

Abstract
Cannabinoid receptor antagonists have been utilized extensively in vivo as well as in vitro, but their selectivity has not been fully examined. We investigated activation of sensory neurons by two cannabinoid antagonists – AM251 and AM630. AM251 and AM630 activated trigeminal (TG) sensory neurons in a concentration-dependent fashion (threshold 1 μM). AM251 and AM630 responses are mediated by the TRPA1 channel in a majority (90–95%) of small-to-medium TG sensory neurons. AM630 (1–100 μM), but not AM251, was a significantly more potent agonist in cells co-expressing both TRPA1 and TRPV1 channels. We next evaluated AM630 and AM251 effects on TRPV1- and TRPA1-mediated responses in TG neurons. Capsaicin (CAP) effects were inhibited by pre-treatment with AM630, but not AM251. Mustard oil (MO) and WIN55,212-2 (WIN) TRPA1 mediated responses were also inhibited by pre-treatment with AM630, but not AM251 (25uM each). Co-treatment of neurons with WIN and either AM630 or AM251 had opposite effects: AM630 sensitized WIN responses, whereas AM251 inhibited WIN responses. WIN-induced inhibition of CAP responses in sensory neurons was reversed by AM630 pre-treatment and AM251 co-treatment (25μM each), as these conditions inhibit WIN responses. Hindpaw injections of AM630 and AM251 did not produce nocifensive behaviors. However, both compounds modulated CAP-induced thermal hyperalgesia in wild-type mice and rats, but not TRPA1 null-mutant mice. AMs also partially regulate WIN inhibition of CAP-induced thermal hyperalgesia in a TRPA1-dependent fashion. In summary, these findings demonstrate alternative targets for the cannabinoid antagonists, AM251 and AM630, in peripheral antihyperalgesia which involve certain TRP channels.
Keywords: pain, TRP, cannabinoids, peripheral
Introduction
Cannabinoids exert profound peripherally meditated thermal and mechanical antinociception and antihyperalgesia in several animal pain models (Calignano et al., 1998; Ibrahim et al., 2003; Johanek et al., 2001; Johanek and Simone, 2004; Khasabova et al., 2008; LaBuda et al., 2005; Malan et al., 2001; Richardson et al., 1998). There is an agreement that cannabinoids utilize multiple pathways to evoke peripheral antinociceptive and antihyperalgesic activities. These pathways are mediated via either metabotropic CB1 (Agarwal et al., 2007; Richardson et al., 1998), CB2 receptors (Malan et al., 2003), or ionotropic transient receptor potential (TRP) channels (Akopian et al., 2009; Akopian et al., 2008; Patwardhan et al., 2006b; Sagar et al., 2004). Cannabinoids appear to exert this peripheral antinociception and antihyperalgesia by either directly inhibiting sensory neurons (Ahluwalia et al., 2000; Akopian et al., 2008; Patwardhan et al., 2006b) or modulating sensory neuron function indirectly via recruitment of non-neuronal peripheral cells such as keratinocytes (Ibrahim et al., 2005), mast cells (Jonsson et al., 2006; Samson et al., 2003), or macrophages (McCoy et al., 1999).
Peripheral mechanisms of cannabinoid actions have been evaluated using both pharmacological (Johanek and Simone, 2004; Malan et al., 2001; Richardson et al., 1998) and genetic approaches (Agarwal et al., 2007; Akopian et al., 2008; Ibrahim et al., 2006). It is important to note that the local injection of CB1 and CB2 receptor antagonists often occur at relatively high concentrations (high μM-low mM range) in several pain models (Fox et al., 2001; Ibrahim et al., 2005; Ibrahim et al., 2006; Malan et al., 2001). In contrast, in vitro binding assay demonstrated that IC50 for AM251, a CB1 antagonist and AM630, a CB2 antagonist are ≈8 nM and ≈31 nM, respectively (Gatley et al., 1996; Hosohata et al., 1997). The specificity of these antagonists beyond CB1 and CB2 has not been evaluated in detail. This is an important question, as additional actions of these antagonists on ion channels involved in nociception could lead to a non-CB1/CB2 mechanism for antagonizing cannabinoid-mediated inhibition of peripheral nociceptors. This concern is supported by the observation that the cannabinoid agonist, WIN 55, 212-2, produced equivalent antinociception in wild-type (WT) and CB1−/− mice (Ibrahim et al., 2006). In contrast, the antinociceptive effects of WIN55, 212-2 were blocked by the CB1 receptor-selective antagonist SR141716A (Bridges et al., 2001; Fox et al., 2001). One possibility for this discrepancy is that the SR141716A could be non-selective for CB1 at high doses. It is possible that TRP channels could also be modulated by cannabinoid antagonists. Thus, an antagonist of the putative anandamide transporter, AM404, gates TRPV1 (Zygmunt et al., 2000). In this study, we investigated activation of sensory neurons by a wide range of concentrations of the frequently used cannabinoid antagonists, AM251 (for CB1) and AM630 (for CB2).
Methods
Animals and primary sensory neuron culture
Breeding colonies for TRPA1 and TRPV1 channel null-mutant mice were provided by Dr. Kevin Kwan and The Jackson Laboratory, respectively (Caterina et al., 2000; Kwan et al., 2006). TRPA1 null-mutant mice were generated on the B6129P1/F2J background. Sprague-Dawley rats, 45–60 days old, were obtained from a commercial breeder (Charles River Laboratories, Inc., Wilmington, MA or Harlan, Indianapolis, IN, USA). All experiments conformed to protocols approved by the University Texas Health Science Center at San Antonio (UTHSCSA) Animal Care and Use Committee (IACUC). We followed guidelines issued by the National Institutes of Health and the Society for Neuroscience to minimize the number of animals used.
The animals were deeply anaesthetized with isoflurane and subsequently sacrificed. The trigeminal ganglia (TG) sensory neuron culture was generated as previously described (Akopian et al., 2007; Salas et al., 2009). Neurons were plated at low-density on poly-D-lysine/laminin coated coverslips or plates (Clontech, Palo Alto, CA). Cells were maintained in the presence or absence of 100 ng/ml NGF-7.02S (Harlan, Indianapolis, IN) as specified for each experiment. Ca2+-imaging and patch clamp electrophysiology were performed 24–72 h after plating.
Constructs and Heterologous Expression in CHO cells
Expression plasmids of TRPV1 (accession number - NM031982) in pcDNA3 (Invitrogen, Carlsbad, CA) and TRPA1 (NM177781) in pcDNA5/FRT (Invitrogen) were used. Expression constructs with a visual marker (green fluorescent protein expressing pEGFP-N1 from Clontech) were delivered into Chinese hamster ovary (CHO) cells using PolyFect (Qiagen, Valencia, CA) according to manufacturers’ protocols. CHO cells were subjected to experimental procedures within 24–48 h after transfection.
CGRP release assay
CGRP release assay of TG neurons was performed as previously described (Patwardhan et al., 2005; Patwardhan et al., 2006a). Briefly, after two initial washes, a 15 min baseline sample was collected. Cultured TG neurons were then pre-treated or co-treated with drugs for 15 min and samples were collected after exposure to WIN. All the supernatants were collected for analysis of iCGRP content by radioimmunoassay (RIA). The basal release was typically 6–8 fmol per well. RIA was performed as previously (Garry et al., 1994; Patwardhan et al., 2006a). Primary antibody against CGRP (final dilution 1:1,000,000) was kindly donated by Dr. M.J. Iadarola (NIDCR/NIH).
Ca2+ Imaging in TG neurons and CHO cells
The Ca2+ imaging experiments and ratiometric data conversion were performed as previously described (Akopian et al., 2007). The net changes in Ca+2 influx were calculated by subtracting the basal [Ca+2]i (mean value collected for 60 s prior to addition of the first compound) from the peak [Ca+2]i value achieved after exposure to the drugs. Ca2+ accumulations above 50 nM were considered positive. This minimal threshold criterion was established by application of 0.1% DMSO as a vehicle.
Electrophysiology
Recordings were made in whole-cell perforated patch voltage clamp (holding potential (Vh) of −60mV) configuration at 22–24° C from the somata of small-to-medium sized cultured TG neurons (15–40 pF) or CHO cells. Data were acquired and analyzed using an Axopatch 200B amplifier and pCLAMP9.0 software (Molecular Devices). Recording data were filtered at 0.5 kHz and sampled at 2 kHz. Access resistance (Rs) was compensated (40–80%) where appropriate up to the value of 13–18 MΩ. Data were rejected when Rs changed >20% during recording, leak currents were >50pA, or input resistance was <200 MΩ. Currents were considered positive when their amplitudes were 5-fold bigger than displayed noise (in root mean square).
Standard external solution (SES) contained (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 D-glucose and 10 HEPES, pH 7.4. The pipette solution consisted of (in mM): 140 KCl, 1 MgCl2, 1 CaCl2, 10 EGTA and 10 HEPES pH 7.3. The pipette solution for the perforated patch configurations consisted of (in mM): 140 KCl, 1 MgCl2, 10 HEPES pH 7.3 and 250 μg/ml amphotericin B (Sigma, St. Louis, MO). Drugs were applied using a fast, pressure-driven and computer controlled 8-channel system (ValveLink8; AutoMate Scientific, San Francisco, CA).
Behavioral assays
Sprague-Dawley rats, 45–60-days old, and wild-type (WT) and TRPA1 null-mutant (TRPA1 KO) mice were used in behavioral assays. Two types of behavior assays were conducted. First, drug-induced nocifensive behavior was measured by observations of licking and flinching behavior over a 15 min time period. Licking and flinching was represented as spent time by the animals during the behavior (i.e. licking and flinching). Drug concentrations are specified in the “Legends to figures”.
Second, capsaicin (CAP)-induced thermal hyperalgesia was utilized as a pain model (Patwardhan et al., 2006b). Thermal withdrawal latencies were measured using methods described previously (Hargreaves et al., 1988). The vehicle for cannabinoids and CAP was 5% DMSO and 5% Tween-80 (Johanek et al., 2001). After habituation and collection of basal withdrawal latencies, animals were injected ipl with the indicated drug combination at −15 min, then injected with CAP (10μg for rats; 1μg for mice) at time point “0”, with measurement of paw withdrawal latencies at 5 and 10 min for evaluation of CAP induced thermal hyperalgesia. Thermal responses in mice were measured at 5 min points after CAP administration. All responses were collected by observers blinded to treatment allocation.
Data analysis
GraphPad Prism 4.0 (GraphPad, San Diego, CA) was used for statistical analysis. The data in Figs were given as mean ± standard error of the mean (SEM), with the value of n referring to the number of analyzed cells or trials for each group. All experiments were performed at least in triplicate. A significant difference between groups was assessed by one-way analysis of variance (ANOVA) with Bonferroni’s multiple comparison post-hoc test. In studies comparing two groups, data were analyzed using a paired or unpaired t-test. A difference was accepted as significant when p<0.05, <0.01 or <0.001 and are identified by *, ** and ***, respectively.
Results
Cannabinoid antagonists AM251 and AM630 activate TG sensory neurons
The application of cannabinoid receptor antagonists AM251 and AM630 (10 μM each) activated a robust Ca2+ accumulation in a subset (≈35–40%) of TG neurons. The AM251 and AM630-evoked Ca2+ influxes into TG sensory neurons were concentration-dependent, and fitted using Hill’s equation (Fig 1A). The EC50 for AM251 and AM630 were 7.37 μM and 15.6 μM, respectively, although AM630 exhibited about four-fold increased efficacy compared with AM251. We next evaluated whether the presence of NGF (100 ng/ml) in culture media altered the magnitude of AM251 and AM630 responses. Figure 1B demonstrates that the 72h-exposure of TG neurons to NGF significantly increased AM251 and AM630 responses.
Figure 1. Activation of TG sensory neurons by AM251 and AM630.
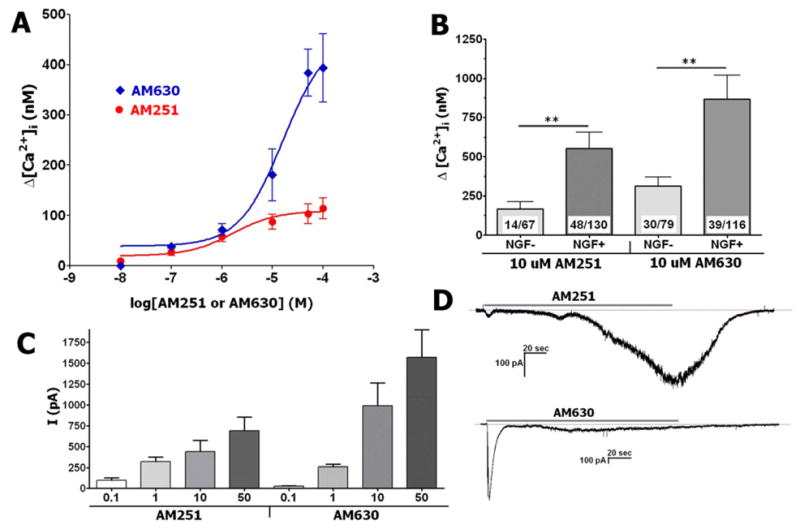
(A) Concentration response curves for AM251 and AM630-evoked Ca2+ accumulation in TG neurons. Drugs were applied for 3 min. TG sensory neurons were cultured for up to 24 h in presence of NGF, n=30–53. (B) Regulation of AM251 and AM630 responses by NGF. TG sensory neurons were cultured for 72 h in the absence or presence of NGF (100 ng/ml). Numbers of analyzed and responsive neurons are indicated. (C) Peak of AM251- and AM630-generated whole-cell currents (Vh=−60 mV) in TG sensory neurons. Drug concentrations (in μM) are indicated. TG sensory neurons were cultured for up to 24 h in presence of NGF, n=6–8. (D) Typical traces recorded during application of AM251 and AM630 (10 μM each) to 24 h-cultured TG neurons. Durations of particular drug applications (3 min) are marked with horizontal bars.
To independently replicate the findings by Ca2+-imaging, AM251- and AM630-gated whole-cell currents (IAM251 and IAM630) were measured. A wide range of concentrations (0.1–50 μM) of AM251 and AM630 are able to generate currents in TG sensory neurons (Fig 1C). The activation threshold for IAM251 and IAM630 were 0.1 and 1 μM, respectively, although IAM630 was substantially larger than IAM251 at concentrations above 10 μM (Fig 1C). Further, IAM630 had visibly faster activation and desensitization kinetics (Fig 1D). Altogether, AM630 and AM251 are able to activate a subset of TG neurons with different efficacies.
AM251 and AM630 activate TRPA1
Since certain cannabinoids activate TRPV1 and TRPA1 channels (Akopian et al., 2009), we evaluated whether AM251 and AM630 also act on these channels. Whole-cell recordings collected from CHO cells transfected with either TRPA1 or TRPV1 confirmed that AM251 and AM630 are able to activate TRPA1 (Fig 2A), but not TRPV1 channels (Fig 2B). TRPV1-expressing CHO cells also did not respond to saturated concentrations of AM251 and AM630 (100 μM; data not shown). In negative control experiments, the application of AM251 and AM630 (25 μM each) were not able to trigger currents in either mock or GFP transfected CHO cells (data not shown).
Figure 2. AM251 and AM630 activate TRPA1 in TG sensory neurons.
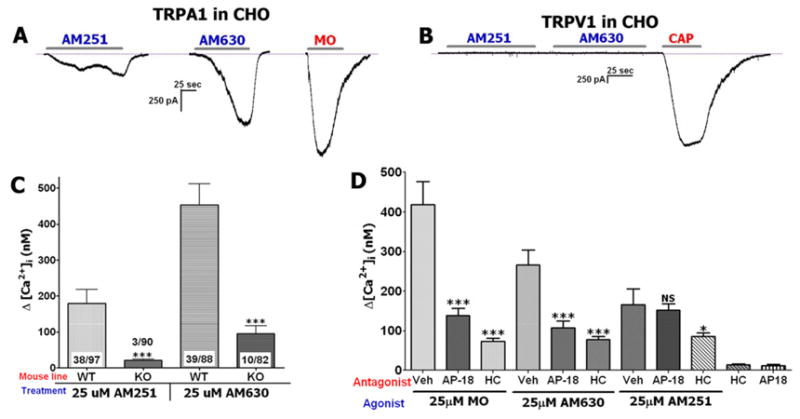
(A) Representative trace shows that AM251 and AM630 (25 μM each) activate TRPA1-expressing CHO cells. Mustard oil (MO; 25 μM) was used as a positive control stimulus. AM251 and AM630 were applied to separate cells to avoid potential cross-desensitization. 8 of 8 (8/8) TRPA1-expressing CHO cells responded (>50 pA current) to AM630 and 8/10 to AM251. (B) Representative trace shows that AM251 and AM630 (25 μM each) do not activate TRPV1-expressing CHO cell. Capsaicin (CAP; 100 nM) was used as a positive control stimulus. Recording was conducted on 14 TRPV1-expressing CHO cells. No cells responded to AM251 (n=8) or AM630 (n=6). Every TRPV1-expressing cell was CAP responsive. Durations of particular drug applications are marked with horizontal bars. (C) AM251 and AM630 do not generate Ca2+ influx into TG neurons from TRPA1 null mutant mice (KO). WT indicates TG cultures from wild-type mice, n=20–39. (D) AM251 and AM630-triggered Ca2+ accumulation in TG sensory neurons was substantially blocked by two TRPA1 specific antagonists, AP-18 (25 μM) and HC030031 (HC; 25 μM). MO was used as a positive control for HC blockage, n=36–67.
Prior studies have demonstrated that cannabinoids can activate other TRP channels (Akopian et al., 2009; Qin et al., 2008). Therefore, we next examined whether TRPA1 is solely responsible for mediating AM251 and AM630 responses in sensory neurons. Figure 2C illustrates that AM251 and AM630-generated Ca2+ influx was eliminated in almost all tested neurons in TRPA1 null-mutant (KO) mice. Several large diameter sensory neurons exhibited small [Ca2+]i accumulation (<200 nM) induced by 25 μM AM251 and AM630 (Fig 2C), although this population comprised <5–10% of sampled TG neurons. AM251 and AM630 responses were also effectively blocked by the TRPA1 specific antagonist HC030031 (HC; 25 μM; Fig 2D) (Hinman et al., 2006). To address the question of whether AM251 and AM630 act on the same, or closely located, sites of the TRPA1 channel, another TRPA1-specific antagonist AP-18 was employed (Petrus et al., 2007). AP-18 effectively blocked AM630 as well as MO (positive control), but not AM251 responses (Fig 2D). This observation implies that AM251 and AM630 could work on two different TRPA1 sites. It should be noted that HC did not completely block MO (25 μM) responses and this finding confirms previous reports (Kremeyer et al., 2010). Altogether, AM251 and AM630 responses are mediated by the TRPA1 channel in a majority of TG small-to-medium sensory neurons.
AM251 and AM630 activation of TRPA1 is modulated by TRPV1
TRPA1 is almost exclusively present in the TRPV1-positive subset of TG sensory neurons (Diogenes et al., 2007; Obata et al., 2005). These channels are able to form a complex in sensory neurons (Staruschenko et al., 2010). Therefore, we evaluated whether TRPA1 activation by a wide-range of AM251 and AM630 concentrations depends upon the presence of the TRPV1 channel. Figure 3A indicates that AM251, at concentrations of 0.1–25 μM, activates TRPA1 and TRPV1/TRPA1 expressing CHO cells with similar magnitudes. However, at a high concentration (100 μM), AM251 generates greater Ca2+ influx in CHO cells co-expressing TRPV1 and TRPA1 (Fig 3A). The EC50 for AM251 activation of TRPV1/TRPA1 co-expressing cells is ≈21 μM. In contrast, AM630 is a substantially more effective agonist for TRPV1/TRPA1 co-expressing cells throughout all tested concentrations (1–100 μM; Fig 3B). Thus, the Emax for AM630 is 8–9-fold greater in TRPV1/TRPA1 versus TRPA1 expressing cells. Interestingly, this type of effect has previously been reported for another TRPA1 agonist, namely the cannabinoid AM1241, which produces greater responses in TRPV1/TRPA1-coexpressing cells compared to TRPA1-containing cells (Akopian et al., 2008). The EC50 for AM630 is comparable in value in both TRPA1 and TRPV1/TRPA1 expressing CHO cells (2 and 4.6 μM, respectively). Altogether, AM630 is a more effective agonist for TRPV1/TRPA1 containing CHO cells as compared to AM251.
Figure 3. AM251 and AM630 activation of TRPA1 is modulated by TRPV1.

(A) Concentration-response curves for AM251 in TRPA1 and TRPA1/TRPV1-expressing CHO cells, n=25–48. (B) Concentration-response for AM251 in TRPA1 and TRPA1/TRPV1-expressing CHO cells, n=15–52.
Effects of AM251 and AM630 on TRPA1- and TRPV1-mediated responses
To understand the cellular effects of AM251 and AM630 on sensory neurons, we investigated whether these cannabinoid antagonists could alter the responsiveness of TRPV1 and TRPA1 channels. The AM251 and AM630 activation of TRPA1 elevates [Ca2+]i in TG neurons, possibly leading to a Ca2+-dependent pharmacological desensitization of TRPV1 (Akopian et al., 2007; Jeske et al., 2006; Koplas et al., 1997). Since TRPA1 and TRPV1 undergo interaction (Salas et al., 2009; Staruschenko et al., 2010), the co-application of AM251 and AM630 could in principle alter TRPV1-mediated responses due to putative allosteric changes in the TRPA1 channel. Therefore, we examined the modulation of CAP responses by pre-treatment as well as co-treatment with AM251 and AM630 (25 μM each). Figure 4A demonstrates that co-treatment of neurons by these cannabinoid antagonists produced no statistically significant changes in [Ca2+]i accumulation in TG neurons. In contrast, pre-treatment for 5 min of sensory neurons by AM630, but not AM251, significantly inhibited CAP-evoked Ca2+ influx (p<0.01; Fig 4B). It could be noted that AM’s were washed away prior to agonist (i.e. CAP) application. This difference in AM251 and AM630 actions could be explained by the amount of Ca2+ influx triggered by these drugs. Thus, AM630 elevates [Ca2+]i to a greater degree than AM251 (Fig 1A). In summary, only pre-treatment with AM630 leads to inhibition of CAP responses in TG neurons.
Figure 4. Effect of AM251 and AM630 on TRPV1 and TRPA1-mediated responses.

(A) Co-treatments of TG neurons (for 2 min) with Veh+CAP (100 nM), AM251 (25μM)+CAP and AM630 (25μM)+CAP have no-effect on overall [Ca2+]i accumulation, n = 24–38. (B) Pre-treatment (for 5 min) of cultured TG neurons with AM630 (25 μM), but not AM251 (25 μM) attenuated CAP (100 nM)-evoked [Ca2+]i accumulation. CAP was applied for 1 min. Wash times between antagonists and CAP applications are 2 min, n=31–41; error bars = SEM **p<0.01. (C) Ca2+ accumulation generated by WIN55, 212–2 (WIN; 25 μM) and mustard oil (MO; 25μM) pre-treated with vehicle or indicated concentrations of AM251, n=22–68; NS is non-significant. (D) Ca2+ accumulation generated by WIN (25 μM) and MO (25μM) pre-treated with vehicle or indicated concentrations of AM630, n=22–60; error bars = SEM **p<0.01.
Pre-treatment with AM251 and AM630 could change the responsiveness of TRPA1 channels, since these cannabinoid antagonists activate the TRPA1 channel (Akopian et al., 2007; Ruparel et al., 2008). Further, co-treatment with AM251 and AM630 could also change the activity of TRPA1 due to allosteric mechanisms (see reviews (Conn et al., 2009; Leach et al., 2007). To examine these possibilities, we evaluated the effects of pre-treatment and co-treatment with AM251 and AM630 on subsequent TRPA1-mediated responses. We first assessed the effect of pre-treatment of TG neurons with AM251 and AM630 (100nM and 25 μM) on MO- and WIN 55,212 (WIN) evoked responses in TG neurons. Pre-treatment with 100nM AM251 had no effect on MO and WIN responses (Fig 4C). However, pre-treatment with AM630 produced a significant inhibition of both MO- and WIN-evoked [Ca2+]i accumulation (Fig 4D).
We next studied the effects of AM251 and AM630 co-treatment and pre-treatment on WIN responses using CGRP release from cultured TG neurons as an independent experimental approach. Similar to the results in our prior study (Fig 4D), pre-treatment of TG neurons with AM630 has blocked WIN-evoked CGRP release (Fig 5A). Importantly, the timing of AM630 administration qualitatively altered neuronal responsiveness to WIN, with a 15 min pretreatment period required to detect an inhibitory response. This pattern was not observed when both compounds were simultaneously administered. Instead, the co-treatment of AM630 and WIN resulted in an increase in CGRP release that was significantly greater than that observed with either compound alone (Fig 5A). This stimulatory interaction is concentration-dependent with a threshold of ≈10 μM (Fig 5C).
Figure 5. Effects of pre-treatment and co-treatment with AM251 and AM630 on WIN-induced CGRP release.

(A) Pre-treatment with AM630 reduces WIN-evoked CGRP release, while co-treatment of TG neurons with AM630 and WIN increase overall CGRP release. (B) Pre-treatment with AM251 does not affect WIN-evoked CGRP release, while co-treatment of neurons with AM251 and WIN substantially decreases overall CGRP release. Concentrations of drugs (AM251, AM630 and WIN) are indicated. Pre-treatment times with AM251 and AM630 are 15 min. WIN treatment and co-treatment with AM251 or AM630 was also for 15 min. Wash times 2 min each with two changes of Hanks solution, n = 6; error bars = SEM **p<0.01, ***p<0.001. (C) Co-treatment of WIN with indicated concentrations of AM630 increase overall CGRP release in a concentration-dependent fashion. (D) In contrast, co-treatment of WIN with indicated concentrations of AM251 decrease overall CGRP release in a concentration-dependent fashion, n = 6; error bars = SEM, *p<0.05, **p<0.01. “B” indicates baseline release of CGRP.
In contrast, AM251 had no effect with pre-treatment, but was a more effective inhibiter of WIN responses after co-application (Fig 5B). The inhibitory threshold of AM251 was ≈10 μM, and a statistically significant effect was achieved at >25 μM (Fig 5B and 5D). It could be noted that AM251 by itself generated CGRP release of < 10 fmol. Altogether, these results imply that AM251 and AM630 modulate TRPA1-mediated responses by different mechanisms; AM630 inhibits TRPA1 via desensitization (achieved by pre-treatment), while AM251 possibly exerts suppression via allosteric regulation of the TRPA1 channel, that in turn leads to suppression of WIN responses.
Modulation of WIN-induced inhibition of capsaicin responses by AM251 and AM630
Our prior studies have demonstrated that WIN inhibits CAP responses via TRPA1 activation in sensory neurons (Akopian et al., 2008), that triggers a phosphatase 2B (calcineurin) signaling pathway (Patwardhan et al., 2006b). Therefore, we tested the hypothesis that suppression of WIN responses by AM251 or AM630 in sensory neurons could block WIN-induced inhibition of CAP responses.
As seen in Figure 6A (and representative trace in Fig 6B), the application of WIN (25 μM) inhibited CAP-evoked current (ICAP) in TG neurons. However, neither pre-treatment with 1 μM AM630 (Fig 6A and 6C) nor co-treatment with 1 μM of AM251 reversed WIN-induced inhibition of CAP responses (Fig 6A); even though these concentrations would saturate binding to the metabotropic CB1 and CB2 receptors (Gatley et al., 1996; Pertwee et al., 1995). Based upon the results presented in Fig 4 and Fig 5, WIN responses can be reduced by either pre-treatment with AM630 (25μM) or co-treatment with AM251 (25μM). Conversely, pre-treatment with 25 μM of AM630 (Fig 6D) or co-treatment with WIN and 25 μM of AM251 (Fig 6E) almost completely reversed the WIN inhibition of ICAP in TG neurons (Fig 6A). These effects were also observed in CHO cells containing TRPA1 and TRPV1 (data not shown), indicating that the effects cannot be attributed to CB1 or CB2 since CHO cells do not express these receptors. Collectively, these data indicate that at concentrations of 25 μM, pre-treatment with AM630 (Fig 6D) or co-treatment with WIN and AM251 (Fig 6E) prevents WIN inhibition of CAP responses in sensory neurons (Fig 6A).
Figure 6. AM251 and AM630 reverse WIN-induced inhibition of CAP-gated currents (ICAP) in sensory neurons.

(A) Pre-treatment with AM630 (25 μM) and co-treatment of AM251 (25 μM) with WIN (25 μM) reverse WIN-induced inhibition of CAP (300 nM)-gated currents (ICAP) in TG neurons. Pretreatment with either 1μM of AM251 or AM630 does not affect WIN (25μM) inhibition. Concentration of the drugs and numbers of CAP responsive neurons are indicated. Numbers of WIN and CAP responsive neurons are indicated. Error bars = SEM, *p<0.05, **p<0.01. (B) A representative trace demonstrates inhibition of ICAP (300 nM) by WIN in TG neurons. (C) A representative trace demonstrates the sequence of drug applications and no effect of 1μM AM630 pre-treatment on WIN-inhibition of ICAP in TG neurons. (D) A representative trace demonstrates the sequence of drug applications and a reverse of WIN-inhibition of ICAP by 25μM AM630 pre-treatment in TG neurons. (E) A representative trace demonstrates the sequence of drug applications and a reverse of WIN-inhibition of ICAP with 25μM AM251 co-treatment in TG neurons. Durations of particular drug applications are marked with horizontal bars.
Effects of AM251 and AM630 on nocifensive behaviors and thermal hyperalgesia
It is well documented that activation of sensory neurons does not necessarily cause nociception in animals. Thus, certain cannabinoids and other ligands generate slowly developing inward currents in sensory neurons via TRP channels (particularly TRPV1 and TRPA1), but do not evoke nociception (Akopian et al., 2008; Liu et al., 1997; Price et al., 2004a, b). Therefore, we first evaluated nociception evoked by AM251 and AM630. Figure 7A shows that unlike CAP (10μg), the hindpaw injection of AM251 and AM630 (30μg each) are not able to induce nociceptive behaviors. In addition, injections of AM251 and AM630 (30μg each) in hind paws of WT mice did not produced statistically significant thermal hyperalgesia (Fig 7B; last two columns).
Figure 7. Effects of AM251 and AM630 on nociception and CAP-induced thermal hyperalgesia.

(A) Nocifensive behavior was induced by AM251 and AM630 (30μg each). CAP (1μg) was used as a positive control. Measurements were collected during 15 min by a blinded observer and consisted of spontaneous flinching and licking of the hindpaw, n=5–8. (B) Modulation of CAP-induced thermal hyperalgesia in wild-type (WT) and TRPA1 null-mutant (TRPA1 KO) mice by AM251 and AM630. AM251 (30 μg) was co-injected with CAP (1μg), and AM630 (30 μg) was injected 15 min prior to CAP administration into the hind paw. Thermal responses were measured 5 min after CAP administration. Error bars = SEM. ***p<0.001 (n=5–7). NS is non-significant.
The injection of CAP triggers a prompt thermal hyperalgesia in animals and humans (Culp et al., 1989; LaMotte et al., 1992; Simone et al., 1991). We have demonstrated that CAP responses can be significantly reduced by pre-treatment with AM630, but not co-treatment with AM251 (Fig 4A and 4B). Here, we examined whether pre-treatment with AM630 and co-treatment with AM251 can reduce CAP-induced thermal hyperalgesia. Figure 7B illustrates that unlike AM251, AM630 is able to significantly attenuate CAP-induced thermal hyperalgesia in WT mice. It was hypothesized that this effect could be mediated by TRPA1 channel. However, these antagonists can target other receptors. To evaluate the role of TRPA1 in vivo, modulation of CAP-induced thermal hyperalgesia by AM251 and AM630 was evaluated in TRPA1 KO mice. Both cannabinoids, AM251 and AM630 were not able to alter CAP-induced thermal hyperalgesia (Fig 7B). These data support in vitro results (Fig 4A and 4B), and indicate that TRPA1 is involved in modulation of CAP-induced thermal hyperalgesia by AM630.
CAP effect on thermal hyperalgesia can be significantly inhibited by the application of cannabinoids, including WIN55,212 (Ibrahim et al., 2006; Johanek et al., 2001; Patwardhan et al., 2006b; Quartilho et al., 2003). To infer the involvement of CB1 or CB2 receptors in these types of experiments, AM251 and AM630 have been employed as prototypical antagonists. In agreement with several reports (Fox et al., 2001; Ibrahim et al., 2006; Johanek et al., 2001; Johanek and Simone, 2004), we found that local (ipl) injection of WIN inhibited CAP-induced thermal hyperalgesia and this inhibition was partially reversed by both AM251 and AM630 (Fig 8A and 8B; see black, brown and green lines). Moreover, pre-treatment with AM630, but not AM251 can inhibit CAP responses in sensory neurons (Fig 4A and 4B). Accordingly, 15 min-pretreatment with AM630, but not AM251 (30μg each) significantly attenuated CAP-evoked thermal hyperalgesia in rats (Fig 8A and 8B; see black and red lines) as well as mice (Fig 7B). Interestingly, the AM630 and AM251 actions were reversed by TRPA1 antagonist HC030031 (HC; Fig 8A and 8B; see red and blue lines). These results are in accordance with data generated in TRPA1 KO mice (Fig 7B). WIN, AM251 and AM630 activate TRPA1 channels (Fig 1–3; (Akopian et al., 2008). Therefore, it could be difficult to interpret the effect of HC on combined pre-treatments with WIN and AM251 or AM630. Nevertheless, HC led to a more pronounced action of WIN with either AM251 or AM630. Thus, WIN+AM251+HC and WIN+AM630+HC exhibited full, rather than partial reversal of WIN-induced inhibition of CAP-evoked thermal hyperalgesia than was achieved by AM251 and AM630 (Fig 8A and 8B; see green and purple lines). Altogether, AM251 and AM630 may exert their peripheral effects by not only inhibiting CB1 and CB2, but also activating TRPA1 channels and subsequently desensitizing TRPA1 as well as TRPV1 channels.
Figure 8. Effects of AM251 and AM630 on nociception and CAP-induced thermal hyperalgesia.

(A) Modulation of CAP-induced thermal hyperalgesia by AM251 in combination with WIN and HC030031 (HC; TRPA1 inhibitor), n=6–7. (B) Modulation of CAP-induced thermal hyperalgesia by AM630 in combination with WIN and HC. AM251 and AM630 were co-applied with WIN and HC (total volume 50μl), 15 min prior to injection of CAP. Error bars = SEM, *p<0.05, **p<0.01 (n=5–6). The dose of each drug is indicated.
Discussion
There is broad agreement that cannabinoids are profoundly capable of producing peripherally mediated anti-nociception and anti-hyperalgesia in many pain models. Several alternative hypotheses on the mechanism(s) mediating these effect have been proposed, and, importantly, are based at least in part upon interpreting the effects of CB1/CB2 receptor antagonists (Johanek and Simone, 2004; Malan et al., 2003; Rice et al., 2002). These interpretations rely on the assumption that the antagonists are strictly specific over defined concentration ranges. It could be noted that although the nM concentrations of AM251 and AM630 are required to block CB1 or CB2 receptors, mM concentrations of these compounds were used in local or systemic injections. Thus, the initial injected concentration of cannabinoid antagonists used in behavior experiments ranges from 300 uM to as much as 2 mM (Ibrahim et al., 2006; Johanek and Simone, 2004; Malan et al., 2001; Oshita et al., 2005). The CB1 and CB2 antagonists, AM251 and AM630, have been used extensively to characterize the role of metabotropic CB1 and CB2 receptors in cannabinoid studies (see Introduction). Proper interpretation of these results, of course, requires knowledge of the specificity of these compounds. Given the structural similarity that many cannabinoid agonists have with TRP agonists, it is possible that CB1/CB2 antagonists might also interact with TRP channels. Therefore, we examined whether CB1/CB2 antagonists activate sensory neurons, modulate WIN- and CAP-evoked responses and affect cannabinoid inhibition of CAP responses and CAP-induced thermal hyperalgesia.
Our results indicate that, at low uM concentrations, both AM251 and AM630, activate TG neurons. The activation of sensory neurons by lipophilic compounds is well documented (Qin et al., 2008; Zygmunt et al., 2002; Zygmunt et al., 1999). It appears that TRPV1 and TRPA1 channels are the main receptors involved in many noxious stimulatory responses (Akopian et al., 2008; Huang et al., 2002; Jordt et al., 2004; Qin et al., 2008; Zygmunt et al., 1999). Some of these lipophilic agents also serve as antagonists, such as AM404 (Zygmunt et al., 2000). The TRPA1 channel mediates almost entirely the measured AM251 and AM630 responses in sensory neurons. Furthermore, it is clearly evident that AM251 and AM630 have distinct pharmacological properties. First, they have a different efficacy and potency. Second, AM630 is a more potent activator of TRPA1 when the channel is co-expressed with TRPV1. A similar effect has been observed previously for the CB2 agonist AM1241 (Akopian et al., 2008). Third, AM630 responses are blocked by both TRPA1 antagonists, HC and AP-18, while only HC is an effective inhibitor of AM251 responses in sensory neurons. This implies that AM630 and AM251 could act on different TRPA1 channel domains.
As activators of the TRPA1 channel, AM251 and AM630 could potentially inhibit subsequent responses mediated by agonists to TRPA1 and TRPV1 (Akopian et al., 2007; Koplas et al., 1997; Ruparel et al., 2008). In support of this point, we demonstrated that pre-treatment of sensory neurons with AM630 (at >25 μM) inhibits CAP, WIN, and MO responses. AM630 could produce pharmacological cross-desensitization of the TRPV1 channel via Ca2+ influx into sensory neurons (Akopian et al., 2007; Koplas et al., 1997; Ruparel et al., 2008). Interestingly, AM630 greatly enhanced the activation of TRPA1 after co-application with the agonist WIN. The effect of AM630 as an antagonist or amplifier of WIN responses may be dictated by the relative efficacy of both agonists as well as their sites of actions.
Unlike AM630, AM251 pre-treatment is not able to desensitize WIN, MO, and CAP responses. These results are consistent with the observation that AM251 generates a relatively low amount of Ca2+ influx, which may not be enough to trigger pharmacological desensitization of TRPA1 and TRPV1 channels (Akopian et al., 2007; Koplas et al., 1997). However, it appears that AM251 can significantly attenuate WIN responses when co-applied together. One of the possibilities is that AM251 allosterically modulates a putative WIN-binding site on TRPA1. Allosteric modulations of GPCR as well as channel functions are a well recognized phenomenon (Conn et al., 2009; Hansen et al., 2010; Leach et al., 2007; Mony et al., 2009; Smith and Simpson, 2003). An alternative possibility is that AM251 may be a partial agonist and thus act as a functional antagonist.
Our results demonstrated that pre-treatment with AM630 or co-treatment with AM251 reverses WIN-induced inhibition of CAP responses in sensory neurons. These effects could have several interpretations. First, it is possible that AM630 and AM251 inhibit WIN activation of the TRPA1 channel, a known mechanism for WIN inhibition of capsaicin effects (Akopian et al., 2008; Patwardhan et al., 2006b). Second, it is possible that AM251 and AM630 could block WIN activation of CB1 or CB2 receptors. This interpretation is based the assumption that CB1 and CB2 are co-expressed with TRPV1 in sensory neurons. This assumption remains controversial since CB1 is minimally co-expressed with TRPV1 in sensory neurons, and CB2 has not been detected in non-inflamed sensory neurons (Bridges et al., 2003; Khasabova et al., 2004; Khasabova et al., 2002; Price et al., 2003). Moreover, we can duplicate these observations using cell expression systems devoid of CB1 and CB2 receptors (i.e., CHO). In addition, WIN does not inhibit CAP responses in sensory neurons at concentrations sufficient to activate CB1 or CB2 (Akopian et al., 2008; Patwardhan et al., 2006b; Price et al., 2004a). Finally, the application of 1 μM of AM251 and AM630 did not reverse WIN inhibition of CAP responses, despite being sufficient to completely block CB1/CB2 receptors (Fig. 6). Taken together, these data provide greater support for interpretation that AM251 and AM630 reverse WIN-induced CAP inhibition in sensory neurons due to attenuation of WIN activation of the TRPA1 channel.
We evaluated the in vivo relevance of our findings in two pain models: nocifensive behavior and CAP-evoked thermal hyperalgesia. Despite their ability to activate sensory neurons, AM251 and AM630 have not produced visible nociception after injection into the hind paw. This phenomenon was observed previously for several TRPV1 and TRPA1 activating compounds including anandamide, WIN55,212-2, 12-phenylacetate 13 acetate 20-homovanillate, zingerone, Δ9THC and olvanil (Jordt et al., 2004; Liu et al., 1997; Liu et al., 1998; Liu et al., 2000; Price et al., 2004a, b). There are two possible explanations of such an effect. First, effective in vivo concentrations of certain compounds (especially lipophilic) may not be sufficient to activate sensory neurons to the extent required to elicit nociceptive behaviors. Thus, WIN and MO (50uM each) generate approximately the same magnitude of currents in sensory neurons (Salas et al., 2009). In contrast, CGRP release from skin preparations is several folds greater for MO than for WIN (Patwardhan et al., 2006b; Ruparel et al., 2008). Second, it was suggested that activation of sensory neurons by these compounds in vivo could have slow kinetics that may lead to deactivation of voltage-gated channels which are essential for action potential generation and propagation, and subsequent nociception (Liu et al., 1997; Liu et al., 2000).
The CAP-evoked thermal hyperalgesia was selected for two reasons: first, desensitization of CAP-responses can be examined, and second, peripheral WIN-induced inhibition of CAP-evoked hyperalgesia has been well characterized (Ibrahim et al., 2006; Johanek et al., 2001; Patwardhan et al., 2006b; Quartilho et al., 2003). The results from the behavior experiment confirmed that AM630, but not AM251, can desensitize TRPV1 channels via a TRPA1 dependent pathway. Further, the inhibition of TRPA1 resulted in a pronounced effect of AM251 and AM630 on WIN inhibition of CAP-evoked hyperalgesia (Fig. 8). It could be noted that use HC in this experiment is complicated by the fact that HC affects WIN responses too (Akopian et al., 2008). Interestingly, AM251 and AM630 only partially (but statistically significantly) reversed WIN inhibition of CAP-evoked hyperalgesia (Fig. 8; (Johanek et al., 2001; Patwardhan et al., 2006b; Quartilho et al., 2003). The results on modulation of CAP responses by AM251 is in agreement with some reports (Johanek et al., 2001; Oshita et al., 2005), but not others (Fioravanti et al., 2008). Thus, it was suggested that constitutive activity of CB1 maintains TRPV1 in a sensitized state (Fioravanti et al., 2008). However, the attenuation of the TRPV1 responses in CB1 null-mutant mice has not been observed in several independent reports (Agarwal et al., 2007; Akopian et al., 2008; Ibrahim et al., 2006). In summary, these results indicate that the cannabinoid antagonists AM251 and AM630 may produce their observed effects via interaction with either metabotropic or ionotropic cannabinoid receptors (Akopian et al., 2009).
Research Highlight.
We studied activation of sensory neurons by cannabinoid antagonists AM251 and AM630
AM251 and AM630 responses are mediated by the TRPA1 channel in sensory neurons.
AM630, but not AM251 responses are modulated by the TRPV1 channel.
AM251 and AM630 inhibit capsaicin and mustard oil responses via different mechanisms.
These findings demonstrate that TRP channels are targets for cannabinoid antagonists.
Acknowledgments
We would like to thank Jie Li for technical assistance, Dr. David Julius (UCSF, San Francisco, CA) and Dr. Ardem Patapoutian (The Scripps Research Institute, San Diego, CA) for kindly provided TRPV1 and TRPA1 expression constructs used in this study. Research was supported by NIH grants DE017696 and DE019311 (to ANA) and DA19585 (to KMH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, Rubino T, Michalski CW, Marsicano G, Monory K, Mackie K, Marian C, Batkai S, Parolaro D, Fischer MJ, Reeh P, Kunos G, Kress M, Lutz B, Woolf CJ, Kuner R. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahluwalia J, Urban L, Capogna M, Bevan S, Nagy I. Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience. 2000;100:685–688. doi: 10.1016/s0306-4522(00)00389-4. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Ruparel NB, Jeske NA, Hargreaves KM. Transient receptor potential TRPA1 channel desensitization in sensory neurons is agonist dependent and regulated by TRPV1-directed internalization. J Physiol. 2007;583:175–193. doi: 10.1113/jphysiol.2007.133231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian AN, Ruparel NB, Jeske NA, Patwardhan A, Hargreaves KM. Role of ionotropic cannabinoid receptors in peripheral antinociception and antihyperalgesia. Trends Pharmacol Sci. 2009;30:79–84. doi: 10.1016/j.tips.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian AN, Ruparel NB, Patwardhan A, Hargreaves KM. Cannabinoids desensitize capsaicin and mustard oil responses in sensory neurons via TRPA1 activation. J Neurosci. 2008;28:1064–1075. doi: 10.1523/JNEUROSCI.1565-06.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges D, Ahmad K, Rice AS. The synthetic cannabinoid WIN55,212–2 attenuates hyperalgesia and allodynia in a rat model of neuropathic pain. Br J Pharmacol. 2001;133:586–594. doi: 10.1038/sj.bjp.0704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges D, Rice AS, Egertova M, Elphick MR, Winter J, Michael GJ. Localisation of cannabinoid receptor 1 in rat dorsal root ganglion using in situ hybridisation and immunohistochemistry. Neuroscience. 2003;119:803–812. doi: 10.1016/s0306-4522(03)00200-8. [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culp WJ, Ochoa J, Cline M, Dotson R. Heat and mechanical hyperalgesia induced by capsaicin. Cross modality threshold modulation in human C nociceptors. Brain. 1989;112 ( Pt 5):1317–1331. doi: 10.1093/brain/112.5.1317. [DOI] [PubMed] [Google Scholar]
- Diogenes A, Akopian AN, Hargreaves KM. NGF up-regulates TRPA1: implications for orofacial pain. J Dent Res. 2007;86:550–555. doi: 10.1177/154405910708600612. [DOI] [PubMed] [Google Scholar]
- Fioravanti B, De Felice M, Stucky CL, Medler KA, Luo MC, Gardell LR, Ibrahim M, Malan TP, Jr, Yamamura HI, Ossipov MH, King T, Lai J, Porreca F, Vanderah TW. Constitutive activity at the cannabinoid CB1 receptor is required for behavioral response to noxious chemical stimulation of TRPV1: antinociceptive actions of CB1 inverse agonists. J Neurosci. 2008;28:11593–11602. doi: 10.1523/JNEUROSCI.3322-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox A, Kesingland A, Gentry C, McNair K, Patel S, Urban L, James I. The role of central and peripheral Cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain. 2001;92:91–100. doi: 10.1016/s0304-3959(00)00474-7. [DOI] [PubMed] [Google Scholar]
- Garry MG, Richardson JD, Hargreaves KM. Sodium nitroprusside evokes the release of immunoreactive calcitonin gene-related peptide and substance P from dorsal horn slices via nitric oxide-dependent and nitric oxide-independent mechanisms. J Neurosci. 1994;14:4329–4337. doi: 10.1523/JNEUROSCI.14-07-04329.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatley SJ, Gifford AN, Volkow ND, Lan R, Makriyannis A. 123I-labeled AM251: a radioiodinated ligand which binds in vivo to mouse brain cannabinoid CB1 receptors. Eur J Pharmacol. 1996;307:331–338. doi: 10.1016/0014-2999(96)00279-8. [DOI] [PubMed] [Google Scholar]
- Hansen KB, Furukawa H, Traynelis SF. Control of assembly and function of glutamate receptors by the amino-terminal domain. Mol Pharmacol. 2010;78:535–549. doi: 10.1124/mol.110.067157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hinman A, Chuang HH, Bautista DM, Julius D. TRP channel activation by reversible covalent modification. Proc Natl Acad Sci U S A. 2006;103:19564–19568. doi: 10.1073/pnas.0609598103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosohata K, Quock RM, Hosohata Y, Burkey TH, Makriyannis A, Consroe P, Roeske WR, Yamamura HI. AM630 is a competitive cannabinoid receptor antagonist in the guinea pig brain. Life Sci. 1997;61:PL115–118. doi: 10.1016/s0024-3205(97)00596-1. [DOI] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, Miller JD, Davies SN, Geppetti P, Walker JM, Di Marzo V. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci U S A. 2002;99:8400–8405. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Deng H, Zvonok A, Cockayne DA, Kwan J, Mata HP, Vanderah TW, Lai J, Porreca F, Makriyannis A, Malan TP., Jr Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci U S A. 2003;100:10529–10533. doi: 10.1073/pnas.1834309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Porreca F, Lai J, Albrecht PJ, Rice FL, Khodorova A, Davar G, Makriyannis A, Vanderah TW, Mata HP, Malan TP., Jr CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc Natl Acad Sci U S A. 2005;102:3093–3098. doi: 10.1073/pnas.0409888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Rude ML, Stagg NJ, Mata HP, Lai J, Vanderah TW, Porreca F, Buckley NE, Makriyannis A, Malan TP., Jr CB2 cannabinoid receptor mediation of antinociception. Pain. 2006;122:36–42. doi: 10.1016/j.pain.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Jeske NA, Patwardhan AM, Gamper N, Price TJ, Akopian AN, Hargreaves KM. Cannabinoid WIN 55,212–2 regulates TRPV1 phosphorylation in sensory neurons. J Biol Chem. 2006;281:32879–32890. doi: 10.1074/jbc.M603220200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanek LM, Heitmiller DR, Turner M, Nader N, Hodges J, Simone DA. Cannabinoids attenuate capsaicin-evoked hyperalgesia through spinal and peripheral mechanisms. Pain. 2001;93:303–315. doi: 10.1016/S0304-3959(01)00336-0. [DOI] [PubMed] [Google Scholar]
- Johanek LM, Simone DA. Activation of peripheral cannabinoid receptors attenuates cutaneous hyperalgesia produced by a heat injury. Pain. 2004;109:432–442. doi: 10.1016/j.pain.2004.02.020. [DOI] [PubMed] [Google Scholar]
- Jonsson KO, Persson E, Fowler CJ. The cannabinoid CB2 receptor selective agonist JWH133 reduces mast cell oedema in response to compound 48/80 in vivo but not the release of beta-hexosaminidase from skin slices in vitro. Life Sci. 2006;78:598–606. doi: 10.1016/j.lfs.2005.05.059. [DOI] [PubMed] [Google Scholar]
- Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- Khasabova IA, Harding-Rose C, Simone DA, Seybold VS. Differential effects of CB1 and opioid agonists on two populations of adult rat dorsal root ganglion neurons. J Neurosci. 2004;24:1744–1753. doi: 10.1523/JNEUROSCI.4298-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabova IA, Khasabov SG, Harding-Rose C, Coicou LG, Seybold BA, Lindberg AE, Steevens CD, Simone DA, Seybold VS. A decrease in anandamide signaling contributes to the maintenance of cutaneous mechanical hyperalgesia in a model of bone cancer pain. J Neurosci. 2008;28:11141–11152. doi: 10.1523/JNEUROSCI.2847-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabova IA, Simone DA, Seybold VS. Cannabinoids attenuate depolarization-dependent Ca2+ influx in intermediate-size primary afferent neurons of adult rats. Neuroscience. 2002;115:613–625. doi: 10.1016/s0306-4522(02)00449-9. [DOI] [PubMed] [Google Scholar]
- Koplas PA, Rosenberg RL, Oxford GS. The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J Neurosci. 1997;17:3525–3537. doi: 10.1523/JNEUROSCI.17-10-03525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremeyer B, Lopera F, Cox JJ, Momin A, Rugiero F, Marsh S, Woods CG, Jones NG, Paterson KJ, Fricker FR, Villegas A, Acosta N, Pineda-Trujillo NG, Ramirez JD, Zea J, Burley MW, Bedoya G, Bennett DL, Wood JN, Ruiz-Linares A. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron. 2010;66:671–680. doi: 10.1016/j.neuron.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ, Corey DP. TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair-cell transduction. Neuron. 2006;50:277–289. doi: 10.1016/j.neuron.2006.03.042. [DOI] [PubMed] [Google Scholar]
- LaBuda CJ, Koblish M, Little PJ. Cannabinoid CB2 receptor agonist activity in the hindpaw incision model of postoperative pain. Eur J Pharmacol. 2005;527:172–174. doi: 10.1016/j.ejphar.2005.10.020. [DOI] [PubMed] [Google Scholar]
- LaMotte RH, Lundberg LE, Torebjork HE. Pain, hyperalgesia and activity in nociceptive C units in humans after intradermal injection of capsaicin. J Physiol. 1992;448:749–764. doi: 10.1113/jphysiol.1992.sp019068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–389. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Liu L, Lo Y, Chen I, Simon SA. The responses of rat trigeminal ganglion neurons to capsaicin and two nonpungent vanilloid receptor agonists, olvanil and glyceryl nonamide. J Neurosci. 1997;17:4101–4111. doi: 10.1523/JNEUROSCI.17-11-04101.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Szallasi A, Simon SA. A non-pungent resiniferatoxin analogue, phorbol 12-phenylacetate 13 acetate 20-homovanillate, reveals vanilloid receptor subtypes on rat trigeminal ganglion neurons. Neuroscience. 1998;84:569–581. doi: 10.1016/s0306-4522(97)00523-x. [DOI] [PubMed] [Google Scholar]
- Liu L, Welch JM, Erickson RP, Reinhart PH, Simon SA. Different responses to repeated applications of zingerone in behavioral studies, recordings from intact and cultured TG neurons, and from VR1 receptors. Physiol Behav. 2000;69:177–186. doi: 10.1016/s0031-9384(00)00200-6. [DOI] [PubMed] [Google Scholar]
- Malan TP, Jr, Ibrahim MM, Deng H, Liu Q, Mata HP, Vanderah T, Porreca F, Makriyannis A. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. 2001;93:239–245. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- Malan TP, Jr, Ibrahim MM, Lai J, Vanderah TW, Makriyannis A, Porreca F. CB2 cannabinoid receptor agonists: pain relief without psychoactive effects? Curr Opin Pharmacol. 2003;3:62–67. doi: 10.1016/s1471-4892(02)00004-8. [DOI] [PubMed] [Google Scholar]
- McCoy KL, Matveyeva M, Carlisle SJ, Cabral GA. Cannabinoid inhibition of the processing of intact lysozyme by macrophages: evidence for CB2 receptor participation. J Pharmacol Exp Ther. 1999;289:1620–1625. [PubMed] [Google Scholar]
- Mony L, Kew JN, Gunthorpe MJ, Paoletti P. Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br J Pharmacol. 2009;157:1301–1317. doi: 10.1111/j.1476-5381.2009.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Katsura H, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Tominaga M, Noguchi K. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest. 2005;115:2393–2401. doi: 10.1172/JCI25437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshita K, Inoue A, Tang HB, Nakata Y, Kawamoto M, Yuge O. CB(1) cannabinoid receptor stimulation modulates transient receptor potential vanilloid receptor 1 activities in calcium influx and substance P Release in cultured rat dorsal root ganglion cells. J Pharmacol Sci. 2005;97:377–385. doi: 10.1254/jphs.fp0040872. [DOI] [PubMed] [Google Scholar]
- Patwardhan AM, Berg KA, Akopain AN, Jeske NA, Gamper N, Clarke WP, Hargreaves KM. Bradykinin-induced functional competence and trafficking of the delta-opioid receptor in trigeminal nociceptors. J Neurosci. 2005;25:8825–8832. doi: 10.1523/JNEUROSCI.0160-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan AM, Diogenes A, Berg KA, Fehrenbacher JC, Clarke WP, Akopian AN, Hargreaves KM. PAR-2 agonists activate trigeminal nociceptors and induce functional competence in the delta opioid receptor. Pain. 2006a;125:114–124. doi: 10.1016/j.pain.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Patwardhan AM, Jeske NA, Price TJ, Gamper N, Akopian AN, Hargreaves KM. The cannabinoid WIN 55,212–2 inhibits transient receptor potential vanilloid 1 (TRPV1) and evokes peripheral antihyperalgesia via calcineurin. Proc Natl Acad Sci U S A. 2006b;103:11393–11398. doi: 10.1073/pnas.0603861103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee R, Griffin G, Fernando S, Li X, Hill A, Makriyannis A. AM630, a competitive cannabinoid receptor antagonist. Life Sci. 1995;56:1949–1955. doi: 10.1016/0024-3205(95)00175-6. [DOI] [PubMed] [Google Scholar]
- Petrus M, Peier AM, Bandell M, Hwang SW, Huynh T, Olney N, Jegla T, Patapoutian A. A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Mol Pain. 2007;3:40. doi: 10.1186/1744-8069-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Helesic G, Parghi D, Hargreaves KM, Flores CM. The neuronal distribution of cannabinoid receptor type 1 in the trigeminal ganglion of the rat. Neuroscience. 2003;120:155–162. doi: 10.1016/S0306-4522(03)00333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Patwardhan A, Akopian AN, Hargreaves KM, Flores CM. Cannabinoid receptor-independent actions of the aminoalkylindole WIN 55,212–2 on trigeminal sensory neurons. Br J Pharmacol. 2004a;142:257–266. doi: 10.1038/sj.bjp.0705778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Patwardhan A, Akopian AN, Hargreaves KM, Flores CM. Modulation of trigeminal sensory neuron activity by the dual cannabinoid-vanilloid agonists anandamide, N-arachidonoyl-dopamine and arachidonyl-2-chloroethylamide. Br J Pharmacol. 2004b;141:1118–1130. doi: 10.1038/sj.bjp.0705711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, Neeper MP, Liu Y, Hutchinson TL, Lubin ML, Flores CM. TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J Neurosci. 2008;28:6231–6238. doi: 10.1523/JNEUROSCI.0504-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartilho A, Mata HP, Ibrahim MM, Vanderah TW, Porreca F, Makriyannis A, Malan TP., Jr Inhibition of inflammatory hyperalgesia by activation of peripheral CB2 cannabinoid receptors. Anesthesiology. 2003;99:955–960. doi: 10.1097/00000542-200310000-00031. [DOI] [PubMed] [Google Scholar]
- Rice AS, Farquhar-Smith WP, Nagy I. Endocannabinoids and pain: spinal and peripheral analgesia in inflammation and neuropathy. Prostaglandins Leukot Essent Fatty Acids. 2002;66:243–256. doi: 10.1054/plef.2001.0362. [DOI] [PubMed] [Google Scholar]
- Richardson JD, Kilo S, Hargreaves KM. Cannabinoids reduce hyperalgesia and inflammation via interaction with peripheral CB1 receptors. Pain. 1998;75:111–119. doi: 10.1016/S0304-3959(97)00213-3. [DOI] [PubMed] [Google Scholar]
- Ruparel NB, Patwardhan AM, Akopian AN, Hargreaves KM. Homologous and heterologous desensitization of capsaicin and mustard oil responses utilize different cellular pathways in nociceptors. Pain. 2008;135:271–279. doi: 10.1016/j.pain.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar DR, Smith PA, Millns PJ, Smart D, Kendall DA, Chapman V. TRPV1 and CB(1) receptor-mediated effects of the endovanilloid/endocannabinoid N-arachidonoyl-dopamine on primary afferent fibre and spinal cord neuronal responses in the rat. Eur J Neurosci. 2004;20:175–184. doi: 10.1111/j.1460-9568.2004.03481.x. [DOI] [PubMed] [Google Scholar]
- Salas MM, Hargreaves KM, Akopian AN. TRPA1-mediated responses in trigeminal sensory neurons: interaction between TRPA1 and TRPV1. Eur J Neurosci. 2009;29:1568–1578. doi: 10.1111/j.1460-9568.2009.06702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson MT, Small-Howard A, Shimoda LM, Koblan-Huberson M, Stokes AJ, Turner H. Differential roles of CB1 and CB2 cannabinoid receptors in mast cells. J Immunol. 2003;170:4953–4962. doi: 10.4049/jimmunol.170.10.4953. [DOI] [PubMed] [Google Scholar]
- Simone DA, Sorkin LS, Oh U, Chung JM, Owens C, LaMotte RH, Willis WD. Neurogenic hyperalgesia: central neural correlates in responses of spinothalamic tract neurons. J Neurophysiol. 1991;66:228–246. doi: 10.1152/jn.1991.66.1.228. [DOI] [PubMed] [Google Scholar]
- Smith AJ, Simpson PB. Methodological approaches for the study of GABA(A) receptor pharmacology and functional responses. Anal Bioanal Chem. 2003;377:843–851. doi: 10.1007/s00216-003-2172-y. [DOI] [PubMed] [Google Scholar]
- Staruschenko A, Jeske NA, Akopian AN. Contribution of TRPV1-TRPA1 interaction to the single channel properties of the TRPA1 channel. J Biol Chem. 2010;285:15167–15177. doi: 10.1074/jbc.M110.106153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Andersson DA, Hogestatt ED. Delta 9-tetrahydrocannabinol and cannabinol activate capsaicin-sensitive sensory nerves via a CB1 and CB2 cannabinoid receptor-independent mechanism. J Neurosci. 2002;22:4720–4727. doi: 10.1523/JNEUROSCI.22-11-04720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Chuang H, Movahed P, Julius D, Hogestatt ED. The anandamide transport inhibitor AM404 activates vanilloid receptors. Eur J Pharmacol. 2000;396:39–42. doi: 10.1016/s0014-2999(00)00207-7. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]