and raw mass spectra are shown for two representative peptides, residues 1–34 (m/z= 938.9, +5 charge state) (C) and residues 204–232 (m/z=811.4, +4 charge state) (D). Data for WT β are shown in red and data for the monomer are shown in blue. In the deuterium incorporation curves for the two peptides, the inset shows the location of the peptide, highlighed in magenta and the two sets of lines indicate the components of the EX1 distributions, i.e. the lower curve is for the lower mass component and the upper curve is for the higher mass component. The mass spectra corresponding to the exchange time-point closest to the approximate half-life of the unfolding events are displayed in black.
Figure 5.

Residues involved in the mutations to form monomeric β clamp show significant differences in deuterium incorporation between WT β clamp and β clamp monomer. Two peptides are highlighted in the crystal structure of β clamp in red in WT β (A) and in blue in the β monomer (B). Deuterium uptake and raw mass spectra are shown for residues 262–282 (m/z=478.5, +5 charge state for WT and m/z=461.7, +5 charge state for β monomer, double mutation I272/A, L273A) (C) and residues 284–306 (m/z=887.8, +3 charge state) (D). Data for the WT are shown in red and data for the monomer are shown in blue. In the deuterium incorporation curves for the two peptides, the inset shows the location of the peptide, highlighed in red and the two sets of lines indicate the components of the EX1 distributions as in Figure 4. The mass spectra corresponding to the exchange time-point closest to the approximate half-life of the unfolding events are displayed in black.
Two representative peptides that display obvious bimodal distributions in WT and monomeric β clamp are shown in Figure 4C and D. Peptide 1–34 is in Domain I while peptide 204–232 is in Domain II. In both of these peptides, as in all of the others with EX1 kinetics (Figure 4A, B), there is an EX1 unfolding event happening simultaneously with EX2 kinetics. For example, in peptide 1–34 from WT β clamp, the deuterium level steadily increases during time points 10 s to 1 h with no indication of EX1 kinetics. Then at about 1 h of labeling, an EX1 kinetic signature begins to appear and persists through 6 h of labeling. To analyze the data, the centroid of the single peak in EX2 or the bimodal peaks in EX1 were determined. When EX1 was apparent, the data were fit with two Gaussian distributions (48) and the deuterium level of each distribution determined separately. The deuterium incorporation graphs (top of Fig 4C,D) split into two curves, one curve for the lower mass component of the EX1 distribution and one curve for the higher mass component. The same kind of processing was performed for all peptides with bimodal distributions (Figure S4 of the Supporting Information). EX1 kinetics were synchronized between all the peptides that displayed them, meaning the unfolding was coordinated among the distinct regions and had the same approximate half-life regardless of the position in the structure.
There was a significant difference between the WT β clamp and the monomer variant in the time at which EX1 related unfolding occurred. For example, in the two peptides of the WT β clamp (Figure 4C and D, labeled in red), the spectra show a single Gaussian distribution until ~1 h. Starting from 1 h, a higher mass distribution appeared and the intensities increased with increasing labeling time. In contrast, in the β monomer, higher mass distributions appeared after only 10 minutes of labeling and the lower mass distribution disappeared generally after 2 h of labeling (Figure 4C and D, labeled in blue). Based on the data that were obtained, we estimate the half-life of unfolding for the intact beta clamp to be approximately 3.5 hours and for the monomer, approximately 45 minutes. This phenomenon suggests that for β clamp, the partial unfolding event is at least four times faster in the monomer.
In addition, the size of the region undergoing cooperative unfolding can be determined from spectra with bimodal patterns. The mass difference between the centroids of the two isotope distributions in WT β clamp residues 1–34 was ~10 Da and it was also ~10 Da in the β monomer. The mass difference between the two envelopes in residues 204–232 was also very similar (~11 in both protein forms) and similar in almost all of the other peptides for which EX1 kinetics were observed (exceptions discussed below). So although the two β proteins experience the unfolding events at different times, the number of residues involved in the unfolding event is similar.
As is evident in Figure 4C,D, the entire isotope distribution continues to increase in mass while at the same time, the EX1 unfolding event occurs. That is, the appearance of two distributions indicative of partial unfolding is concurrent with a gradual mass shift to higher m/z throughout the period before, during and after the bimodal pattern. This observation indicates that the partial unfolding did not occupy the entire region of each peptide but rather some parts of the peptide were undergoing EX2 kinetics and some parts were undergoing EX1 kinetics. For example, in residues 1–34, the m/z increase of the lower isotope distribution from 939.3 to 942.0 in a 6 h-labeling reaction corresponds to a 8-Da shift, which implies that at least eight residues are involved in exchange through EX2 in this peptide that has 30 maximum exchangeable hydrogens. The lower isotope distribution of the peptide representing residues 204–232 exhibit an increase in m/z from 811.9 to 812.9 corresponding to a 3-Da shift from EX2. Some regions of the two proteins exchanged purely by EX2 kinetics (see Supporting information Figure S5). Mixed kinetics (EX2 and EX1 in the same peptide) can easily be observed with HXMS and in the case of the proteins here, shows that both exchange through EX2 and exchange through EX1 were the same magnitude for WT β clamp and β clamp monomer. Again, the only difference was the rate of partial cooperative unfolding displayed by EX1 signatures. In summary, both β proteins displayed an unfolding event under physiological conditions. In WT β clamp, primarily Domain I displayed EX1 kinetics and the half-life of the unfolding motion was ~3.5 h. In comparison, the monomeric variant of β exhibited very different characteristics in which more of the peptides in all three domains had EX1 kinetics with a shorter half-life of unfolding around 45 minutes.
Monomer mutation causes conformational changes in addition to unfolding events
In most peptides with observed EX1 kinetics, the deuterium incorporations of the lower mass distribution were similar between the WT and monomeric β clamp; however, two peptides in the area of the mutation in the monomeric β clamp (residues 262–282 and 284–306) showed significant differences (Figure 5). For residues 262–282 in the β clamp, the m/z of the lower mass envelope increased from 478.7 to 479.5 at 10 min, corresponding to a ~1.5-Da shift; while in the β monomer, the peptide increased from 461.9 to 463.4 corresponding to a ~4-Da shift. This comparison indicates that approximately three residues that were protected in the WT β clamp became exposed to deuterium in the β monomer. Similarly, in residues 284–306 there was a rapid exchange of up to 5 hydrogens exposed to solvent during the local fluctuations in β monomer in the first 10 min, however, there was only one hydrogen exchanged in WT β clamp during that time. These data suggest that the double mutation causes a significant conformational change in the dimer interface of the β clamp even before the unfolding events occur. Additionally, the unfolding half-life of the two peptides in WT β clamp was ~2.5 h (highlighted in black in the mass spectra of Figure 5), which is faster than the ~3.5-h half-life of other EX1 peptides in the WT β clamp.
DISCUSSION
HXMS is useful for monitoring protein conformational dynamics with the ability to follow multiple exchange mechanisms simultaneously. Here we observed both EX1 and EX2 kinetics in the investigation of the β clamp and its monomeric variant. Specifically, we found evidence of local unfolding in Domain I. Using HXMS, we found that the dynamics of the β clamp are such that the β clamp dimer interface may open spontaneously and so it is likely not always a closed ring in solution.
Crystal structures show that the β clamp forms a head-to-tail closed ring; however, in solution, we observe that the β clamp displayed unusual dynamics. In total, 16 peptides of the β clamp, most of them in Domain I, underwent large scale unfolding with t1/2 of 3.5 h, which suggests that the β clamp is highly flexible, especially Domain I. The time required to replicate the E. coli chromosome is approximately 40 min (52). Our observed half-life of unfolding is similar to the half-life of the β clamp on DNA of ~2 h (33). These observations both support the idea that the cell requires a specific mechanism, which is facilitated by the δ subunit of the clamp loader complex, to recycle β clamps in order to efficiently complete replication (33). It is also possible that the intermediate conformations adopted by the dynamic β clamp facilitate distinct interactions with different proteins through induced-fit mechanisms. Finally, a dynamic clamp could be important for dynamic processivity, one model of polymerase switching in which there is a constant and rapid exchange of DNA polymerases at during replication (53).
This phenomenon that Domain I specifically undergoes unfolding may well explain the observation that the three domains of the β clamp have strikingly similar structures but unrelated sequences; the different sequences may dictate their different dynamics. Based on our data, Domain I could control the opening of the β clamp by dissociation from Domain III of its partner protomer. Molecular dynamics simulations of the eukaryotic sliding clamp PCNA revealed a large conformational change from a planar ring to a right-handed spiral in a 10-ns simulation (16). In a longer simulation of 40–90 ns, Adelman et al. also observed that PCNA underwent in-plane relaxation, with oscillation distances of ~15 Å, as well as out-of-plane fluctuations in which PCNA adopted both right- and left-handed spirals (54). In addition, the T4 bacteriophage clamp has been shown to be an open trimer in solution (55). However, in the previous simulation of β clamp dimer and its monomer, the β clamp dimer was found to be stable for up to 2.2-ns while the β clamp monomer underwent a rapid structural relaxation to a more open conformation on the same timescale (23).
The protein-protein interactions involving the processivity clamp are central to a regulatory network linking DNA replication, repair, and other cellular processes (31). At the core of the network are competitive interactions, which use the same peptide-binding cleft formed by Leu177, Pro242, Val247, Val360 and Met362 located between Domains II and III (23, 31, 56) (Figure 1) for binding DNA polymerases, such as pol II, pol III (22), pol IV (DinB) (30), and pol V (UmuD’2C) (25, 27). The consensus sequence in those DNA polymerases is QL[SD]LF (31). Such a simple motif seems insufficient to facilitate so many different and competitive interactions. Our observations provide a possible role for the β clamp in mediating these competitive interactions. The specific dynamics of the β clamp may induce a subtle conformational change in the hydrophobic cleft binding site for this motif and facilitate the interactions of the β clamp with different polymerases and other proteins that all bind to the same site. Furthermore, this flexibility could enable communication between the β clamp protomers, as it has been observed that proteins can compete for binding to the clamp and that protein binding can induce conformational changes in the clamp (28–29, 57–58).
As reported the δ subunit binds the β clamp at the hydrophobic cleft located between Domains II and III (23), which is relatively far from the dimer interface. Therefore, another one or more subunits of the clamp loader complex may assist δ in stabilizing the opened state of the β clamp. Docking of β onto the δ subunit of the crystal structure of γ3δδ’ suggests that γ may also bind to β (59). Leu et al. also found that γ and χ, and possibly δ’, also bind β, but they bind β more weakly than δ binds β (60).
The head-to-tail dimer interface of the β clamp is stabilized mostly by hydrophobic interactions; the monomeric variant contains two amino acid replacements (I272A/L273A, Figure 1) that disrupt the dimer interface and result in a stable monomer (32). The conformational comparison between WT β clamp and its monomeric variant provides insight into the role of the dimer ring in the opening mechanism and in the interactions of the β clamp with other partner proteins. For example, the β monomer has a higher affinity (at least 50-fold) for the δ subunit of the clamp loader complex compared to that of WT β clamp (32); moreover, the β monomer could be co-crystallized with δ but the WT β clamp could not (23). The crystal structure of the β monomer variant bound to the δ subunit of the clamp loader was superimposed with the crystal structure of WT β, revealing that the β monomer does not bend into the half-circle shape that would represent half of the complete β clamp dimer and instead relaxes to a shallower crescent shape (23). This structural difference suggests that the β ring is under spring tension in the closed state (23). Our data agree with this observation that the β monomer displays a less compact structure than WT β but also suggest that the difference between WT β clamp and the β monomer may be caused by the mutation itself, rather than by the binding of the δ subunit. Compared with the WT β clamp, there are more peptides in all three domains of the β monomer that display unfolding with a half-life of ~45 mins, which is faster than the unfolding event in the WT β clamp, a half-life of ~3.5 h (Figures 4, 5). The higher deuterium incorporation, especially in Domain III which is protected in the WT β clamp by the dimer interface (Figure S4 in the Supporting Information), demonstrates that the mutation disrupts the stable dimer interface and allows the monomeric variant to adopt a more relaxed structure.
We find that in particular Domain I of the β clamp is highly flexible (Figure 6), which suggests that the β clamp could open spontaneously in solution and which raises the question of the function of δ subunit. The δ subunit from the γ complex clamp loader is the only subunit that directly interacts with the β clamp and δ alone is capable of removing the β clamp from circular DNA molecules (33, 61). The δ subunit may actively open the clamp or may stabilize or trap a transiently open conformation of the β clamp (23, 62). We observe that the β clamp is very dynamic and therefore our data are not inconsistent with a model in which the function of the δ subunit is to capture and hold the β clamp in an open conformation, or possibly to open further a partially open clamp, as has been suggested in the T4 system (55). This raises the question of how the β clamp remains closed on DNA. There are a number of electrostatic interactions between basic residues on the protein and the backbone of DNA, spanning 10 base pairs of double-stranded DNA, that are likely to contribute to the stability of the clamp on DNA (17, 63).
Figure 6.

A model of β clamp opening mechanism. Based on our HXMS data, Domain I (green) is the most dynamic region compared with Domain II (blue) and Domain III (pink). Highly dynamic Domain I is at the dimer interface, which must be disrupted during clamp loading and therefore our findings are consistent with a model in which the β clamp is partially open in solution.
Moreover, interaction with other partner proteins, such as replicative DNA polymerase, would also be expected to stabilize the clamp on DNA.
The major observations of this report, coupled with previous findings, form a consistent model of β opening (Figure 6). In these experiments we could not differentiate the two identical protomers of the β clamp. However, previous disulfide cross-linking experiments indicate that the β clamp only needs to open at one of the two dimer interfaces to be loaded onto DNA (61). We observe that Domain I (Figure 6, green) displayed a high degree of dynamics. We propose a model in which the δ subunit of the clamp loader complex captures the highly dynamic β clamp in an open state. Moreover, the high degree of flexibility in Domain III could be one mechanism by which the β clamp fine-tunes interactions with its multiple protein partners and thereby regulates DNA replication and repair. We are currently determining the generality of our observations by applying the HX MS technique to other clamp proteins from other species.
Supplementary Material
Abbreviations used
- HXMS
hydrogen exchange mass spectrometry
- WT
wild-type
- pol III
polymerase III
- t1/2
half-life
- nt
nucleotide
- E. coli
Escherichia coli
- IPTG
isopropyl-β-D-thiogalactoside
- PMSF
phenylmethanesulfonyl fluoride
- FWHM
full-width at half-maximum
- Hepes
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- EDTA
ethylenediaminetetraacetic acid
- DTT
dithiothreitol
- TCEP
tris(2-carboxyethyl)phosphine
- MWCO
molecular weight cutoff
Footnotes
We are pleased to acknowledge generous financial support from a New Faculty Award from the Camille & Henry Dreyfus Foundation (PJB), the NSF (CAREER Award, MCB-0845033 to PJB), the NIH (R01-GM086507 to JRE), a research collaboration with the Waters Corporation, and the NU Office of the Provost. PJB is a Cottrell Scholar of the Research Corporation for Science Advancement.
Supporting Information. The mass analysis of the β clamp, peptide maps, deuterium uptake curves of individual peptides, and example analysis of EX2 kinetics. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kornberg A, Baker T. DNA replication. 2. W. H. Freeman and Company; New York: 1992. [Google Scholar]
- 2.McHenry CS. Chromosomal replicases as asymmetric dimers: studies of subunit arrangement and functional consequences. Mol Microbiol. 2003;49:1157–1165. doi: 10.1046/j.1365-2958.2003.03645.x. [DOI] [PubMed] [Google Scholar]
- 3.Maki H, Maki S, Kornberg A. DNA Polymerase III holoenzyme of Escherichia coli. IV. The holoenzyme is an asymmetric dimer with twin active sites. J Biol Chem. 1988;263:6570–6578. [PubMed] [Google Scholar]
- 4.Naktinis V, Onrust R, Fang L, O’Donnell M. Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. II. Intermediate complex between the clamp loader and its clamp. J Biol Chem. 1995;270:13358–13365. [PubMed] [Google Scholar]
- 5.Johnson A, O’Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- 6.Kelman Z, O’Donnell M. DNA polymerase III holoenzyme: structure and function of a chromosomal replicating machine. Annu Rev Biochem. 1995;64:171–200. doi: 10.1146/annurev.bi.64.070195.001131. [DOI] [PubMed] [Google Scholar]
- 7.Ollivierre JN, Silva MC, Sefcikova J, Beuning PJ. Polymerase switching in response to DNA damage. In: Williams MC, Maher ILJ, editors. Biophysics of DNA-Protein Interactions: From Single Molecules to Biological Systems. Springer; New York, NY: 2010. [Google Scholar]
- 8.Maki H, Kornberg A. The polymerase subunit of DNA polymerase III of Escherichia coli. II. Purification of the alpha subunit, devoid of nuclease activities. J Biol Chem. 1985;260:12987–12992. [PubMed] [Google Scholar]
- 9.O’Donnell ME, Kornberg A. Complete replication of templates by Escherichia coli DNA polymerase III holoenzyme. J Biol Chem. 1985;260:12884–12889. [PubMed] [Google Scholar]
- 10.Stukenberg PT, Studwell-Vaughan PS, O’Donnell M. Mechanism of the sliding beta-clamp of DNA polymerase III holoenzyme. J Biol Chem. 1991;266:11328–11334. [PubMed] [Google Scholar]
- 11.Bloom LB. Dynamics of loading the Escherichia coli DNA polymerase processivity clamp. Crit Rev Biochem Mol Biol. 2006;41:179–208. doi: 10.1080/10409230600648751. [DOI] [PubMed] [Google Scholar]
- 12.Ellison V, Stillman B. Opening of the clamp: an intimate view of an ATP-driven biological machine. Cell. 2001;106:655–660. doi: 10.1016/s0092-8674(01)00498-6. [DOI] [PubMed] [Google Scholar]
- 13.Zhuang Z, Ai Y. Processivity factor of DNA polymerase and its expanding role in normal and translesion DNA synthesis. Biochim Biophys Acta. 2010;1804:1081–1093. doi: 10.1016/j.bbapap.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neuwald AF. Evolutionary clues to DNA polymerase III beta clamp structural mechanisms. Nucleic Acids Res. 2003;31:4503–4516. doi: 10.1093/nar/gkg486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Indiani C, O’Donnell M. The replication clamp-loading machine at work in the three domains of life. Nat Rev Mol Cell Biol. 2006;7:751–761. doi: 10.1038/nrm2022. [DOI] [PubMed] [Google Scholar]
- 16.Kazmirski SL, Zhao Y, Bowman GD, O’Donnell M, Kuriyan J. Out-of-plane motions in open sliding clamps: molecular dynamics simulations of eukaryotic and archaeal proliferating cell nuclear antigen. Proc Natl Acad Sci U S A. 2005;102:13801–13806. doi: 10.1073/pnas.0506430102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Georgescu RE, Kim SS, Yurieva O, Kuriyan J, Kong XP, O’Donnell M. Structure of a sliding clamp on DNA. Cell. 2008;132:43–54. doi: 10.1016/j.cell.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong XP, Onrust R, O’Donnell M, Kuriyan J. Three-dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell. 1992;69:425–437. doi: 10.1016/0092-8674(92)90445-i. [DOI] [PubMed] [Google Scholar]
- 19.Oakley AJ, Prosselkov P, Wijffels G, Beck JL, Wilce MC, Dixon NE. Flexibility revealed by the 1.85 A crystal structure of the beta sliding-clamp subunit of Escherichia coli DNA polymerase III. Acta Crystallogr D Biol Crystallogr. 2003;59:1192–1199. doi: 10.1107/s0907444903009958. [DOI] [PubMed] [Google Scholar]
- 20.Scouten Ponticelli SK, Duzen JM, Sutton MD. Contributions of the individual hydrophobic clefts of the Escherichia coli beta sliding clamp to clamp loading, DNA replication and clamp recycling. Nucleic Acids Res. 2009;37:2796–2809. doi: 10.1093/nar/gkp128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maul RW, Ponticelli SK, Duzen JM, Sutton MD. Differential binding of Escherichia coli DNA polymerases to the beta-sliding clamp. Mol Microbiol. 2007;65:811–827. doi: 10.1111/j.1365-2958.2007.05828.x. [DOI] [PubMed] [Google Scholar]
- 22.Dohrmann PR, McHenry CS. A bipartite polymerase-processivity factor interaction: only the internal beta binding site of the alpha subunit is required for processive replication by the DNA polymerase III holoenzyme. J Mol Biol. 2005;350:228–239. doi: 10.1016/j.jmb.2005.04.065. [DOI] [PubMed] [Google Scholar]
- 23.Jeruzalmi D, Yurieva O, Zhao Y, Young M, Stewart J, Hingorani M, O’Donnell M, Kuriyan J. Mechanism of processivity clamp opening by the delta subunit wrench of the clamp loader complex of E. coli DNA polymerase III. Cell. 2001;106:417–428. [PubMed] [Google Scholar]
- 24.Lopez de Saro FJ, Georgescu RE, Goodman MF, O’Donnell M. Competitive processivity-clamp usage by DNA polymerases during DNA replication and repair. EMBO J. 2003;22:6408–6418. doi: 10.1093/emboj/cdg603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beuning PJ, Sawicka D, Barsky D, Walker GC. Two processivity clamp interactions differentially alter the dual activities of UmuC. Mol Microbiol. 2006;59:460–474. doi: 10.1111/j.1365-2958.2005.04959.x. [DOI] [PubMed] [Google Scholar]
- 26.Sutton MD, Duzen JM, Maul RW. Mutant forms of the Escherichia coli beta sliding clamp that distinguish between its roles in replication and DNA polymerase V-dependent translesion DNA synthesis. Mol Microbiol. 2005;55:1751–1766. doi: 10.1111/j.1365-2958.2005.04500.x. [DOI] [PubMed] [Google Scholar]
- 27.Becherel OJ, Fuchs RP, Wagner J. Pivotal role of the beta-clamp in translesion DNA synthesis and mutagenesis in E. coli cells. DNA Repair (Amst) 2002;1:703–708. doi: 10.1016/s1568-7864(02)00106-4. [DOI] [PubMed] [Google Scholar]
- 28.Duzen JM, Walker GC, Sutton MD. Identification of specific amino acid residues in the E. coli beta processivity clamp involved in interactions with DNA polymerase III, UmuD and UmuD’. DNA Repair (Amst) 2004;3:301–312. doi: 10.1016/j.dnarep.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 29.Wagner J, Etienne H, Fuchs RP, Cordonnier A, Burnouf D. Distinct beta-clamp interactions govern the activities of the Y family PolIV DNA polymerase. Mol Microbiol. 2009;74:1143–1151. doi: 10.1111/j.1365-2958.2009.06920.x. [DOI] [PubMed] [Google Scholar]
- 30.Bunting KA, Roe SM, Pearl LH. Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the beta-clamp. EMBO J. 2003;22:5883–5892. doi: 10.1093/emboj/cdg568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dalrymple BP, Kongsuwan K, Wijffels G, Dixon NE, Jennings PA. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc Natl Acad Sci U S A. 2001;98:11627–11632. doi: 10.1073/pnas.191384398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart J, Hingorani MM, Kelman Z, O’Donnell M. Mechanism of beta clamp opening by the delta subunit of Escherichia coli DNA polymerase III holoenzyme. J Biol Chem. 2001;276:19182–19189. doi: 10.1074/jbc.M100592200. [DOI] [PubMed] [Google Scholar]
- 33.Leu FP, Hingorani MM, Turner J, O’Donnell M. The delta subunit of DNA polymerase III holoenzyme serves as a sliding clamp unloader in Escherichia coli. J Biol Chem. 2000;275:34609–34618. doi: 10.1074/jbc.M005495200. [DOI] [PubMed] [Google Scholar]
- 34.Smith DL, Deng Y, Zhang Z. Probing the non-covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J Mass Spectrom. 1997;32:135–146. doi: 10.1002/(SICI)1096-9888(199702)32:2<135::AID-JMS486>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 35.Engen JR. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal Chem. 2009;81:7870–7875. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsutsui Y, Wintrode PL. Hydrogen/deuterium exchange-mass spectrometry: a powerful tool for probing protein structure, dynamics and interactions. Curr Med Chem. 2007;14:2344–2358. doi: 10.2174/092986707781745596. [DOI] [PubMed] [Google Scholar]
- 37.Kelman Z, Yao N, O’Donnell M. Escherichia coli expression vectors containing a protein kinase recognition motif, His6-tag and hemagglutinin epitope. Gene. 1995;166:177–178. doi: 10.1016/0378-1119(95)00556-7. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen LH, Jensen DB, Burgess RR. Overproduction and purification of sigma 32, the Escherichia coli heat shock transcription factor. Protein Expr Purif. 1993;4:425–433. doi: 10.1006/prep.1993.1056. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 41.Wales TE, Fadgen KE, Gerhardt GC, Engen JR. High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal Chem. 2008;80:6815–6820. doi: 10.1021/ac8008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geromanos SJ, Vissers JP, Silva JC, Dorschel CA, Li GZ, Gorenstein MV, Bateman RH, Langridge JI. The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependant LC-MS/MS. Proteomics. 2009;9:1683–1695. doi: 10.1002/pmic.200800562. [DOI] [PubMed] [Google Scholar]
- 43.Weis DD, Engen JR, Kass IJ. Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J Am Soc Mass Spectrom. 2006;17:1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 44.Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys. 1983;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 45.Houde D, Arndt J, Domeier W, Berkowitz S, Engen JR. Characterization of IgG1 conformation and conformational dynamics by hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2009;81:2644–2651. doi: 10.1021/ac802575y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arrington CB, Teesch LM, Robertson AD. Defining protein ensembles with native-state NH exchange: kinetics of interconversion and cooperative units from combined NMR and MS analysis. J Mol Biol. 1999;285:1265–1275. doi: 10.1006/jmbi.1998.2338. [DOI] [PubMed] [Google Scholar]
- 47.Engen JR, Smith DL. Investigating the higher order structure of proteins. Hydrogen exchange, proteolytic fragmentation, and mass spectrometry. Methods Mol Biol. 2000;146:95–112. doi: 10.1385/1-59259-045-4:95. [DOI] [PubMed] [Google Scholar]
- 48.Weis DD, Wales TE, Engen JR, Hotchko M, Ten Eyck LF. Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J Am Soc Mass Spectrom. 2006;17:1498–1509. doi: 10.1016/j.jasms.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 49.Englander SW. Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectrom. 2006;17:1481–1489. doi: 10.1016/j.jasms.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferraro DM, Lazo ND, Robertson AD. EX1 hydrogen exchange and protein folding. Biochemistry. 2004;43:587–594. doi: 10.1021/bi035943y. [DOI] [PubMed] [Google Scholar]
- 51.Miranker A, Robinson CV, Radford SE, Aplin RT, Dobson CM. Detection of transient protein folding populations by mass spectrometry. Science. 1993;262:896–900. doi: 10.1126/science.8235611. [DOI] [PubMed] [Google Scholar]
- 52.Churchward G, Bremer H. Determination of deoxyribonucleic acid replication time in exponentially growing Escherichia coli B/r. J Bacteriol. 1977;130:1206–1213. doi: 10.1128/jb.130.3.1206-1213.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang J, Zhuang Z, Roccasecca RM, Trakselis MA, Benkovic SJ. The dynamic processivity of the T4 DNA polymerase during replication. Proc Natl Acad Sci USA. 2004;101:8289–8294. doi: 10.1073/pnas.0402625101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adelman JL, Chodera JD, Kuo IF, Miller TF, 3rd, Barsky D. The mechanical properties of PCNA: implications for the loading and function of a DNA sliding clamp. Biophys J. 2010;98:3062–3069. doi: 10.1016/j.bpj.2010.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Millar D, Trakselis MA, Benkovic SJ. On the solution structure of the T4 sliding clamp (gp45) Biochemistry. 2004;43:12723–12727. doi: 10.1021/bi048349c. [DOI] [PubMed] [Google Scholar]
- 56.Burnouf DY, Olieric V, Wagner J, Fujii S, Reinbolt J, Fuchs RP, Dumas P. Structural and biochemical analysis of sliding clamp/ligand interactions suggest a competition between replicative and translesion DNA polymerases. J Mol Biol. 2004;335:1187–1197. doi: 10.1016/j.jmb.2003.11.049. [DOI] [PubMed] [Google Scholar]
- 57.Furukohri A, Goodman MF, Maki H. A Dynamic Polymerase Exchange with Escherichia coli DNA Polymerase IV Replacing DNA Polymerase III on the Sliding Clamp. J Biol Chem. 2008;283:11260–11269. doi: 10.1074/jbc.M709689200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beuning PJ, Simon SM, Zemla A, Barsky D, Walker GC. A non-cleavable UmuD variant that acts as a UmuD’ mimic. J Biol Chem. 2006;281:9633–9640. doi: 10.1074/jbc.M511101200. [DOI] [PubMed] [Google Scholar]
- 59.Jeruzalmi D, O’Donnell M, Kuriyan J. Crystal structure of the processivity clamp loader gamma complex of E. coli DNA polymerase III. Cell. 2001;106:429–441. doi: 10.1016/s0092-8674(01)00463-9. [DOI] [PubMed] [Google Scholar]
- 60.Leu FP, O’Donnell M. Interplay of clamp loader subunits in opening the beta sliding clamp of Escherichia coli DNA polymerase III holoenzyme. J Biol Chem. 2001;276:47185–47194. doi: 10.1074/jbc.M106780200. [DOI] [PubMed] [Google Scholar]
- 61.Turner J, Hingorani MM, Kelman Z, O’Donnell M. The internal workings of a DNA polymerase clamp-loading machine. EMBO J. 1999;18:771–783. doi: 10.1093/emboj/18.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bloom LB, Goodman MF. A sliding clamp monkey wrench. Nat Struct Biol. 2001;8:829–831. doi: 10.1038/nsb1001-829. [DOI] [PubMed] [Google Scholar]
- 63.Laurence TA, Kwon Y, Johnson A, Hollars CW, O’Donnell M, Camarero JA, Barsky D. Motion of a DNA sliding clamp observed by single molecule fluorescence spectroscopy. J Biol Chem. 2008;283:22895–22906. doi: 10.1074/jbc.M800174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.