

Abstract
Following adhesion of Helicobacter pylori to gastric epithelial cells, intracellular signaling leads to cytokine production, which causes H. pylori-related gastric injury. Two adjacent homologous genes (alpA and alpB), which encode H. pylori outer membrane proteins, are thought to be associated with adhesion and cytokine induction. We co-cultured gastric epithelial cells with wild type H. pylori strains and their corresponding alpA/alpB-deleted mutants (ΔalpAB). Results were confirmed by complementation. Flow cytometry confirmed that AlpAB was involved in cellular adhesion. Deletion of alpAB reduced interleukin (IL)-6 induction in gastric epithelial cells. Deletion of alpAB reduced IL-8 induction with East Asian but not with Western strains. All AlpAB-positive strains tested activated the extracellular signal-regulated kinase, c-Fos, and cAMP-responsive element-binding protein. Activation of the Jun-N-terminal kinase, c-Jun, and NF-κB was exclusive to AlpAB from East Asian strains. ΔalpAB mutants poorly colonized the stomachs of C57BL/6 mice and were associated with lower mucosal levels of KC and IL-6. Our results suggest that AlpAB may induce gastric injury by mediating adherence to gastric epithelial cells and by modulating proinflammatory intracellular signaling cascades. Known geographical differences in H. pylori-related clinical outcomes may relate to differential effects of East Asian and Western types of AlpAB on NF-κB-related proinflammatory signaling pathways.
Attachment of Helicobacter pylori to host gastric epithelial cells is believed to play an important role in bacterial colonization and in cellular and humoral immune responses to infection (1, 2). For example, direct contact between the bacteria and the epithelium is required for the release of inflammatory mediators, such as IL-8,2 which results in the mucosal accumulation of neutrophils (3).
A number of outer membrane proteins (OMPs) have been reported to be adhesins, including BabA (4), SabA (5), OipA (6), HopZ (7), and the adherence-associated proteins (AlpA and AlpB) that are encoded by two adjacent homologous genes (8–10). alpA and alpB are organized in an operon and are co-transcribed. Evidence that they encode an adhesin comes from experiments where preincubation of H. pylori with an antiserum against the AlpA fusion protein blocked H. pylori binding to paraffin-embedded tissue sections (8). Knock-out of alpAB or alpA also decreased adherence to epithelial cells in paraffin-embedded tissues, consistent with the notion that the alpA and alpB gene products are involved in the adherence phenotype (8–10). However, their roles in adherence to gastric epithelial cells in vivo are unclear. Although AlpAB has been reported to be important for colonization of guinea pigs (11), the effect on other animals has not been described.
The function of AlpAB is unclear partly due to conflicting data regarding the effect of alpAB isogenic mutants on IL-8 induction from epithelial cells (10, 12). Previous studies used alpAB mutants constructed by either transposon shuttle mutagenesis of alpA or insertion of the kanamycin resistance cassette into the gene (8–12). In the present study, we constructed precise deletions of the entire alpA and alpB genes (ΔalpAB mutants) and ΔalpAB complementation mutants to exclude the effects of secondary mutations. These mutants were used to examine H. pylori adherence to gastric epithelial cells. We also investigated the effects of alpAB on induction of IL-6 and IL-8 as well as effects the AlpAB-induced signaling cascade on different transcription factors involved in IL-6 or IL-8 promoter activation. Finally, we examined the role of alpAB in colonization and gastric inflammation in C57BL/6 mice.
Experimental Procedures
H. pylori Mutants
We constructed precise whole alpA and alpB-deleted mutants (ΔalpAB) using gene replacement mutagenesis (13). PCR fragments containing the upstream (hp0911; hp number and location from H. pylori strain 26695, GenBank™ accession number AE000511) and downstream (hp0914) sequences of the alpA-alpB genes were amplified and cloned into to the T7Blue vector (Novagen, Madison, WI), resulting in pTHP911 and pTHP914, respectively. We used template DNA from strain TN2GF4 (denoted TN) to construct mutants from East Asian strains and strain ATCC43504 (denoted 43504) to construct mutants from Western strains. A chloramphenicol resistance gene cassette (cat) was inserted at the BamHI site of pTHP911, resulting in pTHP911∷cat. Finally, the blunt fragment of pTHP911∷cat was inserted at the HincII site of pTHP914, resulting in pTHP911/914∷cat. The purified plasmid was used to inactivate chromosomal genes by natural transformation as previously described (14).
Chromosome-based systems were used for complementation assays (15). We selected hp0796 as a target gene, since our previous study confirmed that isogenic hp0796 mutants induced IL-8 from gastric epithelial cells and bound to the cells similar to wild type (WT) strains (6). PCR fragments containing the whole alpA-alpB gene with its promoter were amplified and cloned into the pBluescript II vector (Stratagene, La Jolla, CA), resulting in pBalpAB. We used template DNA from strain TN and 43504. A kanamycin resistance gene cassette (aph-A3) was inserted at the BamHI site of pBalpAB, resulting in pB[alpAB-aph-A3]. PCR fragments containing the whole hp0796 were amplified and cloned into the pBluescript II vector (Stratagene), resulting in pBhp0796. Finally, the blunt fragment of pB[alpAB-aph-A3] was inserted at the Eco47III site of pBhp0796, resulting in pBhp0796∷alpAB-aph-A3. The purified plasmid was used to complement ΔalpAB mutants by natural transformation using cat and ahp-A3 as double selectable markers. PCR and immunoblot for AlpA and AlpB were performed to confirm the absence or presence of the genes and gene products.
The previously described isogenic hopZ mutant (ΔhopZ), whole cag pathogenicity island (PAI)-deleted mutants (Δcag PAI), and hp0796 mutants (Δhp0796) were used as controls for reduced adherence, for reduced ability to induce IL-6 and IL-8, and to compare the effect of complemented mutants, respectively (6, 16, 17).
Cell Lines and Co-culturing
The human gastric epithelial cancer cell lines MKN28 and MKN45 (Riken Cell Bank, Tsukuba, Japan) and AGS (American Type Culture Collection, Manassas, VA) were cultured according to standard procedures. H. pylori were suspended in phosphate-buffered saline and added to the epithelial cell cultures at a multiplicity of infection of 100 for the indicated periods of time.
Immunoblotting
Anti-AlpA (AK214) (8), anti-AlpB (AK262) (9), anti-BabA (AK277) (18), and anti-OipA antisera (AK282) (19) were used as primary antibodies at a 1:5,000 dilution. Anti-CagA and anti-VacA antibodies (Austral Biologicals, San Ramon, CA) were used at a 1:3,000 dilution. Horseradish peroxidase-conjugated protein A (1:3,000) (Bio-Rad) was used as secondary antibody. Blots were developed with the ECL reagents (Amersham Biosciences). The tyrosine phosphorylation status of CagA in infected AGS cells was examined as previously described (10).
To examine cellular signaling, we used phosphospecific antibodies and control total antibodies for the cAMP-responsive element-binding protein (CREB) and ATF-2 at a 1:1,000 dilution (Cell Signaling Technology Inc.). Horseradish peroxidase-coupled anti-rabbit IgG (1:2,000) (Bio-Rad) was used as second antibody.
IL-6 and IL-8 ELISA
IL-6 and IL-8 protein levels produced from human cell lines co-cultured with H. pylori were measured by ELISA (R&D Systems, Minneapolis, MN) as previously described (6, 20).
Phenotypes of Infected Cells
The presence of the hummingbird phenotype in AGS cells co-cultured with H. pylori for 10 h was analyzed by phase-contrast microscopy. When more than 40% of the cells were elongated, the hummingbird phenotype was considered to have been induced.
H. pylori Binding Assays
H. pylori were stained with the red fluorochrome PKH26 (Sigma), as described previously (21). After washing with culture medium, samples were incubated with gastric cells for 90 min at 37 °C. Cells were washed with phosphate-buffered saline and resuspended in 2% paraformaldehyde. Flow cytometry was performed using a FACScan cytometer (BD Biosciences).
Luciferase Plasmids
Human IL-8 promoter firefly luciferase reporter plasmids p162hu.IL8-luc+ and site-directed mutant plasmids with mutations in four sites (interferon-stimulated responsive element (ISRE)-like element, activator protein-1 (AP-1), CCAAT/enhancer-binding protein (C/EBP), or NF- κB) were described previously (17). Full-length human IL-6 promoter firefly luciferase reporter plasmid p1168hu.IL6-luc+ and site-directed mutant plasmids with mutations in four sites (i.e. AP-1, CRE, C/EBP, or NF-κB) were described previously (20). The PathDetect cis-reporting and the PathDetect trans-reporting systems were purchased from Stratagene (La Jolla, CA).
Cell Transfection
Each luciferase reporter vector (2 μg) was transiently transfected into MKN28 cells using Lipofectamine 2000 reagent (Invitrogen). Nine hours after H. pylori treatment, the cells were lysed using passive lysis buffer (Promega, Madison, WI). The luciferase assays were performed using the Dual-Luciferase reporter assay system (Promega). Normalized luciferase activity is presented as firefly luciferase activity/Renilla luciferase activity. The results are presented as -fold increase of luciferase activity in infected cells relative to uninfected controls.
Binding of Transcription Factors
Nuclear extracts of MKN28 cells (uninfected or infected for 90 min) were prepared using hypotonic/nonionic detergent lysis (22). Equal amounts of nuclear extracts were used for electrophoretic mobility shift assay with duplex oligonucleotides for the consensus NF- κB, AP-1, and CREB binding sites (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) as previously described (17, 20). For semiquantitation, density was measured by scanning using Image J 1.36 software from the National Institutes of Health (available on the World Wide Web at rsbweb.nih.gov/ij/). We compared the amount of radioactive probe in the protein-DNA complexes after standardization with free probe. Density is presented as -fold induction compared with uninfected control. In the gel mobility supershift assays, commercial antibodies (Santa Cruz Biotechnology) were added to the binding reactions before adding the probes.
The same nuclear extracts were tested by ELISA using commercially available kits (TransAM™ NF-κB kit for p65 and TransAM™ AP-1 family kit for c-Jun and c-Fos; Active Motif, Carlsbad, CA) according to the manufacturer's instructions.
c-fos and c-jun mRNA Expression by Real Time Quantitative PCR
Total RNA extracted from infected and uninfected cells for 1 h using TRIzol reagent (Invitrogen) was converted to cDNA and then was analyzed by real time RT-PCR. The levels of c-fos and c-jun mRNAs were measured by SYBR green I-based quantitative real time RT-PCR and, following normalization to glyceraldehyde-3-phosphate dehydrogenase mRNA, were expressed as -fold induction relative to the uninfected control, as previously described (23, 24).
Animal Studies
Six-week-old, specific pathogen-free male C57BL/6 mice were used. They were maintained in an air-conditioned room designed for infected animals with a 12-h light, 12-h dark cycle and free access to food and water. No specific pretreatments were used before orogastric H. pylori inoculation or before the animals were sacrificed. Mice were orogastrically inoculated three times (days 0, 1, and 2) with 0.5 ml of the H. pylori inoculum preparation (∼3 × 108 colony-forming units/ml) or sterile brain heat infusion broth (as an uninfected control) through a feeding needle. Eight weeks after inoculation, colonization efficiency and inflammation were measured. At necropsy, the glandular stomach and first part of the duodenum were removed and opened along the greater curvature. Stomachs were divided longitudinally into two parts, and then one-half was bisected longitudinally into two parts. One-half was transferred to 1 ml of sterile phosphate-buffered saline to quantitate H. pylori and measure cytokine protein levels, one-quarter was fixed in 10% neutral buffered formalin for histological examination, and one-quarter was placed into TriZol Reagent (Invitrogen) and stored at −80 °C for cytokine mRNA analysis. The gastric mucosa was separated as much as possible from the underlying muscle by sharp dissection. All experimental protocols were approved by the local Ethical Committee specializing in animal experiments.
The number of bacteria on the gastric mucosa was assessed as previously described (16, 25). The number of colonies per plate was counted and calculated as the colony-forming units per stomach. We also measured mucosal KC and IL-6 levels. Since mice do not possess the IL-8 gene, we selected KC, which is a CXC chemokine whose promoter activity is regulated by binding to CRE and NF-κB binding sites. Mucosal KC levels were measured by ELISA (R&D Systems) as previously described (16). Since mucosal IL-6 levels in infected mice were below the level of detection of the ELISA kit (3.1 pg/ml) irrespective of H. pylori infection, we measured mucosal IL-6 mRNA levels by RT-PCR as previously described (16). IL-6 mRNA levels were presented as a ratio of 10× IL-6 mRNA to β-actin mRNA.
Statistical Methods
Data are presented as mean and S.E. Statistical analysis was performed by the Mann-Whitney rank sum test, nonpaired t test, and Fisher's exact test, depending on the data set of concern. A p value of less than 0.05 was accepted as statistically significant.
Results
Characterization of ΔalpAB Mutants
We used 11 H. pylori strains (TN, JK51, JK34, JK35, CPY2052 (denoted CPY), CA23, RD26, GI2766, 26695, J99, and 43504). TN, JK51, JK34, JK35, and CPY were isolated in Japan; CA23 in Korea; RD26, GI2766, and J99 in the United States; 26695 in the United Kingdom; and 43504 in Australia. Strains JK51, JK34, 26695, and GI2766 were isolated from patients with gastritis; TN, JK35, CPY, RD26, and J99 from patients with peptic ulcers; and CA23 from a patient with gastric cancer. All are cag PAI-positive, vacA s1 (producing vacuolating cytotoxin), and contain functional BabA and OipA. After natural transformation, we picked eight clones of each ΔalpAB mutant from the 11 parental strains. Gene inactivation was confirmed by PCR and immunoblot for AlpA and AlpB (Fig. 1). We confirmed that all ΔalpAB clones produced CagA, VacA, BabA, and OipA (Fig. 1). Since all clones retained the ability to translocate/phosphorylate CagA (Fig. 1) and to induce the hummingbird phenotype (data not shown), there was no possibility that secondary mutations in the cag PAI influenced cytokine induction, which was observed in a previous study (12).
FIGURE 1. Production of several virulence factors in H. pylori (A) and translocation and phosphorylation of CagA (B).
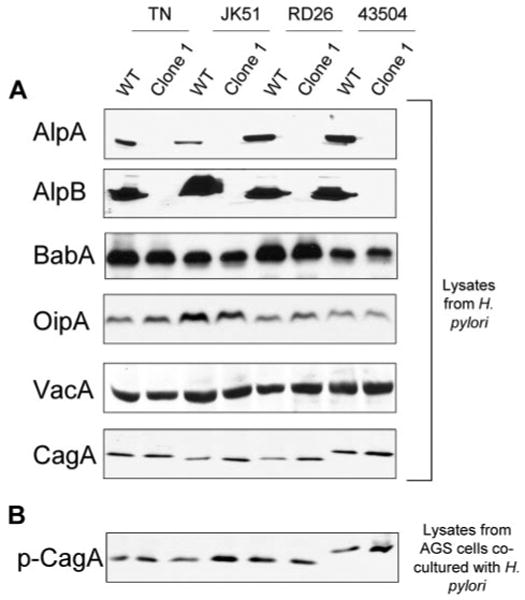
Total lysates of different H. pylori and their corresponding ΔalpAB mutants were separated on 8–12% SDS gels, blotted to nitrocellulose, and incubated with specific antibodies that recognize various H. pylori virulence factors (A). Total lysates of AGS cells co-cultured with H. pylori were separated on 8% SDS gels, blotted to nitrocellulose, and incubated with the phosphotyrosine-specific monoclonal antibody PY99 (B). Typical gels for four parental strains and their ΔalpAB mutants (clone 1) are presented.
Effect of AlpAB on IL-6 and IL-8 Induction
The amount of IL-6 and IL-8 secreted into the gastric epithelial cell culture supernatant reached maximal levels at 24 h after H. pylori infection irrespective of the strain and cell types used (data not shown). Since the levels were similar among clones, we used one clone of each mutant in subsequent experiments. ΔalpAB mutants varied in their ability to induce IL-8 production by gastric epithelial cells in a strain-dependent manner (Fig. 2 for MKN28 cells and supplemental Fig. 1S for MKN45 cells and AGS cells). ΔalpAB mutants from strains isolated in East Asia had ∼50% less IL-8 than their respective parental WT strains. In contrast, the ability to induce IL-8 was similar among parental strains isolated in Western countries and their ΔalpAB mutants.
FIGURE 2. Levels of IL-8 (top) and IL-6 (middle) from MKN28 cells and adherence of H. pylori to MKN28 cells co-cultured with H. pylori (bottom).
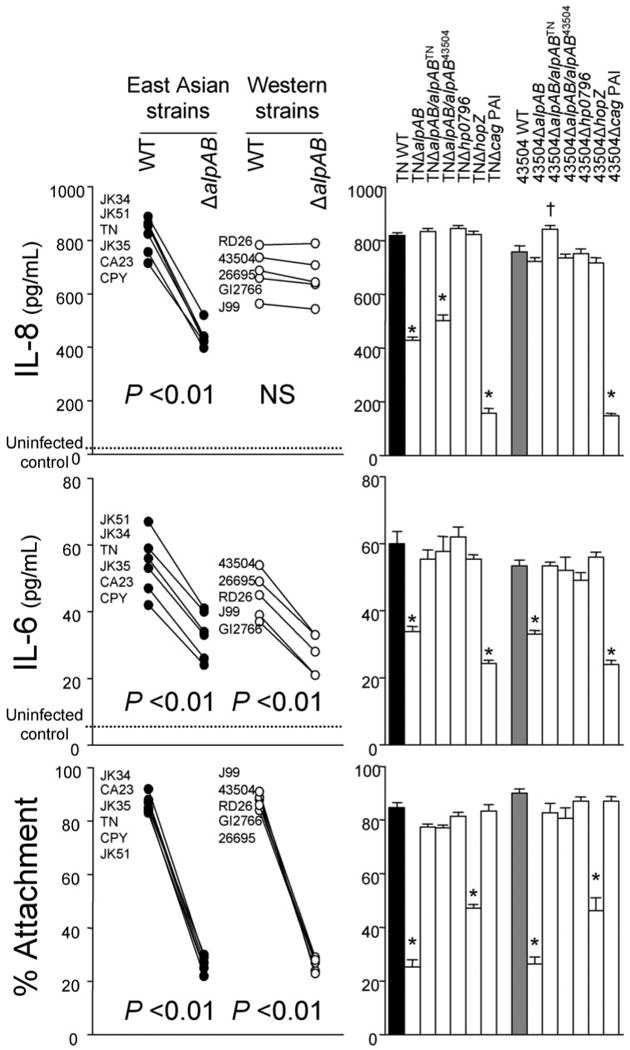
IL-8 and IL-6 levels were measured by ELISA, and adherence was measured by flow cytometry. Left, six East Asian strains and five Western strains were used. After natural transformation, we picked eight clones of each ΔalpAB mutant. Three independent co-cultures were performed for each clone and WT strain, and each was measured in duplicate. Data are expressed as mean of each clone or WT strain. Right, strains TN and 43504 and their mutants were used. Three or four independent co-cultures were performed, and each was measured in duplicate. Data are expressed as mean ± S.E.*,p < 0.01 decreased compared with WT strains; †, p < 0.01 increased compared with WT strains.
To exclude the possibility of polar effects and secondary mutations, complementation of alpAB genes was constructed. alpAB genes from strains TN and 43504 are designated alpABTN and alpAB43504, respectively. As expected, expression of alpABTN in TNΔalpAB restored the ability to induce IL-8 to levels observed in the WT strain (Figs. 2 and 1S). Complementation of 43504ΔalpAB with alpAB43504 did not change its ability to induce IL-8, whereas complementation of 43504ΔalpAB with alpABTN increased the ability to induce IL-8. Complementation of TNΔalpAB with alpAB43504 only partially restored the ability to induce IL-8.
Interestingly, all ΔalpAB clones showed decreased levels of IL-6 induction from MKN28 cells irrespective of the parental strain (Fig. 2). In MKN45 cells and AGS cells, IL-6 levels were below detection irrespective of H. pylori infection. Complementation of ΔalpAB mutants with intact alpAB genes restored the ability to induce IL-6 in strains TN and 43504 (Fig. 2), confirming that AlpAB is involved in IL-6 induction.
Effect of AlpAB on Adherence
Adherence to gastric epithelial cells was decreased with all ΔalpAB mutants irrespective of their other characteristics (Fig. 2 for MKN28 cells and supplemental Fig. 2S for AGS cells and MKN45 cells). The degree of reduction by ΔalpAB mutants was even greater than with ΔhopZ mutants. Complementation of ΔalpAB mutants with intact alpAB genes restored the ability to adhere to the cells (Figs. 2 and 2S), confirming that AlpAB is involved in adherence to epithelial cells.
Effect of AlpAB on Activation of IL-6 and IL-8 Promoters
Luciferase reporter activity of the plasmid containing a −162/+44 fragment of the IL-8 promoter was induced by WT H. pylori strains in MKN28 cells (Fig. 3). The activity decreased when using ΔalpAB mutants from East Asian strains. However, the activity was not changed when using ΔalpAB mutants from Western strains. As expected, the decreased activity associated with TNΔalpAB was restored by complementation with alpABTN but not with alpAB43504. Luciferase reporter activity of the plasmid containing the full-length human IL-6 promoter was also induced by WT H. pylori infection, and ΔalpAB mutants significantly reduced the activity irrespective of the origin of the strain (Fig. 3). Luciferase activity was restored by complementing ΔalpAB mutants with intact alpAB genes for both TN2GF4 and 43504.
FIGURE 3. IL-6 and IL-8 promoter activation following H. pylori infection.
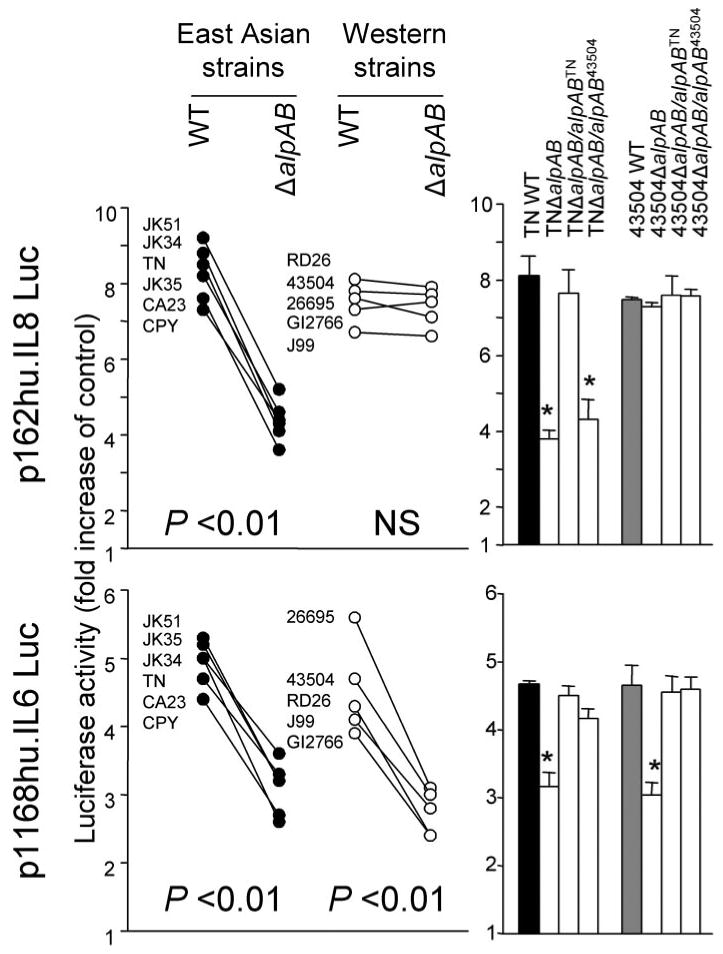
Luciferase reporter activity of a plasmid containing a −162/+44 fragment of the IL-8 promoter and a plasmid containing the full-size human IL-6 promoter was measured. Three independent transfections, each done in triplicate, were performed. MKN28 cells were transiently transfected with 2 μg of plasmid containing IL-8 and IL-6 promoters and 10 ng of Renilla plasmid as an internal control. The cells were then infected with H. pylori for 9 h. Untreated cells served as controls. Normalized luciferase activity induced by H. pylori infection is expressed as -fold increase in H. pylori-infected cells relative to uninfected controls. Data are expressed as mean (left) and mean ± S.E. (right). *, p < 0.01 compared with WT strains. NS, not significant.
Functions of the IL-6 and IL-8 promoters were examined further by mutating each binding site and testing strains TN and 43504 and their respective mutants (Fig. 4). We used site-directed mutant plasmids of the IL-8 −162/+44 fragment with mutations in four sites (ISRE-like element, AP-1, C/EBP, or NF-κB) and site-directed mutant plasmids of full-length human IL-6 promoter with mutations in four sites (i.e. AP-1, CRE, C/EBP, or NF-κB). In agreement with previous studies (17), mutation of the ISRE-like element, AP-1, and NF-κB binding sites in the IL-8 promoter significantly reduced the luciferase activity induced by WT H. pylori (Fig. 4). Mutation of the C/EBP binding site did not affect the luciferase activity irrespective of H. pylori infection. Also in agreement with previous studies (20), mutation of each binding site in the IL-6 promoter significantly reduced the luciferase activity induced by WT H. pylori (Fig. 4). C/EBP had a clearly different effect on the IL-6 and IL-8 promoters during H. pylori infection, since only the C/EBP binding site in the IL-6 promoter was H. pylori-inducible. Mutation of the NF-κB binding sites in the IL-6 and IL-8 promoters almost completely abolished luciferase activity even when induced by WT H. pylori infection, making it difficult to evaluate the roles of AlpAB in NF-κB activation from this experiment.
FIGURE 4. IL-6 and IL-8 promoter activation following H.pylori infection.
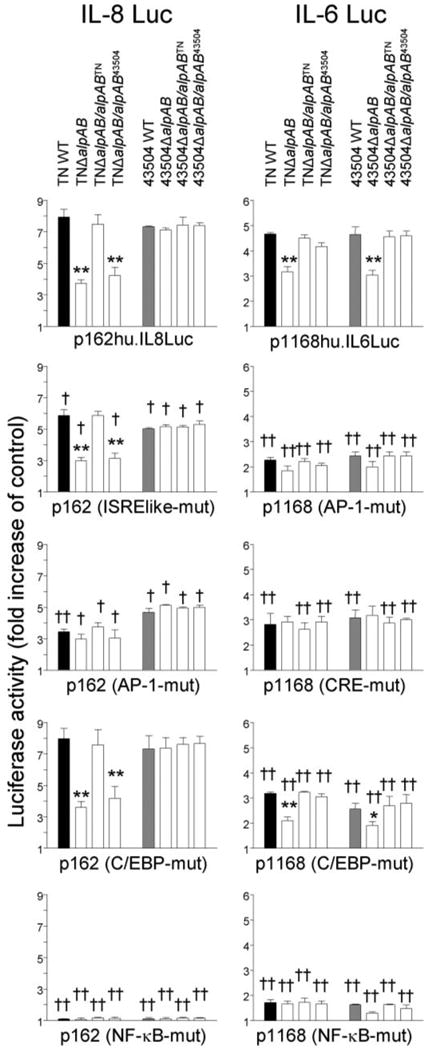
Three independent transfections, each done in triplicate, were performed. Data are expressed as mean ± S.E. MKN28 cells were transiently transfected with 2 μg of plasmids containing various IL-6 and IL-8 promoters and 10 ng of Renilla plasmid as an internal control. The cells then infected with H. pylori for 9 h. Untreated cells served as controls. Normalized luciferase activity induced by H. pylori infection is expressed as -fold increase in H. pylori-infected cells over uninfected controls. *, p < 0.05; **,p < 0.01, compared with WT strains. †, p < 0.05;††, p < 0.01, compared with p162hu.IL8Luc or p1168hu.IL6Luc.
Similar levels of luciferase activity of were observed when plasmids containing mutations of the AP-1 site in the IL-6 and IL-8 promoters were analyzed in WT strains, ΔalpAB mutants, and complemented ΔalpAB mutants using both parental strains TN and 43504. However, since mutation of the AP-1 binding site in the IL-6 and IL-8 promoters reduced the luciferase activity induced by TNΔalpAB or by 43504ΔalpAB, a primary role of AlpAB on AP-1 activation was not confirmed from this experiment. In contrast, mutation of the CRE binding site in the IL-6 promoter affected the luciferase activity induced by WT strains, but not by TNΔalpAB or by 43504ΔalpAB, consistent with AlpAB having a role in activation of CRE binding.
To further examine the effects of ΔalpAB on NF-κB, AP-1, and CRE signal transduction, we used the PathDetect cis-reporting plasmids pNF-κBluc, pAP-1luc, and pCREluc, which contain the luciferase reporter with multiple repeats of the NF-κB, AP-1, and CRE consensus binding sequences (Fig. 5). Luciferase activities of the NF-κB-, AP-1-, and CRE-responsive reporters were induced by each WT H. pylori strain in MKN28 cells. Importantly, NF-κB activity was significantly higher in East Asian WT H. pylori than in Western WT strains (p < 0.01). The activity decreased when using ΔalpAB mutants from East Asian strains but was unchanged using ΔalpAB mutants from Western strains. The decreased activity associated with TNΔalpAB was restored by complementation with alpABTN but not by alpAB43504, confirming that AlpAB from East Asian strains, but not AlpAB from Western strains, was involved in NF-κB activation. In contrast, AP-1 activity was decreased when using ΔalpAB mutants both from East Asian and Western strains. The decrease in activity by TNΔalpAB was completely restored by complementation with alpABTN but only partially restored with alpAB43504. These data suggested that in East Asian strains, AlpAB promotes AP-1 activation, whereas in Western strains, its ability to promote AP-1 activation was partial. All ΔalpAB mutants also failed to induce CRE-dependent luciferase activity, suggesting that AlpAB has a major role in the activation of CRE. In fact, CRE-dependent activation by AlpAB was much greater than that observed with the cag PAI.
FIGURE 5. Transcriptional activity of NF-κB, AP-1, and CRE following H. pylori infection.
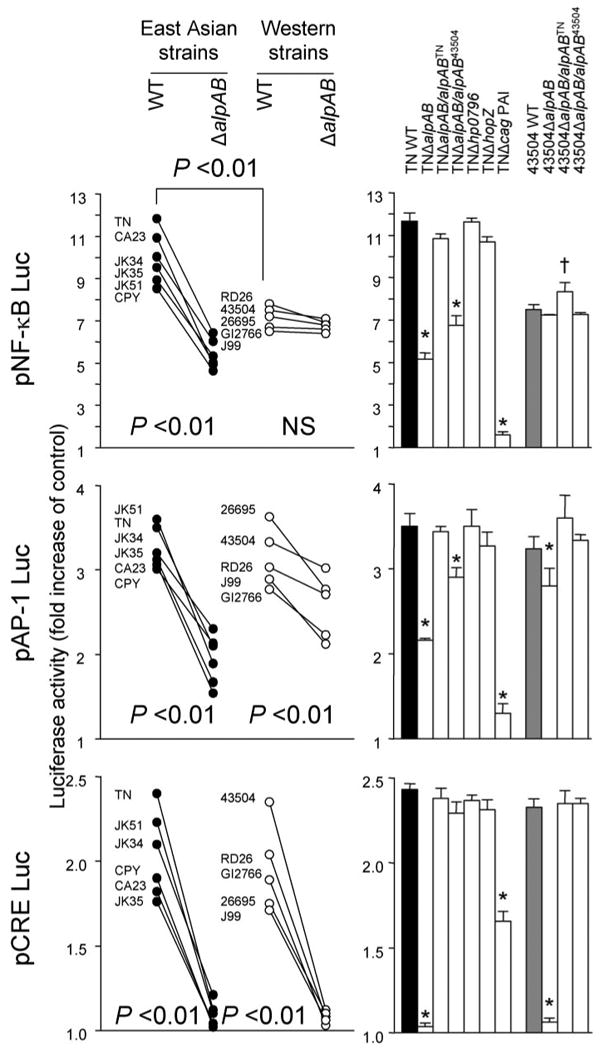
Three independent transfections, each done in triplicate, were performed. MKN28 cells were transiently transfected with 1.2 μg of pNF-κBluc, pAP-1luc, or pCREBluc reporter plasmids separately or together with 10 ng of Renilla plasmid as an internal control. The cells were then infected with H. pylori for 9 h. Untreated cells served as controls. Normalized luciferase activity induced by H. pylori infection is expressed as -fold increase in H. pylori-infected cells relative to uninfected controls. Data are expressed as mean (left) and mean ± S.E. (right). *, p < 0.01 decreased compared with WT strains; †, p < 0.01 increased compared with WT strains.
Effect of AlpAB on Transcription Factor Binding
The above data revealed that AlpAB has a role in the activation of NF-κB, AP-1, and CRE. To confirm our data, we performed electrophoretic mobility shift assay (Fig. 6). Electrophoretic mobility shift assay using strains TN and 43504 showed that both WT strains induced binding complexes for AP-1, CRE, and NF-κB (Fig. 6). Two NF-κB binding complexes (C1 and C2), one AP-1 binding complex (C1), and at least four CRE binding complexes (Cs) were detected. The binding to each site was confirmed to be sequence-specific as shown by competition in the presence of unlabeled wild type oligonucleotide but not in the presence of unlabeled mutated oligonucleotide (data not shown). Super-shift assays showed that p50/p65, c-Fos/c-Jun, and c-Jun/CREB were components of the H. pylori-inducible NF-κB, AP-1, and CRE binding complexes, respectively (supplemental Fig. 3S). Infection with both strains TNΔalpAB and 43504ΔalpAB clearly reduced binding to the CRE and slightly reduced binding to the AP-1 site compared with their parental strains. In contrast, infection with TNΔalpAB, but not with 43504ΔalpAB, reduced binding to the NF-κB site.
FIGURE 6. DNA binding complexes at NF-κB, AP-1, and CRE binding sites following H. pylori infection.
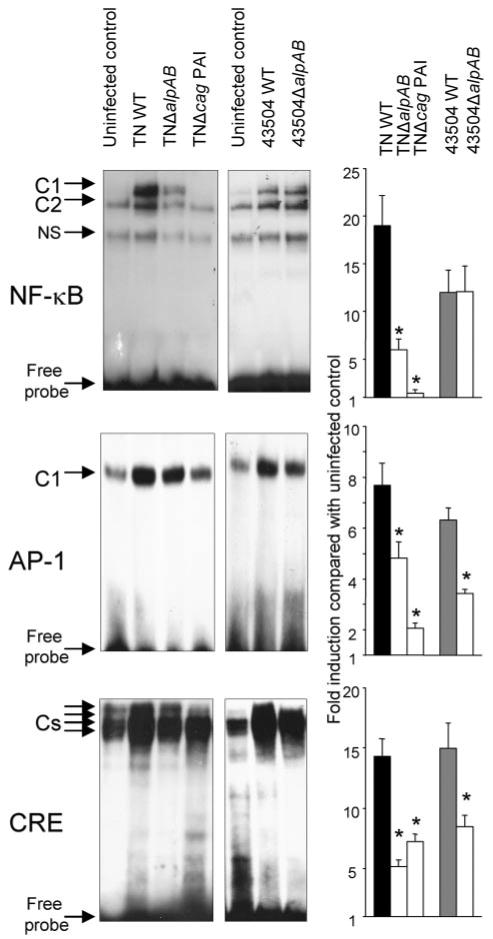
Nuclear extracts were prepared from MKN28 cells either uninfected control (lanes 1 and 5) or infected with WT TN strain, TNΔalpAB, TN2GF4Δcag PAI, WT 43504 strain, and 43504ΔalpAB (lanes 2–4,6, and 7) for 90 min and used for electrophoretic mobility shift assay. Semiquantitation was performed by comparing the amount of radioactive probe in the protein-DNA complexes after standardization with free probe (right). Density was presented as -fold induction over uninfected control. NS, nonspecific.
To further confirm our data, we performed quantitative ELISAs for NF-κB p65, c-Jun, and c-Fos (Fig. 7). ELISA assays confirmed that activation of NF-κB p65 was reduced following deletion of the alpAB gene in East Asian strains. Similar to activation of NF-κB p65, reduced activation of c-Jun by ΔalpAB mutants was also specific for East Asian strains, and activated c-Jun levels were significantly higher using East Asian WT H. pylori than Western WT strains (p < 0.01). Accordingly, decreased activation of NF-κB p65 and c-Jun by TNΔalpAB was only restored by complementation with alpABTN. In contrast, reduced activation of c-Fos by ΔalpAB mutants was observed irrespective of the parental strain and was restored by complementation with intact alpAB genes from both TN and 43504 strains.
FIGURE 7. Activation of NF-κB p65, c-Jun, and c-Fos following H. pylori infection.
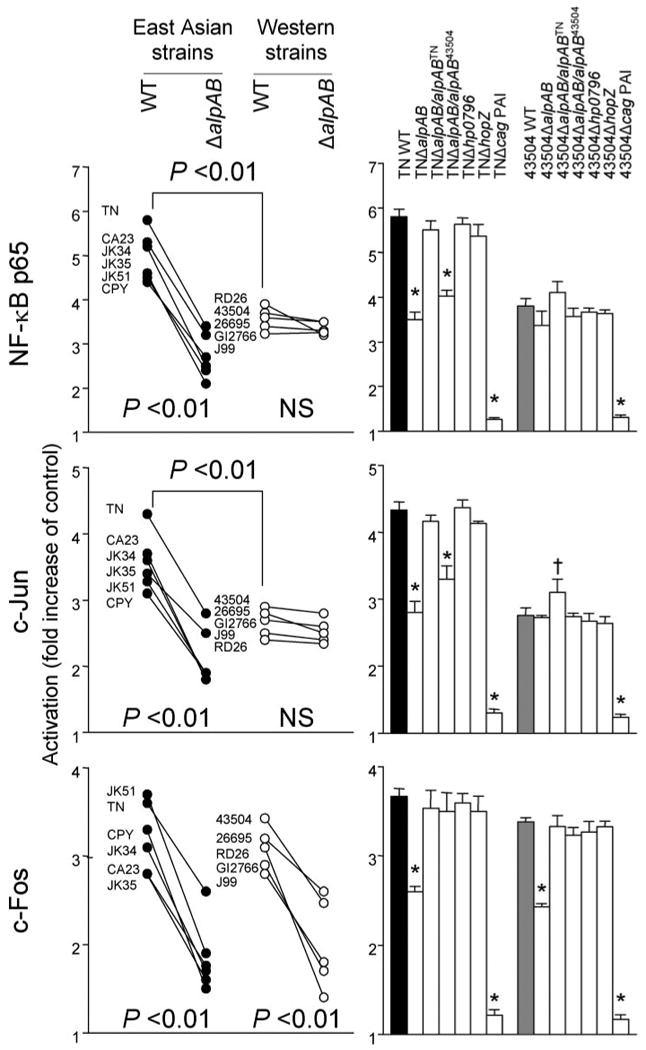
Three independent co-cultures were performed. Nuclear extracts prepared from MKN28 cells (uninfected control or infected with H. pylori for 90 min) were analyzed by ELISA. Data are expressed as mean (left panel) and mean ± S.E. (right panel). *, p < 0.01 decreased compared with WT strains; †, p < 0.01 increased compared with WT strains. NS, not significant.
Role of AlpAB in Expression of Factors That Bind to the AP-1 and CRE Binding Sites
We next measured the levels of c-fos and c-jun mRNA following H. pylori infection in MKN28 cells by real time RT-PCR. Real time RT-PCR analysis showed that peak c-fos and c-jun occurred 1 h after infection irrespective of the strain used (data for 1 h at Fig. 8A). Infection with both East Asian and Western strains of WT H. pylori resulted in marked up-regulation of c-fos and c-jun mRNA expression in MKN28 cells; however, c-jun mRNA levels were significantly higher in East Asian WT H. pylori than in Western WT strains (p < 0.01). Consistent with c-Jun and c-Fos activation measured by ELISA, c-jun mRNA expression was decreased using ΔalpAB mutants from East Asian strains, whereas the levels were not changed with ΔalpAB mutants from Western strains. In contrast, c-fos mRNA expression decreased with ΔalpAB mutants irrespective of the parental strain. Accordingly, decreased c-jun mRNA levels with TNΔalpAB were restored by complementation with alpABTN but were only partially restored with alpAB43504, confirming that AlpAB in East Asian strains, but not in Western strains, was involved in c-jun mRNA expression. The ability of ΔalpAB mutants to reduce c-fos mRNA levels was restored by complementation with intact alpAB genes in both TN and 43504 strains, confirming that AlpAB is involved in c-fos mRNA expression.
FIGURE 8. The expression of c-fos and c-jun mRNAs (A) and CREB phosphorylation (B) in MKN28 cells following H. pylori infection.
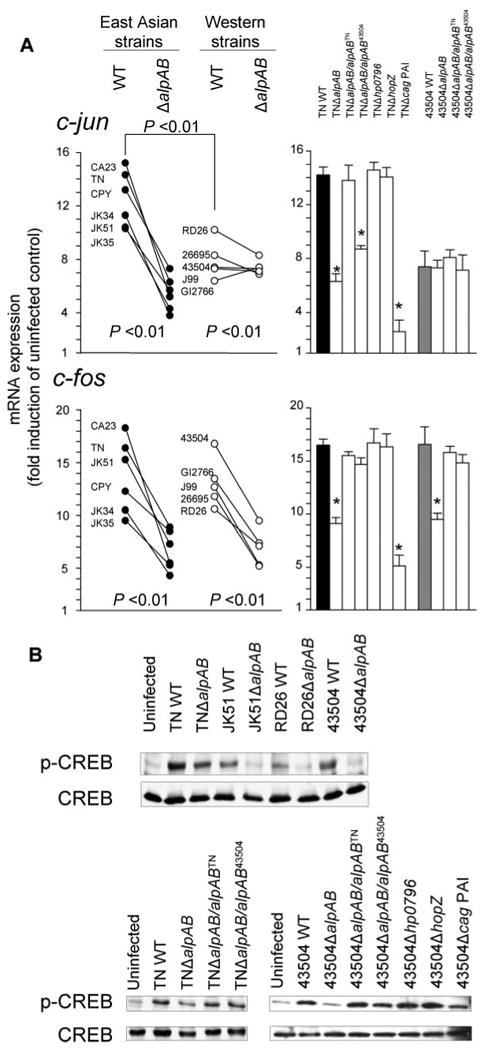
Data at 1 h were presented. A, three independent co-cultures were performed, and each was measured by real time RT-PCR in duplicate. Expression levels were normalized to glyceraldehyde-3-phosphate dehydrogenase mRNA levels and expressed as -fold induction relative to uninfected controls. Data are expressed as mean (left) and mean ± S.E. (right). *, p < 0.01 compared with the WT strain. B, three independent co-cultures were performed, and typical immunoblot results are presented. Typical blots for four parental strains and their ΔalpAB mutants (top) and strains TN and 43504 and their various mutants (bottom) are presented.
We next examined phosphorylation of CREB and ATF-2 proteins following H. pylori infection. Immunoblots revealed that wild type H. pylori infection significantly up-regulated CREB phosphorylation in MKN28 cells with peak levels at 30 min that plateaued at 2 h (data at 1 h are shown in Fig. 8B). Decreased levels of phosphorylated CREB protein were observed in all ΔalpAB infections, and the levels were restored by complementation with intact alpAB genes, suggesting that AlpAB is involved in up-regulating CREB phosphorylation. Phosphorylated ATF-2 protein levels were similar, irrespective of the H. pylori infection in MKN28 cells (data not shown).
Effect of AlpAB on Mitogen-activated Protein Kinase Pathway Activation
The mitogen-activated protein kinase (MAPK) pathway is upstream of CREB, c-Fos, and c-Jun. Therefore, we examined the phosphorylation of MAPKs following H. pylori infection. Recent immunoblot assays have not resolved the relationship between H. pylori infection and phosphorylation of MAPKs (17, 20, 26–31). Thus, we used a different approach employing a reporter gene assay (PathDetect trans-reporting systems) to examine the relationship between the AlpAB and MAPK pathways. In this system, the fusion activator protein consisted of the c-Jun, Elk1, or CHOP transcriptional activation domains fused to the yeast GAL4 DNA binding domain. Following co-transfection of a fusion trans-activator plasmid and the pFR-luc reporter plasmid into mammalian cells, transfected cells were exposed to extracellular stimuli. Activation of Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), or p38 kinase by external cellular stimuli phosphorylate the transcription activation domain of the fusion transactivation protein, thereby inducing transcription of the luciferase gene from the reporter plasmid pFR-luc.
MKN28 cells were co-transfected with the pFR-luc plus the pFA2-Elk1 plasmid. WT H. pylori increased the reporter activity of pFR-luc compared with untreated controls, indicating that H. pylori activated the ERK pathway (Fig. 9). Similarly, co-transfection of MKN28 cells with pFR-luc plus pFA-Jun or pFR-luc plus pFA-CHOP caused increased reporter activity with WT H. pylori, indicating that H. pylori also activates the JNK and p38 pathways.
FIGURE 9. Reporter activities of MAPKs following H. pylori infection.
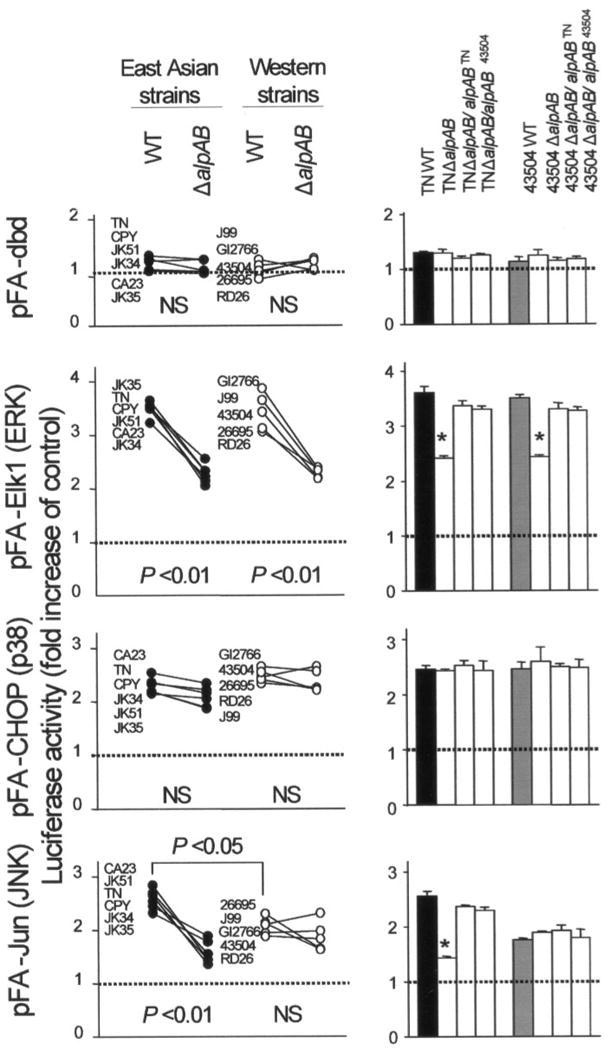
MKN28 cells were co-transfected with 1 μg of the pFR-luc reporter plasmid together with 50 ng of the fusion trans-activator plasmids, pFA2-Jun, pFA2-Elk1, and pFA-CHOP, or the negative control plasmid, pFC-dbd, separately. Transfected cells were subsequently infected with H. pylori for 9 h. Untreated cells served as controls. Normalized luciferase activity induced by H. pylori infection is expressed as -fold increase in H. pylori-infected cells relative to uninfected controls. Three to four independent transfections, each run in duplicate, were performed. Data are expressed as mean (left) and mean ± S.E. (right). *, p < 0.01 compared with the WT strain. NS, not significant.
ΔalpAB mutants from both East Asian strains and Western strains partially suppressed the activity of pFR-luc by pFA2-Elk1, and the activity was restored by complementation of ΔalpAB mutants with an intact alpAB gene, suggesting that AlpAB was involved in activation of the ERK pathway. Similar to the activation of NF-κB and c-Jun, ΔalpAB mutants from East Asian strains also suppressed the activity of the pFR-luc by pFA-Jun. In contrast, ΔalpAB mutants from Western strains did not suppress the activity. Deletions of alpAB genes did not affect the activity of the pFR-luc by pFA-CHOP, suggesting that AlpAB did not activate the p38 pathway. We also confirmed the data using a reporter gene assay by performing an immunoblot for phosphorylation status of ERK, JNK, and p38 (data not shown).
Taken together, AlpAB was involved primarily in activation of the ERK, c-Fos, and CREB in both East Asian and Western strains, whereas AlpAB in East Asian strains, but not Western strains, was involved in activation of the JNK, c-Jun, and NF-κB.
Sequence Differences between East Asian Strains and Western Strains
The above experiments clearly showed that AlpAB in East Asian strains and AlpAB in Western strains play different roles in activation of the JNK/c-Jun/NF-κB pathways and in IL-8 induction. Therefore, we compared the sequences of alpAB genes in the 11 strains we studied, as well as in two additional Western strains deposited in GenBank™. Sequences of the alpA and alpB genes were determined by standard procedures and deposited in GenBank™ (accession numbers AB271157–AB271165). Nucleotide sequences of the alpA and alpB genes clearly differed between East Asian strains and Western strains (supplemental Fig. 4S). Ten positions in AlpA and five positions in AlpB had amino acid sequences specific for either East Asian strains or Western strains (e.g. position 47; alanine in all East Asian strains and threonine in all Western strains) (Fig. 5S).
Role of AlpAB in H. pylori Colonization of C57BL/6 Mice
To examine the effect of alpAB mutants on colonization of the gastric mucosa of mice, we used WT strain TN, TNΔalpAB, and TNΔcag PAI as well as WT strain 43504 and 43504ΔalpAB. Five mice in each group were inoculated with the WT or isogenic mutants, and five mice served as controls. Mice were sacrificed 8 weeks after inoculation.
Both WT strains and TNΔcag PAI colonized mice with similar H. pylori density (Fig. 10). Importantly, 43504ΔalpAB also colonized mice; however, H. pylori density was lower than the parental strain. TNΔalpAB was unable to infect mice. Accordingly, IL-6 mRNA levels and KC protein levels were dramatically decreased in mice infected with 43504ΔalpAB and were similar between mice inoculated with TNΔalpAB and uninfected controls. Both KC and IL-6 induction were regulated by CRE binding in addition to NF-κB binding; thus, the in vivo data correlated well with in vitro data in gastric epithelial cells.
FIGURE 10. H. pylori density and mucosal KC and IL-6 levels in mice 8 weeks after inoculation.
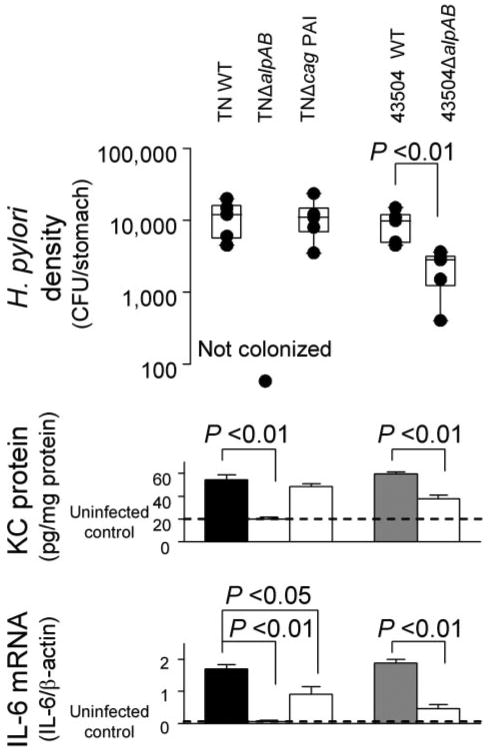
The end of the bars indicates the 25th and 75th percentiles in H. pylori density (top). The median is indicated with a solid line in the box, and the 10th and 90th percentiles are indicated with error bars. Middle, KC protein levels in the gastric mucosa of the mice; bottom, IL-6 mRNA levels in the gastric mucosa of the mice. IL-6 mRNA levels are presented as ratio of 10× IL-6 mRNA to β-actin mRNA. CFU, colony-forming units.
Discussion
Although many studies have investigated the relationship between cag PAI/OipA and signaling transduction pathways (17, 20, 23, 26–33), the effects of other virulence factors on the signaling pathways are largely unknown. In the present study, we found that AlpAB was involved in the stimulation of cellular signaling pathways within gastric epithelial cells, especially the activation of MAPKs, c-Fos, and c-Jun-, CREB-, AP-1-, and NF-κB-related pathways. Importantly, AlpAB in H. pylori from both East Asia and Western countries had the additional ability to activate the ERK, c-Fos, and CREB, whereas only AlpAB from East Asian strains had the ability to activate the JNK, c-Jun, JNK, and NF-κB pathways. Interestingly, we also found that the induction and activation patterns of c-Jun were different from those of c-Fos and that only AlpAB in East Asian strains was involved in c-Jun induction and activation. These facts confirmed that the function of c-Jun was different from that of c-Fos in H. pylori-infected gastric epithelial cells. IL-8 induction was decreased by the deletion of alpAB in East Asian strains but not in Western strains, whereas IL-6 induction was decreased by the deletion of alpAB in all strains studied. Both the IL-6 and IL-8 promoters contain AP-1 and NF-κB binding sites, whereas only the IL-6 promoter contains a CRE binding site. Therefore, binding of c-Fos/CREB to the CRE binding sites should be necessary for maximal IL-6 induction. In contrast, the binding of c-Jun to the AP-1 site and activation of the JNK and NF-κB pathways was important for maximal IL-8 induction. Although c-Fos bound to the AP-1 site in the IL-8 promoter after H. pylori infection, ΔalpAB mutants in Western strains produced IL-8 similar to their parental strains. Thus, our data would suggest that the c-Fos → AP-1 pathway probably does not play an important role in IL-8 induction by H. pylori infection in the gastric epithelial cells.
A previous report showed that ΔalpAB mutants did not induce IL-8 from gastric epithelial cells (12); however, subsequent studies were unable to confirm that finding (10). We previously suggested that ΔalpAB mutants did not induce IL-8 due to the secondary mutations in the cag PAI (10). To confirm the hypothesis, we constructed precise whole alpAB-deleted mutants and complementary ΔalpAB mutants. We confirmed that all ΔalpAB mutants have the potential to induce IL-8, although the ability to induce IL-8 differed between East Asian and Western strains.
AlpAB has been reported to be involved in adherence to human gastric tissue based on adherence of H. pylori to paraffin-embedded tissue sections (8, 9). In the present study, we confirmed that AlpAB was involved in cellular adhesion using live gastric epithelial cells. Although multiple interactions have been reported between H. pylori and the host epithelium (4–7,34,35), it is important that deletion of alpAB alone decreased H. pylori binding to epithelial cells by 60–70%. Interestingly, IL-8 levels in Western strains were independent of the reduction of adherence, although it is well known that adherence of H. pylori to the target cells is required for IL-8 release. However, in our previous study, we confirmed that peak IL-8 levels from gastric epithelial cells co-cultured with H. pylori were similar between using multiplicities of infection of 10 and 100 (17), consistent with the reduction of adherence by 60–70% not influencing IL-8 levels. Experiments in mice showed that although 43504ΔalpAB could colonize mice, the H. pylori density was decreased; TNΔalpAB was unable to infect mice. These results agree with previous studies using guinea pigs (11). It is unknown whether the East Asian type of AlpAB is essential to colonize the mouse gastric epithelium or whether the phenomenon was specific to strains 43504 and TN. Colonization of mice is complicated, and subtle differences in strains, including switch status of several OMPs, have been shown to markedly influence the outcome of animal experiments (16), and not all WT H. pylori will colonize mice. However, it is clear that AlpAB is related to H. pylori density in the stomach irrespective of the regional origin of the H. pylori. Taken together, we confirmed that AlpAB has a function as adhesin both in vitro and in vivo.
Importantly, the ability to activate NF-κB in gastric epithelial cells was significantly higher by East Asian strains than Western strains. We confirmed this phenomenon using 30 strains both from East Asian and Western strain.3 NF-κB in the gastric mucosa is thought to have a central role in regulating genes involved in mucosal inflammatory responses following H. pylori infection. It has long been suspected that NF-κB signaling plays a pivotal role in chronic inflammation-associated malignancies (36–38). Constitutive NF-κB activity has also been observed in a number of human cancers, including gastric cancer (39,40). Moreover, several reports suggest that the main effects of NF-κB on tumor development are exerted at the promotion and progression stages by preventing apoptosis of premalignant cells (reviewed in Refs. 36 and 37). These findings are consistent with the fact that NF-κB activation was higher using strains from East Asia (Japan and Korea), where the prevalence of gastric cancer is extremely high. We found that the different levels of NF-κB activity with H. pylori from East Asia and Western countries depended on AlpAB, and only AlpAB in East Asian strains had the ability to activate the JNK and NF-κB pathways. These data suggest that different functions of AlpAB in different geographic locations might relate to differences in the prevalence of gastric cancer.
The effects of AlpAB varied in a strain-dependent manner in terms of JNK and NF-κB pathways and following IL-8 induction. This has also been observed with CagA (33). CagA has not been thought to be directly involved in IL-8 induction; however, Brandt et al. (33) recently reported that CagA was able to induce IL-8 in a strain-dependent manner (i.e. some cagA mutants induced IL-8, whereas others did not). The mechanisms for these differences are still unknown, and the differences were also observed among Western strains. In contrast, we found that the effects of AlpAB were clearly different between East Asian and Western strains, and accordingly, their structures were also different. Recent reports have suggested that binding of CagA to SHP-2 was stronger in strains with the East Asian CagA structure than those with the Western CagA structure. Only East Asian CagA has intact SHP-2 binding motifs (41). In the case of AlpAB, we found that 10 positions in AlpA and five positions in AlpB had amino acid sequences specific for either East Asian strains or Western strains; however, we could not associate any of these positions specifically to the JNK and NF-κB pathways. Since AlpAB are OMPs and are not likely to enter cells, AlpAB probably interacts with cell surface receptors. Probably, the combination of more active East Asian CagA types (e.g. differences in SHP-2 binding) and more active signaling by AlpAB adhesion might have a synergistic effect in terms of JNK and NF-κB pathways, inflammation, and induction of gastric malignancies. Further experiments using mutants of the East Asian AlpAB-specific structure will be required to examine possible interactions between AlpAB and cell surfaces and to possibly provide insights into the mechanism of H. pylori induction of gastric injury and cancer.
Supplementary Material
Footnotes
This work was supported by National Institutes of Health Grants DK62813 (to Y. Y.), DK50669 (to V. E. R.), and DK56338 (which funds the Texas Gulf Coast Digestive Diseases Center), the Veterans Affairs Merit Review Program (to D. Y. G.), and Deutsche Forschungsgemeinschaft Grant HA2697/9-1 (to R. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBank™/EBI Data Bank with accession number(s) AB271157–AB271165.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
The abbreviations used are: IL, interleukin; AP-1, activator protein-1; C/EBP, CCAAT/enhancer-binding protein; CRE, cAMP-responsive element; CREB, CRE-binding protein; ERK, extracellular signal-regulated kinase; ISRE, interferon-stimulated responsive element; JNK, Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; OMP, outer membrane protein; PAI, pathogenicity island; ELISA, enzyme-linked immunosorbent assay; WT, wild type; RT, reverse transcription.
H. Lu, J. Y. Wu, D. Y. Graham, and Y. Yamaoka, unpublished observation.
References
- 1.Evans DJ, Jr, Evans DG. Helicobacter. 2000;5:183–195. doi: 10.1046/j.1523-5378.2000.00029.x. [DOI] [PubMed] [Google Scholar]
- 2.Odenbreit S. Int J Med Microbiol. 2005;295:317–324. doi: 10.1016/j.ijmm.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Aihara M, Tsuchimoto D, Takizawa H, Azuma A, Wakebe H, Ohmoto Y, Imagawa K, Kikuchi M, Mukaida N, Matsushima K. Infect Immun. 1997;65:3218–3224. doi: 10.1128/iai.65.8.3218-3224.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Borén T. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- 5.Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, ltraja S, Wadstrom T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarstrom L, Borén T. Science. 2002;297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamaoka Y, Kwon DH, Graham DY. Proc Natl Acad Sci U S A. 2000;97:7533–7538. doi: 10.1073/pnas.130079797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peck B, Ortkamp M, Diehl KD, Hundt E, Knapp B. Nucleic Acids Res. 1999;27:3325–3333. doi: 10.1093/nar/27.16.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Odenbreit S, Till M, Hofreuter D, Faller G, Haas R. Mol Microbiol. 1999;31:1537–1548. doi: 10.1046/j.1365-2958.1999.01300.x. [DOI] [PubMed] [Google Scholar]
- 9.Odenbreit S, Faller G, Haas R. Int J Med Microbiol. 2002;292:247–256. doi: 10.1078/1438-4221-00204. [DOI] [PubMed] [Google Scholar]
- 10.Odenbreit S, Kavermann H, Puls J, Haas R. Int J Med Microbiol. 2002;292:257–266. doi: 10.1078/1438-4221-00205. [DOI] [PubMed] [Google Scholar]
- 11.de Jonge R, Durrani Z, Rijpkema SG, Kuipers EJ, van Vliet AH, Kusters JG. J Med Microbiol. 2004;53:375–379. doi: 10.1099/jmm.0.45551-0. [DOI] [PubMed] [Google Scholar]
- 12.Selbach M, Moese S, Meyer TF, Backert S. Infect Immun. 2002;70:665–671. doi: 10.1128/iai.70.2.665-671.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R. Mol Microbiol. 2001;42:1337–1348. doi: 10.1046/j.1365-2958.2001.02714.x. [DOI] [PubMed] [Google Scholar]
- 14.Heuermann D, Haas R. Mol Gen Genet. 1998;257:519–528. doi: 10.1007/s004380050677. [DOI] [PubMed] [Google Scholar]
- 15.McDaniel TK, Dewalt KC, Salama NR, Falkow S. Helicobacter. 2001;6:15–23. doi: 10.1046/j.1523-5378.2001.00001.x. [DOI] [PubMed] [Google Scholar]
- 16.Yamaoka Y, Kita M, Kodama T, Imamura S, Ohno T, Sawai N, Ishimaru A, Imanishi J, Graham DY. Gastroenterology. 2002;123:1992–2004. doi: 10.1053/gast.2002.37074. [DOI] [PubMed] [Google Scholar]
- 17.Yamaoka Y, Kudo T, Lu H, Casola A, Brasier AR, Graham DY. Gastroenterology. 2004;126:1030–1043. doi: 10.1053/j.gastro.2003.12.048. [DOI] [PubMed] [Google Scholar]
- 18.Yamaoka Y, Ojo O, Fujimoto S, Odenbreit S, Haas R, Gutierrez O, El-Zimaity HM, Reddy R, Arnqvist A, Graham DY. Gut. 2006;55:775–781. doi: 10.1136/gut.2005.083014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kudo T, Nurgalieva ZZ, Conner ME, Crawford S, Odenbreit S, Haas R, Graham DY, Yamaoka Y. J Clin Microbiol. 2004;42:2279–2281. doi: 10.1128/JCM.42.5.2279-2281.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu H, Wu JY, Kudo T, Ohno T, Graham DY, Yamaoka Y. Mol Biol Cell. 2005;16:4954–4966. doi: 10.1091/mbc.E05-05-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beswick EJ, Bland DA, Suarez G, Barrera CA, Fan X, Reyes VE. Infect Immun. 2005;73:2736–2743. doi: 10.1128/IAI.73.5.2736-2743.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brasier AR, Jamaluddin M, Casola A, Duan W, Shen Q, Garofalo RP. J Biol Chem. 1998;273:3551–3561. doi: 10.1074/jbc.273.6.3551. [DOI] [PubMed] [Google Scholar]
- 23.Wu JY, Lu H, Sun Y, Graham DY, Cheung HS, Yamaoka Y. Cancer Res. 2006;66:5111–5120. doi: 10.1158/0008-5472.CAN-06-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gan L, Doroudi R, Hagg U, Johansson A, Selin-Sjogren L, Jern S. FEBS Lett. 2000;477:89–94. doi: 10.1016/s0014-5793(00)01788-9. [DOI] [PubMed] [Google Scholar]
- 25.Sawai N, Kita M, Kodama T, Tanahashi T, Yamaoka Y, Tagawa Y, Iwakura Y, Imanishi J. Infect Immun. 1999;67:279–285. doi: 10.1128/iai.67.1.279-285.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kudo T, Lu H, Wu JY, Graham DY, Casola A, Yamaoka Y. Infect Immun. 2005;73:7602–7612. doi: 10.1128/IAI.73.11.7602-7612.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naumann M, Wessler S, Bartsch C, Wieland B, Covacci A, Haas R, Meyer TF. J Biol Chem. 1999;274:31655–31662. doi: 10.1074/jbc.274.44.31655. [DOI] [PubMed] [Google Scholar]
- 28.Wessler S, Hocker M, Fischer W, Wang TC, Rosewicz S, Haas R, Wiedenmann B, Meyer TF, Naumann M. J Biol Chem. 2000;275:3629–3636. doi: 10.1074/jbc.275.5.3629. [DOI] [PubMed] [Google Scholar]
- 29.Wessler S, Rapp UR, Wiedenmann B, Meyer TF, Schoneberg T, Hocker M, Naumann M. FASEB J. 2002;16:417–419. doi: 10.1096/fj.01-0766fje. [DOI] [PubMed] [Google Scholar]
- 30.Keates S, Sougioultzis S, Keates AC, Zhao D, Peek RM, Jr, Shaw LM, Kelly CP. J Biol Chem. 2001;276:48127–48134. doi: 10.1074/jbc.M107630200. [DOI] [PubMed] [Google Scholar]
- 31.Meyer-ter-Vehn T, Covacci A, Kist M, Pahl HL. J Biol Chem. 2000;275:16064–16072. doi: 10.1074/jbc.M000959200. [DOI] [PubMed] [Google Scholar]
- 32.Naumann M, Crabtree JE. Trends Microbiol. 2004;12:29–36. doi: 10.1016/j.tim.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 33.Brandt S, Kwok T, Hartig R, Konig W, Backert S. Proc Natl Acad Sci U S A. 2005;102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beswick EJ, Das S, Pinchuk IV, Adegboyega P, Suarez G, Yamaoka Y, Reyes VE. J Immunol. 2005;175:171–176. doi: 10.4049/jimmunol.175.1.171. [DOI] [PubMed] [Google Scholar]
- 35.Beswick EJ, Pinchuk IV, Minch K, Suarez G, Sierra JC, Yamaoka Y, Reyes VE. Infect Immun. 2006;74:1148–1155. doi: 10.1128/IAI.74.2.1148-1155.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Q, Withoff S, Verma IM. Trends Immunol. 2005;26:318–325. doi: 10.1016/j.it.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 37.Karin M, Greten FR. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 38.Pikarsky E, Ben-Neriah Y. Eur J Cancer. 2006;42:779–784. doi: 10.1016/j.ejca.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 39.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 40.Sasaki N, Morisaki T, Hashizume K, Yao T, Tsuneyoshi M, Noshiro H, Nakamura K, Yamanaka T, Uchiyama A, Tanaka M, Katano M. Clin Cancer Res. 2001;7:4136–4142. [PubMed] [Google Scholar]
- 41.Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, Hatakeyama M. Proc Natl Acad Sci U S A. 2002;99:14428–14433. doi: 10.1073/pnas.222375399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.