

Abstract
The geometric and electronic structures of two mononuclear [(L)CoO2]+ complexes, [(12-TMC)CoO2](ClO4) (1) and [(14-TMC)CoO2](ClO4) (2), have been evaluated using Co K-edge x-ray absorption spectroscopy (XAS) and extended x-ray absorption fine structure (EXAFS) and correlated with density functional theory (DFT) calculations to evaluate the differences in geometric and electronic structure due to changes in the TMC chelate ring size. Co K-edge XAS shows that both 1 and 2 are Co(III) species. Co K-edge EXAFS data show that both 1 and 2 are side-on O2 bound Co(III)-peroxide complexes. A combination of EXAFS and DFT calculations reveal that while the constrained 12-TMC ring in 1 allows for side-on O2 binding to the Co center with ease, the 14-TMC chelate in 2 has to undergo significant distortion of the ring to overcome steric hindrance posed by the four cis-methyl groups of the chelate to allow side-on O2 binding to the Co center. The Ni analogue of 2, [(14-TMC)NiO2]+, has been shown to form an end-on bound Ni(II)-superoxide species. The electronic and geometric factors that determine the different electronic structures of 2 and [(14-TMC)NiO2]+ are evaluated using DFT calculations. The results show that while the sterics of the cis-14-TMC chelate contribute to the geometry of O2 binding and results in an end-on bound Ni(II)-O2- complex in [(14-TMC)NiO2]+, the higher thermodynamic driving force for the oxidation of the Co(II) overcomes this steric constraint, resulting in the stabilization of a side-on bound Co(III)-O22- electronic structure in 2.
1. Introduction
Transition metal active sites of proteins play vital roles in dioxygen binding and reduction in a variety of important biological processes as they facilitate the spin-forbidden interaction between dioxygen and organic matter.1-4 Understanding the mechanism of dioxygen binding and the geometric and electronic structures of the various intermediates generated in the process of dioxygen activation are of key importance in understanding the overall reaction mechanism of metalloproteins. In the case of mononuclear dioxygen activating proteins, the first step involves the formation of a M-O2 (M = metal active site) intermediate, which can either react directly or lead to the formation of a high-valent metal-oxo intermediate. This M-O2 intermediate can potentially assume different O2 binding modes (side-on or end-on), different oxidation states (O2, O2- or O22-), different spin states at the metal center and different exchange stabilizations between the dioxygen moiety and the metal center. Biomimetic efforts have resulted in the synthesis of various M-O2 (M-O) intermediates,5-8 and spectroscopic characterization of these intermediates has been directed towards understanding factors that determine their electronic structure, which can make a key contribution to the reactivity.
Macrocyclic tetradentate N4 ligands, such as 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane (14-TMC), have been effectively used in synthesizing mononuclear metal-oxygen complexes (M = Cr, Mn, Fe and Ni).9-13 Recently, we have successfully synthesized Ni-O2 complexes with a variety of different TMC ligands9,11 in which the ligand is varied from a 12-membered to a 14-membered ring (i.e., 12-TMC to 14-TMC; 12-TMC = 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclododecane). X-ray absorption spectroscopic (XAS) studies and density functional theory (DFT) calculations on [(12-TMC)NiO2] and [(14-TMC)NiO2] have revealed differences in electronic and geometric structures that are associated with large steric hindrance posed by the 14-TMC ring bound to the Ni center with all four methyl groups cis to the O2 binding side (Scheme 1). These differences translate to reactivity differences; while [(12-TMC)NiO2] (side-on bound NiIII-O22-) participates in nucleophilic reactions (aldehyde oxidation), [(14-TMC)NiO2] (end-on bound NiII-O2-) is capable of conducting electrophilic oxidative reactions.11
Scheme 1.
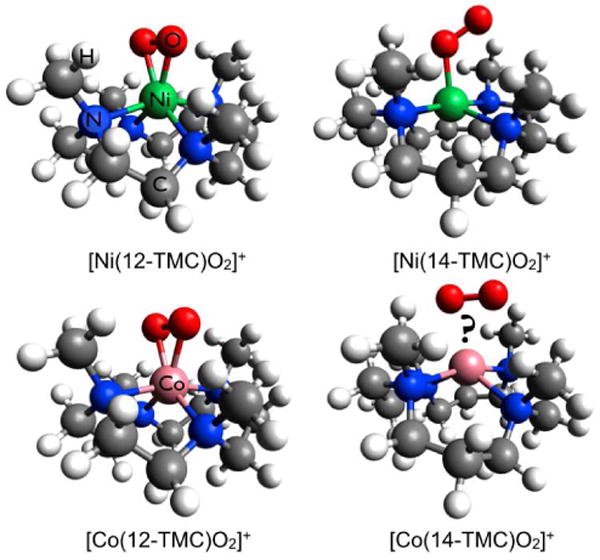
The geometric structures of [Ni(12-TMC)O2]+ (crystal structure), [Ni(14-TMC)O2]+ (obtained from a combination of spectroscopic techniques and DFT calculations) and [Co(12-TMC)O2]+ (crystal structure). The structure of [Co(14-TMC)O2]+ has not been accessible via x-ray diffraction. Color code: Ni(green), Co(pink), O(red), N(blue), C(grey) and H(white)
LCo-O2 complexes are important analogues of biological dioxygen carriers and have been synthesized with porphyrin, salen and N4 macrocyclic ligands.14-16 We have extended these synthetic efforts towards cobalt complexes to characterize reactivity differences of the (TMC)Co-O2 complexes with changing macrocycle ring size.17 In a recent crystal structure of [(12-TMC)CoO2] (1), we have shown a side-on binding mode of the O2 moiety to the Co center (Scheme 1) and showed that 1 is EPR silent. Resonance Raman (rR) data revealed an O-O stretching frequency of 902 cm-1 and Evan's method analysis of the NMR data show that 1 has no unpaired electrons. Together, these data indicate a ground state that is consistent with a Stotal=0, Co(III)-O22- assignment of 1. [(14-TMC)CoO2] (2) has also been synthesized, while no crystal structure or rR data have been accessible, Evans method analysis of the NMR data revealed the presence of unpaired spin on the Co center.
In this study, the differences in electronic and geometric structure between 1 and 2 have been investigated using a combination of Co K-edge XAS, extended x-ray absorption fine structure (EXAFS) and DFT calculations. The factors that lead to the differences in magnetic properties of the two complexes have been evaluated. The role of the macrocyclic ligands in determining the geometric and electronic structure of the Co complexes is evaluated and compared and contrasted to its effects on the (TMC)Ni-O2 complexes. These studies reveal interplay of contributions from steric effects, bonding interaction and the preferential stability of different oxidation states in determining the final electronic and geometric structures.
2. Experimental Section
2.1 Sample Preparation
[Co(12-TMC)O2](ClO4) (1),17 [Co(14-TMC)O2](ClO4) (2),18 [Co(12-TMC)(CH3CN)](ClO4)2 (1pre, precursor of 1)17 and [Co(14-TMC)(CH3CN)](ClO4)2 (2pre, precursor of 2)18 were synthesized as previously described. Caution: Perchlorate salts are potentially explosive and should be handled with care! Since both 1 and 2 are thermally unstable, care was taken to minimize decomposition during loading of the samples into the XAS cells and loading time was minimized. 2 mm lucite XAS cells were used with 37 μm Kapton windows for the solution samples of 1 and 2. The solid samples for 1pre and 2pre were finely ground with BN into a homogeneous mixture and pressed into a 1 mm aluminum spacer between 37 μm Kapton windows. The samples were immediately frozen and stored under liquid N2 conditions. During data collection, the samples were maintained at a constant temperature of ∼10 K using an Oxford Instruments CF 1208 liquid helium cryostat.
2.2 X-ray Absorption Spectroscopy
Co K-edge XAS data were measured on the frozen samples of both the oxygenated complexes, 1 and 2, and their Co(II) precursors, 1pre and 2pre, on the 16 pole, 2 T wiggler beamline 9-3 at the Stanford Synchrotron Radiation Lightsource (SSRL) under standard ring conditions of 3 GeV and 100 mA ring current. A Si(220) double-crystal monochromator was used for energy selection. Other optical components used for the experiments were a Rh-coated harmonic rejection mirror and a cylindrical Rh-coated bent focusing mirror. Data on 1 and 2 were measured in fluorescence mode using a Canberra 30-element solid state Ge detector. Data on 1pre and 2pre were measured in transmission mode using an ionization chamber placed after the sample. Internal energy calibration was accomplished by simultaneous measurement of the absorption of a Co foil placed between two ionization chambers situated after the sample. The first inflection point of the foil spectrum was fixed at 7709.5 eV. The samples were monitored for potential signs of photoreduction throughout the course of data collection. To minimize the effect of beam damage and photoreduction, data were collected on an unexposed region of the sample cell after every 4 scans. Data presented here are 8 scan average spectra for both 1 and 2, a 6 scan average spectra for 1pre and a 4 scan average spectra for 2pre. The data were processed by fitting a second-order polynomial to the pre-edge region and subtracting this from the entire spectrum as background. A three-region spline of orders 2, 3, and 3 was used to model the smoothly decaying post-edge region. The data were normalized in the Pyspline program19 by subtracting the cubic spline and assigning the edge jump to 1.0 at 7730 eV.
Theoretical EXAFS signals χ(k) were calculated by using FEFF (macintosh version 8.4)20-22 and the crystal structure of 1, 1pre and 2pre and a structural model of 2 based on the DFT geometry optimized structure (see section 2.3). The theoretical models were fit to the data using EXAFSPAK.23 The structural parameters varied during the fitting process were the bond distance (R) and the bond variance σ2, which is related to the Debye-Waller factor resulting from thermal motion, and pair-wise static disorder of the absorbing and scattering atoms. The non-structural parameter E0 (the energy at which k = 0) was also allowed to vary but was restricted to a common value for every component in a given fit. The S02 value was set at 0.8 for the entire course of the fit. Coordination numbers were systematically varied in the course of the fit but were fixed within a given fit.
2.3 Electronic Structure Calculations
Gradient-corrected, (GGA) spin-unrestricted, broken-symmetry, density functional calculations were carried out using the ORCA24,25 package on an 8-cpu linux cluster. The Becke8826,27 exchange and Perdew8628 correlation non-local functionals were employed to compare the electronic and geometric structure differences between 1 and 2. The coordinates obtained from the crystal structure of 1 were used as the starting input structure for 1. For 2, the crystal structure of a monomeric [Co(14-TMC)(acac)]3+ species29 was modified to a [Co(14-TMC)O2]+ starting input structure. The core properties basis set CP(PPP) (as implemented in ORCA)24,30 was used on Co and the Ahlrichs' all electron triple-ζ TZVP31,32 basis set was used on all other atoms. A tight convergence criterion was selected. Population analyses were performed by means of Mulliken Population Analysis (MPA). Wave functions were visualized and orbital contour plots were generated in Molden.33 Compositions of molecular orbitals and overlap populations between molecular fragments were calculated using the QMForge.19
3 Results and Analysis
3.1 Co K-edge X-ray Absorption Spectroscopy
The normalized Co K-edge XAS spectra of 1 and 1pre are shown in Figure 1(A). The inset shows the expanded pre-edge region. The pre-edge feature occurs due to a quadrupole-allowed, dipole-forbidden Co 1s→3d transition and is a measure of the ligand field strength at the metal center.34-36 As seen from Figure 1, on going from 1pre to 1, the pre-edge energy position shifts from 7709.2 eV to 7710.1 eV indicating an increase in the ligand field strength in 1. The edge region (∼7715 – 7725 eV) occurs due to the dipole-allowed Co 1s→4p+continuum transition and relative shifts in the edge energy position reflect the change in charge at the Co center (QCo). Furthermore, additional transitions in the rising edge can occur due to charge transfer shakeup processes or multiple scattering effects.37-40 In 1, the edge energy shifts up by ∼1.3 eV (relative to 1pre) to 7720.6, indicating an increase in QCo. This comparison of the Co K-edge XAS data of 1 with its Co(II) precursor 1pre indicates that 1 is one electron oxidized Co(III) species with an increased ligand field strength. The edge-shift combined with rR data,17 which show an O-O stretching frequency of 902 cm-1 in 1, indicate a Co(III)-peroxide electronic structure with the peroxide bound in a side-on fashion to the Co(III). The crystal structure of 1 reveals a six coordinate active site in contrast to the 5-coordinate site in 1pre. The higher coordination number in 1 leads to an increased ligand field strength and is consistent with the ∼0.9 eV pre-edge shift to higher energy.41
Figure 1.
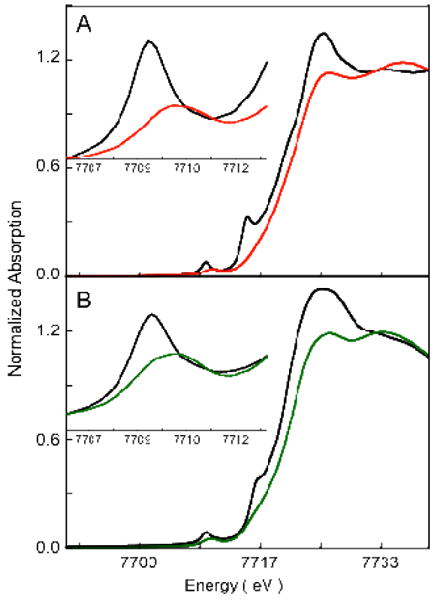
Normalized Co K-edge XAS spectra. A: 1 ( ) and 1pre (
) and 1pre ( ); B: 2 (
); B: 2 ( ) and 2pre (). The insets show the expanded pre-edge region.
) and 2pre (). The insets show the expanded pre-edge region.
The normalized Co K-edge XAS spectra of 2 and 2pre are shown in Figure 1(B). The inset shows the expanded pre-edge region. A similar trend is observed on going from 2pre to 2 as observed on going from 1pre to 1; the pre-edge energy position shifts from 7709.3 to 7710.1 eV, indicating an increase in the ligand field strength in 2. The edge position shifts up from 7719.1 eV in 2pre to 7720.4 eV in 2, indicating an increase in QCo on 2. This increase in the edge energy position indicates that 2 is one electron oxidized species relative to 2pre and is also best described as a Co(III) complex.
3.2 Co K-edge EXAFS
Figure 2(A) shows a comparison of the non-phase shift corrected Fourier Transforms for 1 and 2. The inset shows the EXAFS comparison. The EXAFS analysis of 1 has been published elsewhere17 and the best fit is reproduced in Table 1 for comparison. The comparison shows that on going from 1 to 2, the EXAFS intensity decreases dramatically with a concomitant decrease in the first shell intensity of the Fourier Transforms. Additionally, the first shell in 2 splits and a peak is observed at 1.7 Å (non-phase shift corrected). The second shell intensity decreases almost two-fold relative to that in 1. The best fit to the EXAFS data for 2 are shown in Figure 2(B) and Table 1. The first shell was fit with 2 Co-O components at 1.88 Å and 2 Co-N at 2.10 Å. Attempts to fit the data with 2 additional Co-N components, either at the same distance (2.10 Å) or at longer distances were unsuccessful. The second shell was fit with single and multiple scattering contributions from Co-C (2.9 Å) and the corresponding Co-C-N (3.15 Å) shells. A third shell was required to fit the data with Co-C (3.5 Å) and Co-C-N (3.6 Å) shells. The presence of only 2 short Co-N distances and requirement of third shell single and multiple scattering components indicate the elongation of 2 Co-N bond to significantly longer distances in 2 such that the single scattering component is not required but the combined single and multiple scattering from the ring carbons, which is stronger, is required in the best fit. Thus, the EXAFS data reveal that the 14-TMC ring of 2 has undergone significant distortion with two elongated Co-N distances (evidenced by the presence of long distance multiple scattering), two short Co-N distances and two shorter Co-O distances. In contrast, the EXAFS data and the crystal structure of 1 reveal that the active site has two short Co-O bonds (∼1.88 Å) and four similar Co-N bonds (∼ 2.0 Å).
Figure 2.
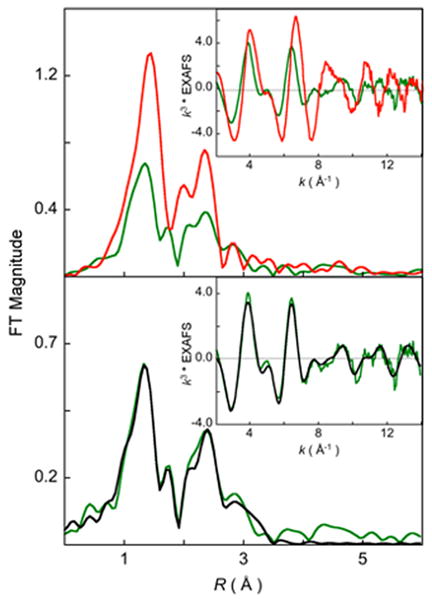
(A) Comparison of the non-phase shift corrected Fourier Transforms for 1 () and 2 (). The inset shows the EXAFS comparison. (B) FEFF best fit results for 2. Data () and fit ().
Table 1. EXAFS Least Squares Fitting Results.
| Complex | Coordination/Path | R(Å)a | σ2(Å2)b | E0 (eV) | F c |
|---|---|---|---|---|---|
| 1 | 2 Co-O | 1.85 | 323 | 1.81 | 0.16 |
| 4 Co-N | 1.99 | 317 | |||
| 8 Co-Cd | 2.84 | 393 | |||
| 24 Co-C-Nd | 3.14 | 382 | |||
| 2 | 2 Co-O | 1.88 | 341 | 2.91 | 0.08 |
| 2 Co-N | 2.10 | 530 | |||
| 4 Co-Cd | 2.90 | 853 | |||
| 8 Co-C-Nd | 3.16 | 853 | |||
| 4 Co-C-Ne | 3.52 | 628 | |||
| 12 Co-C-Ne | 3.57 | 224 | |||
The estimated standard deviations for the distances are in the order of ± 0.02 Å.
The σ2 values are multiplied by 105.
Error is given by Σ[(χobsd − χcalcd)2 k6]/Σ[(χobsd)2k6].
The SS and MS contribution from the carbon atoms adjacent to the two 2.10 Å N atoms.
The longer distance SS and MS distances were required to fit the outer shell of the FT.
3.3 DFT Calculations
3.3.1 [(12-TMC)CoO2]+
Spin unrestricted DFT calculations were performed on the [(12-TMC)CoO2]+ moiety obtained from the crystal structure of 1. [(12-TMC)CoO2]+ can potentially exist in 14 different geometric and electronic configurations: either end-on or side-on binding mode for O2 in combination with S = 0, S = 1 or S = 2 spin states at the cobalt center and S = 1/2 or S = 0 on the O2.42 Geometry optimizations were performed allowing for the calculations to converge to all 14 configurations out of which, six different electronic configurations were achieved. These six configurations were compared to investigate differences in bonding and energies. The energies and structural parameters are given in Table 2. The calculations show that the side-on bound STotal = 0, [(12-TMC)Co(III)O22-]+ electronic structure corresponds to the lowest energy configuration for 1 and is significantly more stable than any other configuration. The DFT calculated geometric and electronic structure of 1 is consistent with the x-ray diffraction, Co K-edge XAS and rR data which are consistent with a Co(III)O22- species. The 1H NMR spectrum of 1 does not show paramagnetic shift over a range from -100 to 300 ppm, which indicates a STotal = 0 in 1, also consistent with the DFT results. It is of interest to note that all the end-on O2 bound species converge to the Co(II)-O2- electronic configuration and so does the high-spin side-on O2 bound species. Only the low-spin STotal = 0, side-on bound species has a Co(III)-O22- ground state, consistent with the optical and x-ray spectroscopy data.43
Table 2. Selected DFT Parameters for [(12-TMC)CoO2]+.
| End-on STotal=0 |
End-on STotal=1 |
End-on STotal=2 |
Side-on STotal=0 |
Side-on STotal=1 |
Side-on STotal=2 |
||
|---|---|---|---|---|---|---|---|
| ΔE (Kcal/mol)a | 0 | -1.3 | 6.6 | -13.6 | -5.1 | 6.5 | |
| Structural Parameters | Co-O | 1.80 | 1.79 | 2.1 | 1.88 | 1.86 | 2.07 |
| O-O | 1.35 | 1.35 | 1.34 | 1.44 | 1.37 | 1.33 | |
| Co-N(N′)b | 2.06(2.11) | 2.06(2.12) | 2.14(2.20) | 2.02(2.05) | 2.07(2.26) | 2.13(2.19) | |
| Spin Densityb Co, O2 |
0.6, -0.6 | 1.3, 0.6 | 2.6, 1.2 | 0, 0 | 1.6, 0.4 | 2.7, 1.2 |
The ΔE is calculated as (E-EEnd-on, S(Total)=0).
The Co-N(N′) distances are the average of two trans Co-N bonds.
The Mulliken spin densities for Co and O2 are shown (the spin densities on both O atoms have been added to represent O2).
3.3.2 [(14-TMC)CoO2]+
Spin unrestricted DFT calculations were performed on a [(14-TMC)CoO2]+ moiety, which was obtained by modifying the crystal structure of the [(14-TMC)Co(acac)]3+ complex. Similar to the [(12-TMC)CoO2]+ calculations above, six ground state configurations were achieved for [(14-TMC)CoO2]+ with a combination of starting end-on and side-on bound geometries with spin-states of S = 0, 1 and 2. The energies and structural parameters of these six configurations are shown in Table 3. In contrast to the [(12-TMC)CoO2]+ calculations, in which the side-on bound STotal = 0 was significantly more stable than any other species, the energies of the six [(14-TMC)CoO2]+ species differ by at most ∼6 kcal/mol. Thus, the energies cannot be used to distinguish between the different electronic configurations in [(14-TMC)CoO2]+.
Table 3. Selected DFT Parameters for [(14-TMC)CoO2]+.
| End-on STotal = 0 |
End-on STotal = 1 |
End-on STotal = 2 |
Side-on STotal = 0 |
Side-on STotal = 1 |
Side-on STotal = 2 |
||
|---|---|---|---|---|---|---|---|
| ΔE (Kcal/mol)a | 0 | -4.1 | 0.52 | -5.1 | -0.5 | 1.0 | |
| Structural Parameters | Co-O | 1.85 | 1.84 | 1.87 | 1.90 | 1.87 | 2.03 |
| O-O | 1.35 | 1.35 | 1.33 | 1.42 | 1.38 | 1.36 | |
| Co-N(N′)b | 2.07(2.18) | 2.08(2.17) | 2.14(2.25) | 2.08(2.13) | 2.15(2.31) | 2.13(2.19) | |
| Spin Densityb Co, O2 |
-0.5, 0.5 | 1.3, 0.7 | 2.6, 1.1 | 0, 0 | 1.8, 0.1 | 2.7, 1.1 |
The ΔE is calculated as (E-EEnd-on, S(Total)=0).
The Co-N(N′) distances are the average of two trans Co-N bonds.
The Mulliken spin densities for Co and O2 are shown (the spin densities on both O atoms have been added to represent O2).
The paramagnetic shift observed in the 1H NMR spectrum of 2 is consistent with an S = 1 ground state (see supporting information).44 Based on the NMR data, either the end-on or side-on bound STotal = 1 species corresponds to the true ground state of 2. DFT calculations show that the end-on bound STotal = 1 species converges to a Co(II)(S=1/2)-O2(S=1/2) (ferromagnetically coupled) ground state, while the side-on bound STotal = 1 species converges to a Co(III)(S=1)-O2(S=0) ground state. The XAS data presented in Figure 1 clearly show that 2 is a Co(III) species and thus confirm that the correct ground state is Co(III)(S=1)-O2(S=0).45
The optimized geometry for 2 in the side-on bound Co(III)(S=1)-O2(S=0) form has 2 Co-O bonds at 1.88 Å, 2 Co-N bonds at 2.15 Å and 2 elongated Co-N bonds at 2.31 Å, indicating a significant distortion in the 14-TMC ring. This distortion is consistent with the EXAFS analysis (Section 3.2 and Scheme 2). Note that the significant elongation of the two Co-N bonds in 2 (2.3 Å is still a bond) is consistent with a distribution of the ligand field at the Co center. This ligand field distribution leads to the stabilization of the intermediate Co(III) S = 1 species. In contrast, most six-coordinate Co(III) complexes have an (S = 0 or 2) ground state.
Scheme 2.
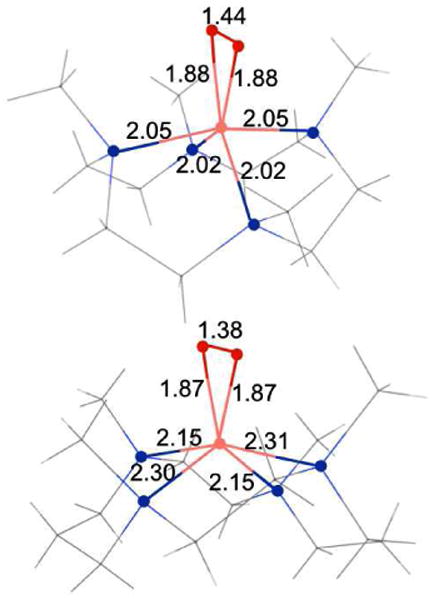
The DFT calculated nearest neighbor distances for 1 (top) and 2 (bottom).
4 Discussion
4.1 Electronic Structure of [Co(12-TMC)O2]+ and [(Co(14-TMC)O2]+
1 and 2 are mononuclear O2 bound Co complexes formed by the addition of excess H2O2 to their starting Co(II) analogues, 1pre and 2pre, respectively, in the presence of base. Understanding the geometric and electronic structures of 1 and 2 is an important step in our overall understanding of the M-O2 bonding at biological metal centers. In this study, the geometric and electronic structures of 1 and 2 have been investigated using Co K-edge and EXAFS techniques coupled with DFT calculations. Comparison of the Co K-edge XAS edge of 1 and 2 to 1pre and 2pre, respectively, show that both 1 and 2 are Co(III) complexes. NMR data show that 1 is diamagnetic, while 2 is paramagnetic with S = 1. Together, these data reveal that the electronic structure of 1 and 2 have Co(III)(S=0)-O22-(S=0) and Co(III)(S=1)-O22-(S=0) ground states, respectively. EXAFS analyses of 1 and 2, coupled with the crystal structure of 1, demonstrates that both are side-on bound Co(III)-O22- species. The experimental data coupled with DFT calculations show that the 4 Co-N bonds in 1 are equidistant at ∼2.0 Å from the Co center. In contrast, 2 has a distorted ligand environment with two Co-N bonds at 2.10 Å, and two elongated Co-N distances (not observed in EXAFS). DFT calculations show that these two Co-N bonds are at ∼2.31 Å and are trans to each other. This alleviates the steric hindrance posed by the methyl groups and allows for the side-on binding of the O2 group. Therefore, the 14-TMC ring in 2 undergoes significant distortion to accommodate the O22- to bind in a side-on fashion, which leads to a decrease in ligand field strength at the Co center and stabilizes its intermediate spin S = 1 ground state.
4.2 Correlations between NiO2 and CoO2 Complexes
An interesting contrast is observed between the O2 bound cobalt and nickel complexes of 12-TMC and 14-TMC. Both [(12-TMC)CoO2]+ (1) and [(12-TMC)NiO2]+ have a side-on O2 bound low-spin M(III)-O22- (M=Co, Ni) ground state. However, while [(14-TMC)CoO2]+ (2) has a side-on O2 bound S=1 Co(III)(S=1)-O22-(S=0) ground state, [(14-TMC)NiO2]+ has an end-on O2 bound S=1/2 Ni(II)(S=1)-O2-(S=1/2) ground state. This difference is best seen in the comparison of the Ni and Co K-edge XAS data of the four complexes as shown in Figure 3. On going from [(12-TMC)NiO2]+ to [(14-TMC)NiO2]+ (Figure 3B), the edge shifts down by 1.8 eV, clearly indicating a dramatic decrease in charge in [(14-TMC)NiO2]+ (i.e, Ni(III) to Ni(II)).46 In addition, Ni K-edge EXAFS analysis revealed that in contrast to [(12-TMC)NiO2]+, which has 2 short Ni-O bonds, [(14-TMC)NiO2]+ has one Ni-O bond. DFT calculations have suggested that the 14-TMC ligand with the 4 cis-methyl groups47 posed a strong steric hindrance to the binding of the O2 in a side-on fashion and only allowed for a relatively hindrance free end-on binding mode with a Ni(II)-O2- electronic configuration. In contrast, in the 12-TMC case, the omission of two ring carbons constrained the ring, leading to an open conformation. This allowed the facile side-on binding of the O2 forming a Ni(III)-O22- species. In contrast to the nickel systems, the similarity in the Co K-edge edge energies for 1 and 2 (Figure 3A) shows that both are Co(III) species and the EXAFS data show that both have side-on bound Co(III)-O22- structures. Thus, despite the steric hindrance posed by the 14-TMC ring, the O2 assumes a side-on binding mode to Co in 2 and to relieve the steric constraints, the 14-TMC ring distorts as observed by experiment and DFT calculations (see section 3.2).
Figure 3.
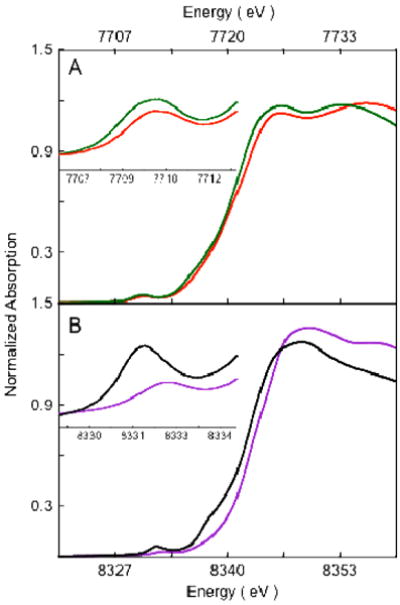
Comparison of the XAS pre-edge and edge shifts. (A): Co K-edge for 1 () and 2 (). (B): Ni K-edge for [(12-TMC)NiO2]+ ( ) and [(14-TMC)NiO2]+ (). Insets show the expanded pre-edge region.
) and [(14-TMC)NiO2]+ (). Insets show the expanded pre-edge region.
To understand this difference in bonding of O2 to Co and Ni in the 14-TMC case, the energy contributions of the different factors that drive side-on versus end-on bonding were evaluated. The conversion of an end-on bound Mn+-O2- to side-on bound M(n+1)+-O22- is affected by different energy terms resulting from a) the oxidation of Mn+, b) the reduction of O2-, c) potential change in exchange stabilization between the Mn+ and O2- spin systems, d) differences in bonding, and e) destabilization due to steric factors. The differences in these energy terms were estimated for the O2n- bound Ni and Co complexes of 14-TMC. To evaluate factor a), DFT geometry optimizations were performed on end-on bound [(14-TMC)Ni(II)O2-]+ and [(14-TMC)Co(II)O2-]+ and the vertical energy changes upon oxidation of these Ni(II) and Co(II) species were calculated.48 The calculations show that the oxidation of the Co(II) species is 11.5 kcal/mol more favorable relative to the Ni(II) species. The relative contribution of factor b) is similar in both the Co and Ni case and can be omitted from consideration. The calculated exchange stabilization in [(14-TMC)Ni(II)O2-]+ is higher than that in [(14-TMC)Co(II)O2-]+ by 1.8 kcal/mol.49 On going from an end-on to a side-on binding mode of O2, both the Co and Ni species gain an additional metal-O bond. A comparison of the Mulliken populations of the valence metal orbitals involved in bonding with the O22- shows only minor relative changes on going from the Ni to the Co complex (see Supporting Information) and can also be omitted from consideration. Thus, the Co species prefers to be in the side-on M(III)-O22- configuration by ∼13.3 kcal/mol relative to the Ni species. The significant stability of the Co(III) oxidation state relative to the Ni(III) oxidation state overcomes the steric hindrance posed by the 4 cis-methyl groups in [(14-TMC)CoO2]+ leading to a distorted 14-TMC ring and a side-on O2 bound Co(III) complex. This difference can be clearly seen in the comparison of the Fourier transforms of the EXAFS data of the Ni/Co, 12- and 14-TMC complexes (Figure 4). The first shell intensity of the two 12-TMC complexes are very similar, while a decrease in intensity is seen on going from [(14-TMC)NiO2]+ to [(14-TMC)CoO2]+, indicating the overall decrease in first shell coordination due to the weakening of two Co-N bonds.
Figure 4.
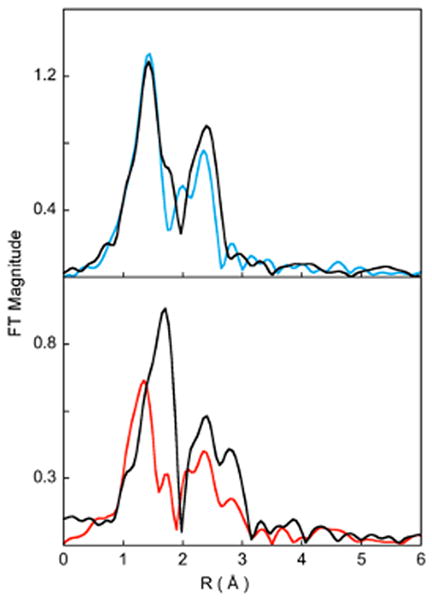
Comparison of the non-phase shift corrected Fourier Transforms for A: 1 () and [(12-TMC)NiO2](ClO4) ( ), B: 2 () and [(14-TMC)NiO2](ClO4) ().
), B: 2 () and [(14-TMC)NiO2](ClO4) ().
In contrast to the 14-TMC systems with steric interactions between the 4 cis-methyl groups, in the steric-free [(12-TMC)NiO2]+ case, the side-on bound Ni(III)O22- form is 8.7 kcal/mol more stable than the end-on O2 bound Ni(II)O2- form. Combining this with the calculated relative stability of the Co(III) case, the steric constraints posed by the 14-TMC ring can be bracketed, as higher than ∼8.7 kcal/mol but less than ∼22 kcal/mol.
In summary, for both the [CoO2]+ and [NiO2]+ systems, end-on O2 binding leads to the stabilization of an [M(II)-O2-]+ electronic structure, while side-on O2 binding leads to the formation of a [M(III)-O22-]+ species. The sterics of the cis-14-TMC chelate contributes to the geometry of O2 binding and therefore the electronic structure for the [Ni(14-TMC)O2]+ complex results in an end-on bound Ni(II)-O2- complex. However, the higher thermodynamic driving force for the oxidation of the Co(II) overcomes this steric constraint, resulting in the stabilization of a side-on bound Co(III)-O22- electronic structure in the 14-TMC complex.
Supplementary Material
Acknowledgments
SSRL operations are funded by the Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology program is supported by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the Department of Energy, Office of Biological and Environmental Research. The research was supported by NRF/MEST of Korea through the CRI and WCU (R31-2008-000-10010-0) Programs (W.N.) and by the NIH DK31450 (E.I.S).
Footnotes
Supporting Information. The DFT geometry optimized coordinated of 1 and 2 in xyz format. DFT calculated molecular orbital contour plots representing the Mulliken spin density of the valence Co d-orbitals in end-on and side-on bound [(12-TMC)CoO2]+ and [(14-TMC)CoO2]+. The 1H NMR spectrum of 2. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Ritimukta Sarangi, Email: ritis@slac.stanford.edu.
Edward I. Solomon, Email: edward.solomon@stanford.edu.
References
- 1.Solomon EI, Sarangi R, Woertink JS, Augustine AJ, Yoon J, Ghosh S. Acc Chem Res. 2007;40:581–591. doi: 10.1021/ar600060t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235–350. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 3.Kovaleva EG, Lipscomb JD. Nat Chem Biol. 2008;4:186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hersleth HP, Ryde U, Rydberg P, Gorbitz CH, Andersson KK. J Inorg Biochem. 2006;100:460–476. doi: 10.1016/j.jinorgbio.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 5.Costas M, Mehn MP, Jensen MP, Que L., Jr Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 6.Lewis EA, Tolman WB. Chem Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 7.Nam W. Acc Chem Res. 2007;40:522–531. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]
- 8.Hatcher LQ, Karlin KD. J Biol Inorg Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 9.Kieber-Emmons MT, Annaraj J, Seo MS, Van Heuvelen KM, Tosha T, Kitagawa T, Brunold TC, Nam W, Riordan CG. J Am Chem Soc. 2006;128:14230–14231. doi: 10.1021/ja0644879. [DOI] [PubMed] [Google Scholar]
- 10.Cho J, Woo J, Nam W. J Am Chem Soc. 2010;132:5958–5959. doi: 10.1021/ja1015926. [DOI] [PubMed] [Google Scholar]
- 11.Cho J, Sarangi R, Annaraj J, Kim SY, Kubo M, Ogura T, Solomon EI, Nam W. Nat Chem. 2009;1:568–572. doi: 10.1038/nchem.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Annaraj J, Suh Y, Seo MS, Kim SO, Nam W. Chem Commun (Camb) 2005:4529–4531. doi: 10.1039/b505562h. [DOI] [PubMed] [Google Scholar]
- 13.Seo MS, Kim JY, Annaraj J, Kim Y, Lee YM, Kim SJ, Kim J, Nam W. Angew Chem Int Ed. 2007;46:377–380. doi: 10.1002/anie.200603414. [DOI] [PubMed] [Google Scholar]
- 14.Hikichi S, Akita M, Moro-Oka Y. Coord Chem Rev. 2000;198:61–87. [Google Scholar]
- 15.Degtyarenko I, Nieminen RM, Rovira C. Biophys J. 2006;91:2024–2034. doi: 10.1529/biophysj.106.083048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norman JA, Pez GP, Roberts DA. In: Oxygen Complexes and Oxygen Activation by Transition Metals. Martell AE, Sawyer DT, editors. Plenum Press; New York: 1988. [Google Scholar]
- 17.Cho, J.; Sarangi, R.; Kang, H. Y.; Lee, J. Y.; Kubo, M.; Ogura, T.; Solomon, E. I.; Nam, W. 2010, submitted.
- 18.Jo Y, Annaraj J, Seo MS, Lee YM, Kim SY, Cho J, Nam W. J Inorg Biochem. 2008;102:2155–2159. doi: 10.1016/j.jinorgbio.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 19.Tenderholt A. Pyspline and QMForge. 2007 [Google Scholar]
- 20.Rehr JJ, Mustre de Leon J, Zabinsky SI, Albers RC. J Am Chem Soc. 1991;113:5135–5140. [Google Scholar]
- 21.Rehr JJ, Albers RC. Red Mod Phys. 2000;72:621–654. [Google Scholar]
- 22.Mustre de Leon J, Rehr JJ, Zabinsky SI, Albers RC. Phys Rev B. 1991;44:4146–4156. doi: 10.1103/physrevb.44.4146. [DOI] [PubMed] [Google Scholar]
- 23.George GN. EXAFSPAK and EDG-FIT. 2000 [Google Scholar]
- 24.Neese F, Olbrich G. Chem Phys Lett. 2002;362:170–178. [Google Scholar]
- 25.Neese F. ORCA: an ab initio, DFT and semiempirical Electronic Structure Package, Version 2.4, Revision 16. 2004 [Google Scholar]
- 26.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 27.Becke AD. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 28.Perdew JP. Phys Rev B. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 29.Simon E, L'Haridon P, Pichon R, L'Her M. Inorg Chim Acta. 1998;282:173–179. [Google Scholar]
- 30.Sinnecker S, Slep LD, Bill E, Neese F. Inorg Chem. 2005;44:2245–2254. doi: 10.1021/ic048609e. [DOI] [PubMed] [Google Scholar]
- 31.Schaefer A, Huber C, Ahlrichs R. J Chem Phys. 1994;100:5829–5835. [Google Scholar]
- 32.Schaefer A, Horn H, Ahlrichs R. J Chem Phys. 1992;97:2571–2577. [Google Scholar]
- 33.Schaftenaar G, Noordik JH. J Comput Aided Mol Des. 2000;14:123–134. doi: 10.1023/a:1008193805436. [DOI] [PubMed] [Google Scholar]
- 34.Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1997;119:6297–6314. [Google Scholar]
- 35.Penner-Hahn JE, Scott RA, Hodgson KO, Doniach S, Desjardins SR, Solomon EI. Chem Phys Lett. 1982;88:595–598. [Google Scholar]
- 36.Shulman RG, Yafet Y, Eisenberger P, Blumberg WE. Proc Natl Acad Sci. 1976;73:1384–1388. doi: 10.1073/pnas.73.5.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kosugi N, Yokoyama T, Asakura K, Kuroda H. Chem Phys. 1984;91:249–256. [Google Scholar]
- 38.Frank P, Benfatto M, Hedman B, Hodgson KO. Inorg Chem. 2008;47:4126–39. doi: 10.1021/ic7021243. [DOI] [PubMed] [Google Scholar]
- 39.Bair RA, Goddard WA. Phys Rev B. 1980;22:2767–2776. [Google Scholar]
- 40.An edge transition is observed at 7714.9 eV in 3 and at 7116.2 eV in 4, which can be attributed to multiple scattering contributions from the linear MeCN group.
- 41.Sarangi R, Aboelella N, Fujisawa K, Tolman WB, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2006;128:8286–8296. doi: 10.1021/ja0615223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The configurations are the side-on and end-on bound forms of: CoII(S=3/2)+O2-(S=1/2), CoIII(S=2)+O22-(S=0), CoIII(S=1)+O22-(S=0), CoII(S=1/2)+O2-(S=1/2), CoII(S=3/2)+O2-(S=-1/2), CoIII(S=0)+O22-(S=0), CoII(S=1/2)+O2-(S=-1/2).
- 43.The bond distances and Mulliken spin densities on the intermediate spin STotal=1 species indicate a mixed Co(III)-O22-/ Co(II)-O2- character in the ground state.
- 44.Note that in an earlier publication 2 was assigned a S=2 ground state based on the observation of paramagnetic NMR data.17 Recent NMR data indicate that the previous assignment was erroneous owing to the presence of extraneous contamination.
- 45.Note that the calculated energy of Co(III)(S=1)-O2(S=0) is higher than the Co(III)(S=0)-O2(S=0) species by 4.6 Kcal/mol. However, the NMR data clearly show that 2 is paramagnetic, excluding the possibility that 2 is Co(III)(S=2)-O2(S=0).
- 46.Sarangi R, Dey M, Ragsdale SW. Biochemistry. 2009;48:3146–56. doi: 10.1021/bi900087w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.FEFF fits to the EXAFS data were consistent with all four methyl groups cis to the O2 binding side.
- 48.The calculations were performed in the appropriate spin states and were checked to confirm that the vertical oxidation only affected the metal center.
- 49.The exchange stabilization energy was obtained from the optimized energies of the high-spin and low-spin states of [(14-TMC)Ni(II)O2-]+ and [(14-TMC)Co(II)O2-]+.50-51
- 50.Noodleman L. J Chem Phys. 1981;74:5737–5743. [Google Scholar]
- 51.Noodleman L, Davidson ER. Chem Phys. 1985;109:131–138. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.