

Abstract
Glutamate is an essential excitatory neurotransmitter regulating brain functions. Excitatory amino acid transporter (EAAT)-2 is one of the major glutamate transporters expressed predominantly in astroglial cells and is responsible for 90% of total glutamate uptake. Glutamate transporters tightly regulate glutamate concentration in the synaptic cleft. Dysfunction of EAAT2 and accumulation of excessive extracellular glutamate has been implicated in the development of several neurodegenerative diseases including Alzheimer’s disease, Huntington’s disease, and amyotrophic lateral sclerosis. Analysis of the 2.5-kb human EAAT2 promoter showed that NF-κB is an important regulator of EAAT2 expression in astrocytes. Screening of approximately 1,040 FDA-approved compounds and nutritionals led to the discovery that many β-lactam antibiotics are transcriptional activators of EAAT2 resulting in increased EAAT2 protein levels. Treatment of animals with ceftriaxone (CEF), a β-lactam antibiotic, led to an increase of EAAT2 expression and glutamate transport activity in the brain. CEF has neuroprotective effects in both in vitro and in vivo models based on its ability to inhibit neuronal cell death by preventing glutamate excitotoxicity. CEF increases EAAT2 transcription in primary human fetal astrocytes (PHFA) through the NF-κB signaling pathway. The NF-κB binding site at −272 position was critical in CEF-mediated EAAT2 protein induction. These studies emphasize the importance of transcriptional regulation in controlling glutamate levels in the brain. They also emphasize the potential utility of the EAAT2 promoter for developing both low and high throughput screening assays to identify novel small molecule regulators of glutamate transport with potential to ameliorate pathological changes occurring during and causing neurodegeneration.
INTRODUCTION
During the last decade, multiple studies have demonstrated that glutamate is the major excitatory neurotransmitter in the brain, with up to 40% of all synapses being glutamatergic (Fairman and Amara, 1999). Although glutamatergic neurons are distributed throughout the nervous system, they are prominently represented in the cerebral cortex and limbic regions of the brain. Glutamate is the primary excitatory amino acid neurotransmitter in the central nervous system (CNS) and is also a potent neurotoxin that may lead to the death of nerve cells. Glutamate released from glutamatergic nerve endings participates in the signaling process through different types of glutamatergic receptors and then must be taken up from the synaptic cleft (Kanai and Hediger, 2003). Accumulation of excess extracellular glutamate and subsequent overstimulation of glutamatergic receptors increase the production of reactive and excitotoxic oxygen/nitrogen species, which induce oxidative stress leading to neuronal death (Ganel and Rothstein, 1999). Thus, the concentration of glutamate in the synaptic cleft and the duration of its action are carefully orchestrated in order to maintain homeostasis and prevent neuronal death. The high-affinity sodium-dependent transporter systems provide the predominant mechanism for uptake of glutamate in the brain and maintain the proper concentration of this potentially excitotoxic amino acid. Increased levels of glutamate are associated with a number of neurological disorders such as epilepsy, stroke and neurodegenerative diseases and dysfunctional glutamate transporters are often the initiating event or part of the cascade leading to brain injury.
Transport and Regulation Mechanisms (Types of Transporters)
Glutamate is the main excitatory neurotransmitter in the CNS, responsible for fast excitatory neurotransmission. Under normal conditions, glutamate released from the presynaptic neuron activates ionotropic glutamate receptors present on the postsynaptic neurons. This results in the influx of Na+ and Ca+2 ions into the cell, leading to membrane depolarization and generation of action potentials. The concentration of glutamate in the synaptic cleft, as well as the resultant activity of the postsynaptic ionotropic glutamate receptors, is tightly regulated by interplay between glutamate release and glutamate clearance (Kanai and Hediger, 2003). Five subtypes of glutamate transporters have been cloned to date (Table 1) (Arriza et al., 1994). Three of these glutamate transporters were originally identified in rat brain: GLAST, GLT-1 and EAAC1. Their human homologues are: EAAT1, EAAT2 and EAAT3, respectively. The two remaining human and rodent subtypes, EAAT4 and EAAT5 share common nomenclature. All five of the transporters are localized in a different way among the various brain structures. GLAST immunostaining and protein expression are most prominent in the cerebellum with moderate levels in other structures such as the hippocampus and the forebrain. In contrast, GLT-1 expression is mainly found in the forebrain regions with minor expression in cerebellum. Both of these transporters represent the most noteworthy “astrocytic” transporters localized on the membrane of astroglia or in Bergmann glia associated with excitatory synapses. EAAT3 is expressed in different regions of the brain, but at very low levels. The remaining transporters, EAAT4 and EAAT5, are only expressed in the cerebellum and retina, respectively (Kanai and Hediger, 2003; Arriza et al., 1994; Beart and O’Shea, 2007).
Table 1.
Main characteristics of known human glutamate transporters.
| Transporter | CNS Locations | Cellular Expression | Properties | References |
|---|---|---|---|---|
| EAAT1 | Highest expression in cerebellum, also detected in cortex, spinal cord | Astrocytes | Major subtype expressed during CNS development. | Storck et al., 1992; Bar- Peled et al., 1997; Furata et al. 1997; Maragakis et al., 2004 |
| EAAT2 | Throughout brain, spinal cord. | Primarily in astrocytes, also: neurons, oligodendrocytes | Responsible for >90% of total Glutamate uptake. | Maragakis et al., 2004; Lauriat et al., 2007; Sheldon et al., 2007 |
| EAAT3 | Throughout the brain, especially cortex, hip- pocampus, cerebellum and basal ganglia | Postsynaptic neuronal terminals, also detected in astrocytes | Membrane expression under dynamic regulation by intracellular kinases and cholesterol. Possibly important in areas of higher neuron:glia ratio | Conti et al., 1998; He et al., 2000; Torres et al., 2003; Nieoullon et al., 2006 |
| EAAT4 | Soma and dendrites of Purkinje cells(cerebellum). Also, hippocampus, neocortex | Postsynaptic neuronal terminals, alsodetected in astrocytes | High Cl− conductance. Regulator of neuronal excitability, counteracting depolarization of neurons | Fairman et al., 1995; Bar-Peled et al., 1997; Hu et al., 2003; Massie et al., 2008 |
| EAAT5 | Retina | Rod photoreceptor and bipolar cells | High Cl− conductance | Fairman et al., 1995; Arriza et al., 1997 |
The EAATs are membrane-bound pumps that closely resemble ion channels. These transporters play the important role of regulating concentrations of glutamate in the extracellular space, maintaining it at low physiological levels that promote biological function without promoting toxicity. After glutamate is released as the result of an action potential, glutamate transporters quickly remove it from the extracellular space to keep its levels low, thereby terminating synaptic transmission (Beart and O’Shea, 2007). The translocation of glutamate over the cell membrane is an energetically unfavorable process, as it takes place against a massive concentration gradient. Thus, the EAATs are referred to as symporters- a term used to describe a transporter protein that transports a substrate by the co- and counter-transport of ions. The transport cycle is initiated by concomitant binding of a substrate molecule, three sodium ions and a proton to an outward-facing conformation of the EAAT. This triggers a conformational cascade resulting in the EAAT adopting an inward-facing conformation from which the sodium ions, protons and the substrates are released into the cytoplasm of the cell. Subsequently, the transporter returns to its outward-facing conformation via the counter-transport of a potassium ion and becomes accessible for a new substrate molecule in the synaptic cleft (Danbolt et al., 1992; Levy et al., 1998; Robinson, 1998). Additionally, the EAATs possess a thermodynamically uncoupled Cl− flux, particularly pronounced for EAAT4 and EAAT5, which combine high Cl− conductance with relatively poor glutamate uptake capacities and thus seem to represent dual transporter/Glu-gated Cl− channels (Fairman et al., 1995).
Glutamate uptake is regulated at multiple levels (Gegelashvili et al., 2000). The expression of the transporter protein is regulated by cAMP, neuronal factors, and in response to various brain injuries (Anderson and Swanson, 2000). For example, GLT1 can be induced in astroglial cultures by the pituitary adenylate cyclase-activating peptide (PACAP), a neuron-derived peptide (Figiel and Engele, 2000). The activity of expressed transporters can be regulated by phosphorylation, sulfhydryl oxidation, arachidonic acid and other factors (Conradt and Stoffel, 1997; Gegelashvili et al., 2000; Anderson and Swanson, 2000). In addition, at least some glutamate transporter subtypes can transit between the intracellular compartment and the membrane surface. EAAT3 has been shown to move to the membrane surface in C6 glioma cells after phorbol ester-mediated protein kinase C (PKC) activation whereas astrocyte GLAST accumulation at the membrane may be inhibited by phorbol ester (Gonzalez et al., 2003). In both instances, the changes in membrane localization of transporters correlate withchanges in glutamate transportactivity.
Astrocytic Roles in Glutamate Transport
Rapid removal of glutamate from the extracellular space is required for the survival and normal function of neurons. Although all CNS cell types express glutamate transporters, astrocytes are the cell type primarily responsible for glutamate uptake (Fairman and Amara, 1999; Kanai and Hediger, 2003). Glutamate uptake by astrocytes is mediated by Na+-independent and Na+-dependent systems. Astrocyte Na+-dependent glutamate transporters were originally cloned from rat brain and termed GLAST and GLT-1. Transporter activity is normally regulated at multiple levels, including protein expression, cell surface trafficking, protein binding, and by phosphorylation and other direct modifications (Anderson and Swanson, 2000; Sonnewald et al. 2002; Bunch et al., 2009).
Neurodegeneration
Neurodegeneration results from the cumulative loss in structure or function of neurons. Neuronal death in these cases can be caused by triggers for programs that lead to cellular demise, or programmed cell death (PCD), such as apoptosis, autophagy, or cytoplasmic/Type III cell death. Processes triggering cell death include defects in protein degradation, reactive oxygen species, calcium dysregulation, mitochondrial dysfunction, and excitotoxicity (Schweichel and Merker, 1973; Clarke, 1990). Cross talk between pathways has been reported (Gonzalez-Polo et al., 2005; Hsieh et al., 2009) and neurodegenerative disease samples can show morphological signs of necrosis (Artal-Sanz et al., 2005, 2006). Thus, the mechanisms responsible for neuronal death may represent more of a continuum rather than a process that can simply be defined categorically.
Mechanisms of Neuronal Death
Apoptosis can occur as a result of stimulation of surface death receptors and proceed via the extrinsic pathway through Caspases-8 or -10 (Salvesen and Dixit, 1997). Alternatively, apoptosis can occur by intrinsic, caspase-9-related pathways triggered by signals from the mitochondria or, in some cases, the endoplasmic reticulum and other organelles (Yuan and Yankner, 1999; Morishima et al., 2002; Rao et al., 2002). Autophagy is considered a response to cellular stress, which can result in cell death after prolonged activation. Autophagy is a complex process and there are now examples where autophagy can actually be protective and situations where it changes from protective to apoptosis (Bhutia et al., 2010a, 2010b). Its hallmark is the catabolism of organelles by enclosing them in membranes, or autophagosomes, then fusing with a lysosome to complete the degradation. The resulting products are then recycled for the recovery of energy and amino acids. Autophagy has been shown to be a compensatory mechanism for the ubiquitin-proteasome degradation system (Pandey et al., 2007). Thus, the impairment of autophagy may have a role in neurodegeneration caused by the aggregation of misfolded proteins. A number of neurodegenerative diseases demonstrate a level of misfolded protein accumulation including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and Amyotrophic Lateral Sclerosis (Kopito and Ron, 2000; Taylor et al., 2002). These misfolded proteins can be triggers for cellular stress responses and cell death, leading to neurodegenerative effects.
Glutamate-Mediated Excitotoxicity
An additional source of neuronal damage is glutamate-mediated excitotoxicity, which is implicated in the pathogenesis of trauma, ischemia, and neurodegenerative diseases. Excessive stimulation of glutamate receptors can have numerous detrimental effects such as calcium homeostasis dysfunction, increased nitric oxide (NO) production, activation of proteases, an increase in cytotoxic transcription factors, and increased free radicals (Want and Qin, 2010). Generally, ion imbalance during excitotoxicity results from defects in gating ions from entering the cytoplasm as well as impairments in pumping ions out of the cytoplasm (Carafoli et al., 2001). Glutamate receptor overstimulation causes postsynaptic neurons to be overloaded by extracellular Ca2+ and Na+ (Rothman and Olney, 1995) as well as intracellular Ca2+ via release from mitochondria. Mechanisms used by the cell to reduce Ca2+ levels, such as calcium pumps (PMCA pump) and exchangers (NCX exchanger), are also impaired (Philipson and Nicoll, 2000; Carafoli et al., 2001). Additionally, Cl− plays an important role in acute excitotoxicity as an influx of Cl−, Na+, and water can cause cell lysis after excessive depolarization of the postsynaptic membrane (Inglefield and Schwartz-Bloom, 1998).
Role of Astrocytes in Neuronal Death and Dysfunction
Astrocytes constitute the majority of glial cells within the brain and may account for up to 50% of the brain’s volume (Tower and Young, 1973). Numerous discoveries continue to uncover a plethora of astrocytic functions. The astrocyte is a versatile cell tactically located between blood vessels and neurons (Kettenmann and Ransom, 2005). Among their functions, astrocytes take up glucose, influence cerebral blood flow (Mulligan and MacVicar, 2004; Takano et al., 2006; Gordon et al., 2008), maintain extracellular potassium (Karwoski et al., 1989), and neurotransmitter levels (Danbolt, 2001), including glutamate. The role of astrocytes in neuronal degeneration has been extensively studied in ALS. Mutations in Cu-Zn superoxide dismutase (SOD1) have been linked to amyotrophic lateral sclerosis (ALS), leading to investigation of its role in astrocytes and its possible connection to motor neuron death. In astrocytes expressing mutant SOD1, ROS and prostaglandin D2, both toxic to motor neurons, were released at higher levels (Di Giorgio et al., 2007, 2008; Nagai et al., 2007; Marchetto et al., 2008). Additionally, mutant SOD1 astrocytes lost the ability to positively regulate motor neuron expression of the GluR2 subunit in AMPA glutamate receptors (Van Damme et al., 2007). This subunit controls the receptor’s level of permeability to Ca2+, a highly important factor in glutamate excitotoxicity. The astrocyte-selective glutamate transporter EAAT2 has been shown to be paramount in keeping extracellular glutamate below excitotoxic levels (Rothstein et al., 1996). Astrocytes in close proximity to the neural synapse permits detection and reaction to increased levels of extracellular neurotransmitters (Volterra and Meldolesi, 2005). When they are damaged in a way that affects their ability to sense or respond to increases in glutamate levels, the microenvironment for nearby neurons is disrupted. This may lead to an acceleration of the degenerative process (Rossi and Volterrab, 2009).
Excitotoxicity in Ischemic Neuronal Damage
In addition to neurodegenerative disease, excitotoxicity may be a factor in ischemic models of neuronal damage (Bruijn et al., 2004). Channel dysregulation leading to Ca2+ influx during ischemia can overload neurons in a manner similar to excitotoxic conditions (Xiong et al., 2004). An important Na+/Ca2+ exchanger, NCX, is cleaved both under ischemic conditions and when exposed to glutamate, leading to neuronal cell death (Philipson and Nicoll, 2000). Hypoxia in brain white matter can lead to loss of oligodendrocytes, which myelinate axons, and subsequent functional deficits (Park et al., 2004). Ischemia followed by reperfusion is known to cause Ca2+ overload and a spike in NO production, which results in apoptotic cell death due to ER stress (Oyadomari et al., 2002). Additionally, NO reacts with O2−, producing peroxynitrite (OONO−) leading to neuron damage (Lipton et al., 1993; Yamauchi et al., 1998). Again, calcium is highly important as its dysregulation can lead to inappropriate activation of calpains, calcium-dependant proteases. Calpains are implicated in damage during ischemia as well as excitotoxic neurodegeneration (Lankiewicz et al., 2000). Polymorphisms in glutamate receptors, such as those found in EAAT2, may also play a role in the vulnerability of neurons post-ischemic insults (Mallolas et al., 2006). COX-2 inhibition has shown promise as a treatment in vitro to combat the effects of overstimulation of glutamate receptors (Carlson, 2003; McCullough et al., 2004). This can be useful as COX-2 upregulation has been identified in neurons and non-neuron cells following cerebral ischemia (Iadecola et al., 1999) and in the spinal cord (Yasojima et al., 1999), hippocampus and cortex (Yokota et al., 2004) of ALS patients.
Cloning and Regulation of EAAT2
EAAT2
The predominant glutamate transporter in the adult brain among the five EAATs (EAAT1-5) is EAAT2, also identified in the rodent as GLT-1, which is primarily expressed in astrocytes although its expression has been observed during development in neurons and oligodendrocytes (Bar-Peled et al., 1997; Furuta et al., 1997; Maragakis et al., 2004; Milton et al., 1997; Sheldon and Robinson, 2007). The normal physiological function of EAAT2 is the clearance of the neurotransmitter glutamate from neuronal synapses in the central nervous systems (Anderson and Swanson, 2000; Lehre and Danbolt, 1998). Impaired glutamate uptake by dysfunction or reduced expression of EAAT2 has been implicated in the pathogenesis of various diseases (Anderson and Swanson, 2000; Blei and Larsen, 1999; Chan and Butterworth, 1999; Couratier et al., 1993; Li et al., 1997; Martin et al., 1997; Pappas et al., 1998; Plaitakis and Caroscio, 1987; Rothstein et al., 1992; Rothstein et al., 1995; Tanaka et al., 1997; Torp et al., 1995; Trotti et al., 2001; Trotti et al., 1999; Wang et al., 2002). Downregulation of EAAT2 followed by accumulation of glutamate in extracellular fluid and neuronal death have been documented in chronic, debilitative neurological disorders of diverse etiology, including ALS (Couratier et al., 1993; Plaitakis and Caroscio, 1987; Rothstein et al., 1992; Rothstein et al., 1995; Trotti et al., 2001; Trotti et al., 1999), Alzheimer’s disease (Li et al., 1997), several forms of epilepsy (Tanaka et al., 1997), ischaemia/stroke (Martin et al., 1997; Torp et al., 1995), HIV-associated dementia (Pappas et al., 1998; Wang et al., 2002), traumatic brain injury (Martin et al., 1997) and hepatic encephalopathy (Blei and Larsen, 1999; Chan and Butterworth, 1999).
Localization
While expression of EAAT4 and EAAT5 is predominantly limited to the cerebellum and retina, respectively, EAAT1, EAAT2 and EAAT3 are widely distributed in the CNS (Table 1) (Arriza et al., 1997; Lehre et al., 1995; Rothstein et al., 1994; Yamada et al., 1996). EAAT3 is mainly expressed in neurons (Rothstein et al., 1994). In the adult CNS glutamate uptake from the synapse is principally controlled by glial transporters, EAAT1 and EAAT2 (Sheldon and Robinson, 2007). Although EAAT1 is mainly expressed in the developmental period of the CNS, its overall expression is considerably lower than that of EAAT2 in the adult CNS (Bar-Peled et al., 1997; Furuta et al., 1997; Maragakis et al., 2004; Storck et al., 1992). EAAT2 is predominantly expressed in astrocytes under normal conditions, although it is also detected in oligodendrocytes and neurons (Bar-Peled et al., 1997; Furuta et al., 1997; Maragakis et al., 2004; Milton et al., 1997; Sheldon and Robinson, 2007). EAAT2 is highly expressed throughout the brain and spinal cord, and glial EAAT2 is responsible for more than 90% of total glutamate uptake (Bar-Peled et al., 1997; Furuta et al., 1997; Maragakis et al., 2004). In addition, the GLT-1 knockout mice showed significant loss of hippocampal neurons, seizures, and 50% mortality at 6 weeks of age (Tanaka et al., 1997). Accordingly, its expression is primarily detected in the cell membrane of astrocytes interdigitated between synapses (Schmitt et al., 2002; Sullivan et al., 2004). However, the function of EAAT2 in oligodendrocytes and neurons remains unclear.
Cloning of the EAAT2 promoter
Based on its significant role in maintaining the normal functioning of the brain, numerous studies have addressed the mechanism(s) regulating expression of human EAAT2 (Collier, 1998; Gegelashvili et al., 1997; Miralles et al., 2001; Mitrovic et al., 1998; Munch et al., 2000; Palmada et al., 2002; Robinson, 1998; Vandenberg et al., 1997; Zelenaia et al., 2000). Rodent GLT-1 mRNA and protein are downregulated by TNF-α and upregulated by TGF-α, EGF and cAMP (Gegelashvili et al., 1997; Schlag et al., 1998; Zelenaia et al., 2000). However, confirmation of potential transcriptional regulation of the human EAAT2 and rodent GLT-1 genes was not possible because of the lack of information about the genomic organization of these genes. Early attempts to identify the promoter region of the human EAAT2 gene were unsuccessful because of the presence of a very large intron (~100kb) separating exon 1 from exon 2. To clone the EAAT2 promoter a sequential progressive genomic scanning cloning (SPGS) approach was employed, in which nylon filter papers with a human genomic BAC library were initially screened by a PCR-amplified and labeled exon 2 probe (Su et al., 2003b). This screening identified genomic fragments containing exon 2 with a large intron between exon 1 and 2. Additional screening using probes containing part of intron 1 provided three clones including the sequences of exon 1 and an additional ~2.5-Kb of the 5′ upstream region (Su et al., 2003b). Sequential analysis of this putative human EAAT2 promoter (EAAT2-Prom) identified five Sp1 sites and GC-rich repeats without a TATA box (Su et al., 2003b). Using a primer extension assay, the transcriptional initiation site of EAAT2 was determined as an adenosine residue located at 283-bp upstream of the translation initiation codon (ATG) (Su et al., 2003b).
Transcriptional Regulation
Previous studies showed that EAAT2 is not expressed in cultured astrocytes, but co-culturing astrocytes with neurons induces its expression (Schlag et al., 1998; Swanson et al., 1997). This effect was also mimicked by treating astrocytes with neuron-conditioned media or with cAMP, epidermal growth factor (EGF) or pituitary adenylate cyclase-activating polypeptide (Fig. 1) (Figiel and Engele, 2000; Gegelashvili et al., 1997; Schlag et al., 1998; Su et al., 2003b; Swanson et al., 1997; Zelenaia et al., 2000). These treatments induce EAAT2 mRNA levels as well as its transcription rate (Sitcheran et al., 2005; Su et al., 2003b; Zelenaia et al., 2000). The studies with pharmacological and genetic inhibitors suggest that these effects depend on the signaling pathways through PI3K and NF-κB (Li et al., 2006; Su et al., 2003b; Zelenaia et al., 2000). In addition, NF-κB directly binds to the EAAT2 promoter, and regulates its transcription (Sitcheran et al., 2005). NF-κB is primarily a transcriptional activator (Baldwin, 1996). However, there are specific examples where it can also act as a transcriptional repressor (Gires et al., 2001; Ke et al., 2001). TNF-α and dioxin negatively regulates the expression of epithelial cell adhesion molecule (EpCAM) and cytochrome P-450 1A1, which is mediated by NF-κB (Gires et al., 2001; Ke et al., 2001). The EAAT2 promoter is a unique model for study of NF-κB function, as NF-κB is required for activation as well as repression of the EAA2 promoter (Sitcheran et al., 2005; Su et al., 2003b). Both N-myc and NF-κB are required for TNF-α-mediated transcriptional repression of EAAT2 (Sitcheran et al., 2005; Su et al., 2003b). On the other hand, NF-κB mediates EGF-, TGF-α- and cAMP-induced EAAT2 promoter activation (Sitcheran et al., 2005; Su et al., 2003b). CEF also induces EAAT2 expression through increasing NF-κB binding to the EAAT2 promoter (Fig. 2) (Lee et al., 2008). These results highlight NF-κB as crucial regulator of EAAT2 expression in response to diverse cellular signals. Recent studies also revealed that the binding of κB-motif binding phosphoprotein to the EAAT2 promoter is critical for the astroglial synaptic function of EAAT2 and that EAAT2 is a PPARγ target gene involved in neuroprotection (Romera et al., 2007; Yang et al., 2009).
Figure 1.
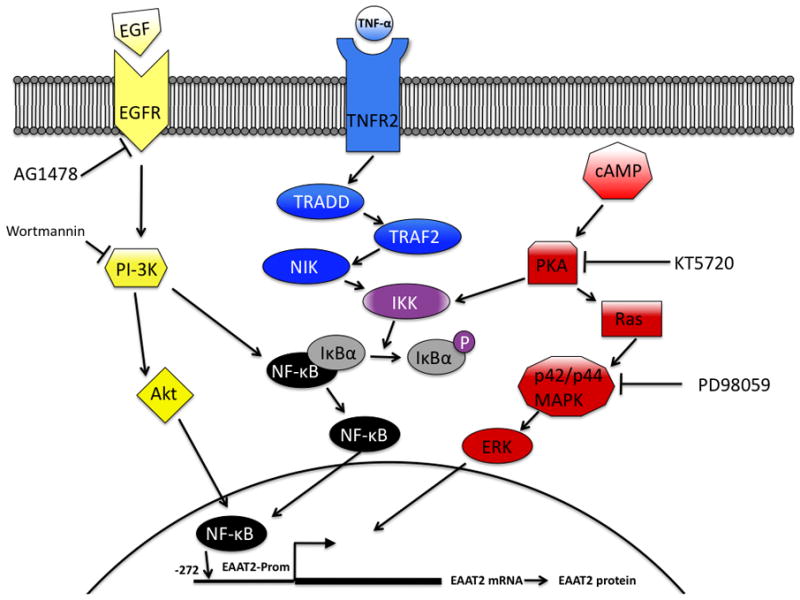
Schematic representation of pathways and inhibitors affecting EAAT2-Prom activity. EGF-R, EGF receptor; TNFR, TNF-α receptor; TRADD, TNF receptor-1-associated death domain protein; TRAF2, TNF receptor-associated factor 2; NIK, NF-κB-induced kinase; IκK, I-κB kinase; IκB, inhibitor of NF-κB; ERK, extracellular signal-regulated kinase.
Figure 2.
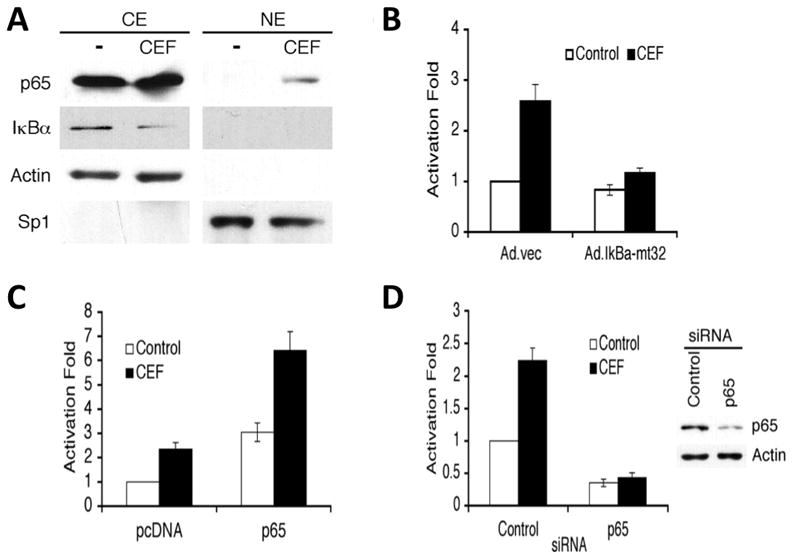
Activation of NF-κB by CEF. A, PHFA were treated with 10 μM for 4 days. Nuclear extracts (NE) and cytoplasmic extracts (CE) were prepared and immunoblotted with the indicated antibodies. B, PHFA were infected with Ad.vec or Ad.IκBα-m32. The infected cells were transfected with 3κB-Luc, containing three tandem NF-κB binding sites. C, PHFA were transfected with the EAAT2Pro-954 and pcDNA or p65 expression vector together with pSV-β-galactosidase plasmid as an internal control. One day after transfection, cells were treated with 10 μM ceftriaxone for 2 days. D, PHFA were transfected with the EAAT2Pro-954 and control siRNA or p65 siRNA together with a pSV-β-galactosidase plasmid as an internal control. P65 siRNA treatment abolished ceftriaxone-mediated induction of EAAT2 promoter activity. (Data from Lee et al., J Biol Chem, 2008)
Screening of FDA-Approved Drugs and Regulation of EAAT2 Expression by β-Lactam Antibiotics
Screening of Compounds Identifies β-Lactam Antibiotics as Inducers of EAAT2
Administration of 1,040 FDA-approved drugs and nutritionals to spinal cord slice cultures derived from 9-day old rats identified compounds that increased activity of EAAT2 and the levels of EAAT2 protein (Rothstein et al., 2005). Each compound was added at 100 μM biweekly and studied in duplicate or triplicate. Rat tissue was collected 5 to 7 days after the drug treatment and examined for expression of the EAAT2 protein using EAAT2 anti-peptide antibodies. Fifteen different β-lactam antibiotics were able to stimulate EAAT2 protein expression by more than two-fold. This increased expression of EAAT2 protein was detected as early as 48 h after drug treatment.
In vitro Effects on EAAT2 Expression
Human EAAT2 promoter linked to firefly luciferase was stably transfected into primary human fetal astrocytes (PHFA) or PHFA immortalized using H-TERT (IM-PHFA) (Su et al., 2003a) and induction of EAAT2 promoter activity by the same active compounds identified using brain slices was examined by comparing relative luciferase activity. The human EAAT2 promoter was significantly activated by CEF, amoxicillin and dibutyryl cyclic AMP while controls such as glutamate and glycine had no effect (Rothstein et al., 2005). This activation was dose-dependent and lasted at least 7 days. The increased expression of EAAT2 was observed as early as 48 h after drug treatment. The results of these experiments suggest that the model system we have developed, either PHFA or IM-PHFA can be used to screen for compounds that selectively upregulate EAAT2 promoter activity (Fig. 3).
Figure 3.
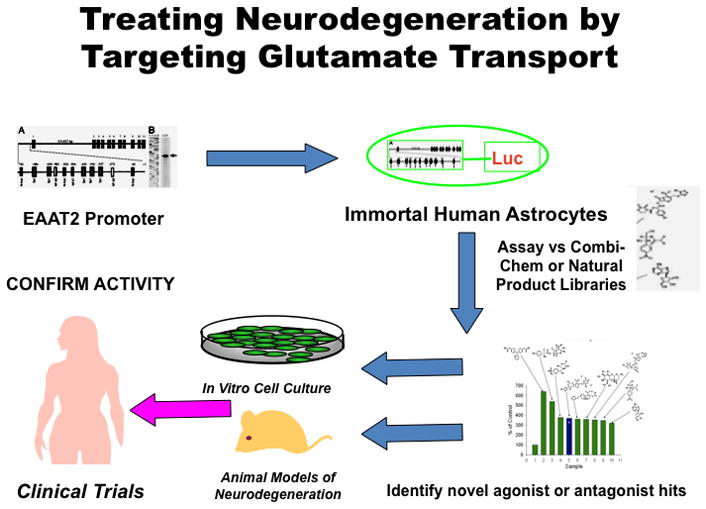
Schematic representation of screening paradigm to identify regulators of EAAT2 expression affecting glutamate transport in the brain. Using the EAAT2 promoter (EAAT2-Prom) linked to a luciferase reporter gene (EAAT2-Luc) we have established Telomerase-immortalized primary human fetal astrocyte (IM-PHFA) clones, expressing EAAT2-Luc (IM-PHFA-EAAT2-Luc). These clones can be used to screen various small molecule libraries, including Combi-Chem, ASINEX and Natural Product Libraries, to identify novel agonist and antagonists of the EAAT2-Prom. Once putative regulators of EAAT2 have been identified they can be tested for activity in in vitro neuronal/astrocyte co-culture systems and animal models of neurodegeneration. Positive compounds can be modified chemically to produce potentially active/safe compounds that pass the blood brain barrier and can be used for clinical trials to define active agents capable of preventing or treating various neurodegenerative conditions.
In vivo Effects on EAAT2 Expression
In vivo studies were performed with normal rats to examine the biological activity of CEF, which was administered i.p. (200 mg/kg) for 5 to 7 days (Rothstein et al., 2005). The effect of CEF on rats was very pronounced in that it increased GLT1 protein expression by three-fold and also increased the level of a splice variant GLT1b in the hippocampus and spinal cord and this effect persisted for three months after treatment. Other glutamate transporters such as GLAST and EAAC1, and EAAT4 were not affected by CEF. When CEF was chronically administered to GLT1-BAC-eGFP promoter reporter mice activation of the EAAT2 promoter was also observed in vivo. Significant increases of reporter expression were detected in astroglial soma and processes throughout the hippocampal CA1 neuropil region in mice. However, neuronal expression of this reporter was not activated by CEF treatment in the brain region. Biochemically, glutamate transport activity was enhanced by treatment with cephalosporin, one of the derivatives of CEF, as evidenced by increased glutamate uptake into the cortical membrane or spinal cord (Rothstein et al., 2005). CEF treatment in vitro and in vivo resulted in a 3-fold increase of EAAT2 protein expression levels and a comparable increase in EAAT2 transporter activity. Increasing expression of the glutamate transporter in cell lines and transgenic mice correlated with enhanced neuroprotection in glia (Guo et al., 2003). Exposure of cultured neurons to a short ischemic preconditioning resulted in subsequent stronger resistance to a severe ischemic condition (Romera et al., 2004). One-hour treatment of cultured neurons under a low oxygen-glucose deprivation (OGD) condition was lethal to neurons due to neuronal injury with toxicity that involved excessive glutamate. However, preconditioning with 5 min treatment of short OGD resulted in a significant increase of resistance to neuronal death, called neuronal protection. This protection was caused in part by increased expression of the EAAT2 protein (Romera et al., 2004). Ischemic preconditioning OGD (5 min) applied 24 h before 1 h OGD mediated neuronal protection. Pretreatment with 1 μM CEF 48 h before a 1 h OGD treatment was also protective, preventing neuronal death in ischemic tolerance (Fig. 4). CEF treatment of spinal cord cultures prevented in a dose-dependent manner threo-hydroxyaspartate (THA)-induced motor neuron loss (Fig. 4) (Rothstein et al., 2005). To test if CEF could alter neurodegenerative changes in a disease model, G37A SOD1 mice were treated with CEF daily. CEF treatment significantly delayed loss of muscle strength and body weight, and increased overall survival of mice by 10 days (Fig. 4). CEF also altered cellular neurodegeneration in vivo and G93A mice receiving CEF at 70 days of age showed a significant prevention of motor neuron loss and reduction of hypercellular gliosis compared to control mice (Rothstein et al., 2005).
Figure 4.
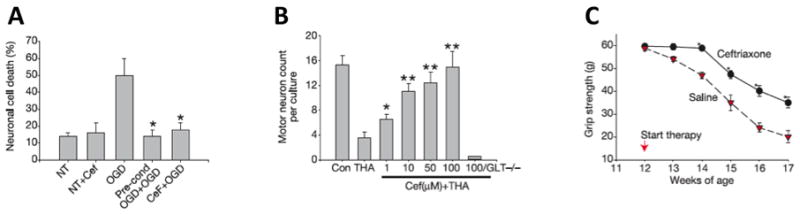
In vitro and in vivo neuroprotection by CEF. A, Oxygen glucose deprivation (OGD) preconditioning or ceftriaxone pre-treatment (1 μM) was protective compared to no treatment. B, CEF treatment of spinal cord cultures prevented threo-hydroxyaspartate (THA)-induced loss of motor neuron but not in GLT1-null mouse tissue. C, CEF treatment delayed loss of muscle strength in G93A SOD1 ALS mice. (Data from Rothstein et al., Nature, 2005)
Mechanisms of EAAT2 Expression Regulation: A Prominent Role for NF-κB
The mechanism by which CEF enhances EAAT2 expression was investigated in primary human fetal astrocytes (PHFA) (Lee et al., 2008). CEF increased EAAT2 transcription in PHFA through the NF-κB signaling pathway. NF-κB binding site at −272 position was critical in ceftriaxone-mediated EAAT2 protein induction. CEF-induced NF-κB activation through degradation of IκBα and induction of p65 nuclear translocation upregulated its’ downstream target EAAT2 (Fig. 2). As a result, CEF increased glutamate uptake by overexpression of EAAT2 across the plasma membrane of astrocytes.
Drug Screening for Potential Regulators of Glutamate Levels
A potential involvement of mammalian target of rapamycin (mTOR) signaling was studied in EAAT2 regulation in astrocytes (Wu et al., 2010). Culturing astrocytes in astrocyte-defined medium including EGF and insulin resulted in enhancement of the levels of the phosphorylated forms of Akt1 and mTOR as well as the expression of EAAT2 protein. Inhibitor treatment against Akt1 decreased mTOR phosphorylation and EAAT2 expression. However, mTOR inhibition resulted in decreased EAAT2 expression, but did not change Akt1 phosphorylation. Colton et al. (2010) reported screening large compound libraries for molecules that can activate translation of EAAT2 transcript with long 5′-UTRs utilizing a cell-based ELISA assay. Their HTS approach resulted in identification of lead compounds that showed dose-dependent increase of EAAT2 protein in vitro. Li et al. (2010) identified through screening of a library of 1040 FDA approved compounds and natural products harmine, a natural beta-carbilone alkaloid, as one activator of EAAT2 promoter that showed increased GLT1 expression and glutamate transporter activity in an animal model. Wu et al. (2010) studied functional relationship in ethanol response in astrocytes between EAAT2 and equilibrative nucleoside transporter (ENT1), a factor known to be important in regulating ethanol effects in brain (Dunwiddie and Masino, 2001). ENT1-specific inhibitor and siRNA treatment resulted in decreased EAAT2 expression and glutamate uptake. Treatment with 100 mM ethanol led to enhanced expression of EAAT2 and glutamate transport activity. The ethanol-mediated increase of EAAT2 mRNA expression was completely abolished by ENT1-specific siRNA treatment, indicating EAAT2 expression is ENT1-dependent in ethanol regulation. Rats that received optimal doses of CEF displayed attenuation of cue-induced relapse to cocaine-seeking behavior due to an increase in GLT1 expression in both prefrontal cortex and nucleus accumbens (Sari et al., 2009). Taken together these data indicate that EAAT2 plays an important role in regulation of alcohol and drug addiction through glutamate transport activity and it may provide unique opportunities for therapeutic intervention.
Conclusions and Future Translational Potential and Directions
Regulation of extracellular glutamate levels in the brain is crucial for maintenance of its normal functions. Abnormalities in this process are implicated in several neurodegenerative diseases including Alzheimer’s disease, Huntington’s disease, and amyotrophic lateral sclerosis. Since the major regulator of extracellular glutamate levels in the brain is the EAAT2 promoter, proper expression and regulation of this promoter is critical for maintaining brain homeostasis and survival of neurons. This review is focused on the critical role of EAAT2 and glutamate in neurodegeneration and the potential use of the EAAT2 promoter for developing screening protocols to identify molecules capable of physiologically and safely regulating glutamate levels in vitro and in vivo in animals, with ultimate applications in humans. We have identified β-lactam antibiotics, such as CEF, as transcriptional activators of EAAT2 that are capable of providing neuronal protection through facilitating glutamate uptake by astroglial cells (Rothstein et al., 2005). This finding suggests potential applications for these types of drugs as therapeutic agents to limit and prevent glutamate excitotoxicity. A thorough understanding of the mechanism(s) underlying transcriptional activation of EAAT2 may help identify potentially new molecules and targets for drug discovery leading to compounds that can ameliorate and potentially prevent neurodegeneration (Lee et al., 2008). Through chemical modeling, it may be possible to develop new derivatives of CEF with enhanced pharmacological and bioactivity properties that can be orally delivered and pass more readily through the blood brain barrier to decrease the severity and progression of specific neurodegenerative diseases. Use of the EAAT2 promoter as a screening paradigm also provides an entry point for identifying potentially new classes of neuroprotective drugs that function by controlling glutamate levels in the synaptic region of neurons (Fig. 3).
Acknowledgments
Contract grant sponsor: National Institutes of Health; Contract grant R01 CA134721 and P01 NS31492 and the Samuel Waxman Cancer Research Foundation P.B.F.; the Goldhirsh Foundation for Brain Cancer Research, the Dana Foundation and the McDonnell Foundation to D.S.; the National Research Foundation Basic Science Research Program (2010-0008219) and Medical Research Center Program (2009-0063466) of the Korean Ministry of Education, Science and Technology to S.G.L.
The present studies were supported in part through NIH NCI grants R01 CA134721, R03 MH093195, P01 NS31492, and the National Foundation for Cancer Research (NFCR) (PBF); the Goldhirsh Foundation for Brain Cancer Research, the Dana Foundation and the McDonnell Foundation (DS); and the National Research Foundation Basic Science Research Program (2010-0008219) and Medical Research Center Program (2009-0063466) of the Korean Ministry of Education, Science and Technology (SGL). DS is the Harrison Endowed Scholar and a Blick Scholar in the VCU Massey Cancer Center and the VCU School of Medicine. PBF holds the Thelma Newmeyer Corman Endowed Chair in Cancer Research in the VCU Massey Cancer Center. We thank Eric L. Howlett for his contributions to our EAAT2 studies.
LITERATURE CITED
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Eliasof S, Kavanaugh MP, Amara SG. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci U S A. 1997;94:4155–4160. doi: 10.1073/pnas.94.8.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artal-Sanz M, Tavernarakis N. Proteolytic mechanisms in necrotic cell death and neurodegeneration. FEBS Lett. 2005;579:3287–3296. doi: 10.1016/j.febslet.2005.03.052. [DOI] [PubMed] [Google Scholar]
- Artal-Sanz M, Samara C, Syntichaki P, Tavernarakis N. Lysosomal biogenesis and function is critical for necrotic cell death in Caenorhabditis elegans. J Cell Biol. 2006;173:231–239. doi: 10.1083/jcb.200511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Bar-Peled O, Ben-Hur H, Biegon A, Groner Y, Dewhurst S, Furuta A, Rothstein JD. Distribution of glutamate transporter subtypes during human brain development. J Neurochem. 1997;69:2571–2580. doi: 10.1046/j.1471-4159.1997.69062571.x. [DOI] [PubMed] [Google Scholar]
- Beart PM, O’Shea RD. Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol. 2007;150:5–17. doi: 10.1038/sj.bjp.0706949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutia SK, Dash R, Das SK, Azab B, Su Z-z, Lee S-G, Grant S, Yacoub A, Dent P, Curiel DT, Sarkar D, Fisher PB. Mechanism of authophagy to apoptosis switch triggered in prostate cancer cells by antitumor cytokine mda-7/IL-24. Cancer Res. 2010a;70:3667–3676. doi: 10.1158/0008-5472.CAN-09-3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutia SK, Kegelman TP, Das SK, Bzab B, Su Z-z, Lee S-G, Sarkar D, Fisher PB. Astrocyte elevated gene-1 induces protective autophagy. Proc Natl Acad Sci USA. 2010b doi: 10.1073/pnas.1009479107. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blei AT, Larsen FS. Pathophysiology of cerebral edema in fulminant hepatic failure. J Hepatol. 1999;31:771–776. doi: 10.1016/s0168-8278(99)80361-4. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Bunch L, Erichsen MN, Jensen AA. Excitatory amino acid transporters as potential drug targets. Expert Opin Ther Targets. 2009;13:719–731. doi: 10.1517/14728220902926127. [DOI] [PubMed] [Google Scholar]
- Carafoli E, Santella L, Branca D, Brini M. Generation, control, and processing of cellular calcium signals. Crit Rev Biochem Mol Biol. 2001;36:107–260. doi: 10.1080/20014091074183. [DOI] [PubMed] [Google Scholar]
- Carlson NG. Neuroprotection of cultured cortical neurons mediated by the cyclooxygenase-2 inhibitor APHS can be reversed by a prostanoid. J Neurosci Res. 2003;71:79–88. doi: 10.1002/jnr.10465. [DOI] [PubMed] [Google Scholar]
- Chan H, Butterworth RF. Evidence for an astrocytic glutamate transporter deficit in hepatic encephalopathy. Neurochem Res. 1999;24:1397–1401. doi: 10.1023/a:1022532623281. [DOI] [PubMed] [Google Scholar]
- Choi JS, Kim HY, Chung JW, Chun MH, Kim SY, Yoon SH, Lee MY. Activation of Src tyrosine kinase in microglia in the rat hippocampus following transient forebrain ischemia. Neurosci Lett. 2005;380:1–5. doi: 10.1016/j.neulet.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol. 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- Collier DA. Abnormal EAAT2 splicing in amyotrophic lateral sclerosis. Mol Psychiatry. 1998;3:298. [PubMed] [Google Scholar]
- Colton CK, Kong Q, Lai L, Zhu MX, Seyb KI, Cuny GD, Xian J, Glicksman MA, Lin CL. Identification of translational activators of glial glutamate transporter EAAT2 through cell-based high-throughput screening: an approach to prevent excitotoxicity. J Biomol Screen. 2010;15:653–662. doi: 10.1177/1087057110370998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradt M, Stoffel W. Inhibition of the high-affinity brain glutamate transporter GLAST-1 via direct phosphorylation. J Neurochem. 1997;68:1244–1251. doi: 10.1046/j.1471-4159.1997.68031244.x. [DOI] [PubMed] [Google Scholar]
- Conti F, DeBiasi S, Minelli A, Rothstein JD, Melone M, et al. EAAC1, a high-affinity glutamate transporter, is localized to astrocytes and gabaergic neurons besides pyramidal cells in the rat cerebral cortex. Cereb Cortex. 1998;8:108–116. doi: 10.1093/cercor/8.2.108. [DOI] [PubMed] [Google Scholar]
- Couratier P, Hugon J, Sindou P, Vallat JM, Dumas M. Cell culture evidence for neuronal degeneration in amyotrophic lateral sclerosis being linked to glutamate AMPA/kainate receptors. Lancet. 1993;341:265–268. doi: 10.1016/0140-6736(93)92615-z. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Storm-Mathisen J, Kanner BI. An [Na+ + K+]coupled L-glutamate transporter purified from rat brain is located in glial cell processes. Neuroscience. 1992;51:295–310. doi: 10.1016/0306-4522(92)90316-t. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Di Giorgio FP, Boulting GL, Bobrowicz S, Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 2008;3:637–648. doi: 10.1016/j.stem.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- Fairman WA, Amara SG. Functional diversity of excitatory amino acid transporters: ion channel and transport modes. Am J Physiol. 1999;277:481–486. doi: 10.1152/ajprenal.1999.277.4.F481. [DOI] [PubMed] [Google Scholar]
- Figiel M, Engele J. Pituitary adenylate cyclase-activating polypeptide (PACAP), a neuron-derived peptide regulating glial glutamate transport and metabolism. J Neurosci. 2000;20:3596–3605. doi: 10.1523/JNEUROSCI.20-10-03596.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci. 1997;17:8363–8375. doi: 10.1523/JNEUROSCI.17-21-08363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganel R, Rothstein JD. Glutamate transporter dysfunction and neuronal death. In: Hannah M, Adelmann GA, Peter J, editors. Ionotropic glutamate receptors in the CNS. Berlin: Springer; 1999. pp. 472–493. [Google Scholar]
- Gegelashvili G, Danbolt NC, Schousboe A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J Neurochem. 1997;69:2612–2615. doi: 10.1046/j.1471-4159.1997.69062612.x. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Dehnes Y, Danbolt NC, Schousboe A. The high-affinity glutamate transporters GLT1, GLAST, and EAAT4 are regulated via different signalling mechanisms. Neurochem Int. 2000;37:163–170. doi: 10.1016/s0197-0186(00)00019-x. [DOI] [PubMed] [Google Scholar]
- Gires O, Kieu C, Fix P, Schmitt B, Munz M, Wollenberg B, Zeidler R. Tumor necrosis factor alpha negatively regulates the expression of the carcinoma-associated antigen epithelial cell adhesion molecule. Cancer. 2001;92:620–628. doi: 10.1002/1097-0142(20010801)92:3<620::aid-cncr1362>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Gonzalez MI, Bannerman PG, Robinson MB. Phorbol myristate acetate-dependent interaction of protein kinase C{alpha} and the neuronal glutamate transporter EAAC1. J Neurosci. 2003;23:5589–5593. doi: 10.1523/JNEUROSCI.23-13-05589.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquere S, Eskelinen EL, Pierron G, Saftig P, Kroemer G. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–3102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456:745–749. doi: 10.1038/nature07525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Janssen WG, Rothstein JD, Morrison JH. Differential synaptic localization of the glutamate transporter EAAC1 and glutamate receptor subunit GluR2 in the rat hippocampus. J Comp Neurol. 2000;418:255–269. [PubMed] [Google Scholar]
- Hediger MA, Welbourne TC. Introduction: glutamate transport, metabolism, and physiological responses. Am J Physiol. 1999;277:F477–F480. doi: 10.1152/ajprenal.1999.277.4.F477. [DOI] [PubMed] [Google Scholar]
- Hsieh YC, Athar M, Chaudry IH. When apoptosis meets autophagy: deciding cell fate after trauma and sepsis. Trends Mol Med. 2009;15:129–138. doi: 10.1016/j.molmed.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu WH, Walters WM, Xia XM, Karmally SA, Bethea JR. Neuronal glutamate transporter EAAT4 is expressed in astrocytes. Glia. 2003;44:13–25. doi: 10.1002/glia.10268. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Forster C, Nogawa S, Clark HB, Ross ME. Cyclooxygenase-2 immunoreactivity in the human brain following cerebral ischemia. Acta Neuropathol (Berl) 1999;98:9–14. doi: 10.1007/s004010051045. [DOI] [PubMed] [Google Scholar]
- Inglefield JR, Schwartz-Bloom RD. Activation of excitatory amino acid receptors in the rat hippocampal slice increases intracellular Cl− and cell volume. J Neurochem. 1998;71:1396–1404. doi: 10.1046/j.1471-4159.1998.71041396.x. [DOI] [PubMed] [Google Scholar]
- Irving EA, Barone FC, Reith AD, Hadingham SJ, Parsons AA. Differential activation of MAPK/ERK and p38/SAPK in neurones and glia following focal cerebral ischaemia in the rat. Brain Res Mol Brain Res. 2000;77:65–75. doi: 10.1016/s0169-328x(00)00043-7. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Hediger MA. The glutamate and neutral amino acid transporter family: physiological and pharmacological implications. Eur J Pharmacol. 2003;479:237–247. doi: 10.1016/j.ejphar.2003.08.073. [DOI] [PubMed] [Google Scholar]
- Karwoski CJ, Lu HK, Newman EA. Spatial buffering of light-evoked potassium increases by retinal Muller (glial) cells. Science. 1989;244:578–580. doi: 10.1126/science.2785716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Rabson AB, Germino JF, Gallo MA, Tian Y. Mechanism of suppression of cytochrome P-450 1A1 expression by tumor necrosis factor-alpha and lipopolysaccharide. J Biol Chem. 2001;276:39638–39644. doi: 10.1074/jbc.M106286200. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Ransom BR. Neuroglia. Oxford: Oxford University Press; 2005. [Google Scholar]
- Kopito RR, Ron D. Conformational disease. Nature Cell Biol. 2000;2:E207–E209. doi: 10.1038/35041139. [DOI] [PubMed] [Google Scholar]
- Lankiewicz S, Marc Luetjens C, Truc Bui N, Krohn AJ, Poppe M, Cole GM, Saido TC, Prehn JH. Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J Biol Chem. 2000;275:17064–17071. doi: 10.1074/jbc.275.22.17064. [DOI] [PubMed] [Google Scholar]
- Lauriat TL, McInnes LA. EAAT2 regulation and splicing: relevance to psychiatric and neurological disorders. Mol Psychiatry. 2007;12:1065–1078. doi: 10.1038/sj.mp.4002065. [DOI] [PubMed] [Google Scholar]
- Lee SG, Su ZZ, Emdad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, Fisher PB. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem. 2008;283:13116–13123. doi: 10.1074/jbc.M707697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15 (3 Pt 1):1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy LM, Warr O, Attwell D. Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J Neurosci. 1998;18:9620–9628. doi: 10.1523/JNEUROSCI.18-23-09620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LB, Toan SV, Zelenaia O, Watson DJ, Wolfe JH, Rothstein JD, Robinson MB. Regulation of astrocytic glutamate transporter expression by Akt: evidence for a selective transcriptional effect on the GLT-1/EAAT2 subtype. J Neurochem. 2006;97:759–771. doi: 10.1111/j.1471-4159.2006.03743.x. [DOI] [PubMed] [Google Scholar]
- Li S, Mallory M, Alford M, Tanaka S, Masliah E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol. 1997;56:901–911. doi: 10.1097/00005072-199708000-00008. [DOI] [PubMed] [Google Scholar]
- Li Y, Sattler R, Yang EJ, Nunes A, Ayukawa Y, Akhtar S, Ji G, Zhang P-W, Rothstein JD. Harmine, a natural beta-carboline alkaloid, upregulates astroglial glutamate transporter expression. Neuropharmacology. 2010 doi: 10.1016/j.neuropharm.2010.10.016. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- Mallolas J, Hurtado O, Castellanos M, Blanco M, Sobrino T, Serena J, Vivancos J, Castillo J, Lizasoain I, Moro MA, Dávalos A. Polymorphism in the EAAT2 promoter is associated with higher glutamate concentrations and higher frequency of progressing stroke. J Exp Med. 2006;203:711–717. doi: 10.1084/jem.20051979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragakis NJ, Dietrich J, Wong V, Xue H, Mayer-Proschel M, Rao MS, Rothstein JD. Glutamate transporter expression and function in human glial progenitors. Glia. 2004;45:133–143. doi: 10.1002/glia.10310. [DOI] [PubMed] [Google Scholar]
- Marchetto MC, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH. Noncell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell. 2008;3:649–657. doi: 10.1016/j.stem.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Brambrink AM, Lehmann C, Portera-Cailliau C, Koehler R, Rothstein J, Traystman RJ. Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann Neurol. 1997;42:335–348. doi: 10.1002/ana.410420310. [DOI] [PubMed] [Google Scholar]
- Massie A, Cnops L, Smolders I, McCullumsmith R, Kooijman R, Kwak S, Arckens L, Michotte Y. High-affinity Na+/K+-dependent glutamate transporter EAAT4 is expressed throughout the rat fore- and midbrain. J Comp Neurol. 2008;511:155–72. doi: 10.1002/cne.21823. [DOI] [PubMed] [Google Scholar]
- McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton ID, Banner SJ, Ince PG, Piggott NH, Fray AE, Thatcher N, Horne CH, Shaw PJ. Expression of the glial glutamate transporter EAAT2 in the human CNS: an immunohistochemical study. Brain Res Mol Brain Res. 1997;52:17–31. doi: 10.1016/s0169-328x(97)00233-7. [DOI] [PubMed] [Google Scholar]
- Miralles VJ, Martinez-Lopez I, Zaragoza R, Borras E, Garcia C, Pallardo FV, Vina JR. Na+ dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) in primary astrocyte cultures: effect of oxidative stress. Brain Res. 2001;922:21–29. doi: 10.1016/s0006-8993(01)03124-9. [DOI] [PubMed] [Google Scholar]
- Mitrovic AD, Amara SG, Johnston GA, Vandenberg RJ. Identification of functional domains of the human glutamate transporters EAAT1 and EAAT2. J Biol Chem. 1998;273:14698–14706. doi: 10.1074/jbc.273.24.14698. [DOI] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Munch C, Schwalenstocker B, Hermann C, Cirovic S, Stamm S, Ludolph A, Meyer T. Differential RNA cleavage and polyadenylation of the glutamate transporter EAAT2 in the human brain. Brain Res Mol Brain Res. 2000;80:244–251. doi: 10.1016/s0169-328x(00)00139-x. [DOI] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieoullon A, Canolle B, Masmejean F, Guillet B, Pisano P, Lortet S. The neuronal excitatory amino acid transporter EAAC1/EAAT3: does it represent a major actor at the brain excitatory synapse? J Neurochem. 2006;98:1007–1018. doi: 10.1111/j.1471-4159.2006.03978.x. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7:335–345. doi: 10.1023/a:1016175429877. [DOI] [PubMed] [Google Scholar]
- Palmada M, Kinne-Saffran E, Centelles JJ, Kinne RK. Benzodiazepines differently modulate EAAT1/GLAST and EAAT2/GLT1 glutamate transporters expressed in CHO cells. Neurochem Int. 2002;40:321–326. doi: 10.1016/s0197-0186(01)00087-0. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Pappas TC, Alagarsamy S, Pollard RB, Nokta M. HIV decreases glutamate transport in SK-N-MC neuroblastoma cells. J Neurovirol. 1998;4:69–79. doi: 10.3109/13550289809113483. [DOI] [PubMed] [Google Scholar]
- Park E, Velumian AA, Fehlings MG. The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J Neurotrauma. 2004;21:754–774. doi: 10.1089/0897715041269641. [DOI] [PubMed] [Google Scholar]
- Philipson KD, Nicoll DA. Sodium-calcium exchange: a molecular perspective. Annu Rev Physiol. 2000;62:111–133. doi: 10.1146/annurev.physiol.62.1.111. [DOI] [PubMed] [Google Scholar]
- Plaitakis A, Caroscio JT. Abnormal glutamate metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1987;22:575–579. doi: 10.1002/ana.410220503. [DOI] [PubMed] [Google Scholar]
- Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE, Ellerby HM. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277:21836–21842. doi: 10.1074/jbc.M202726200. [DOI] [PubMed] [Google Scholar]
- Robinson MB. The family of sodium-dependent glutamate transporters: a focus on the GLT-1/EAAT2 subtype. Neurochem Int. 1998;33:479–491. doi: 10.1016/s0197-0186(98)00055-2. [DOI] [PubMed] [Google Scholar]
- Romera C, Hurtado O, Mallolas J, Pereira MP, Morales JR, Romera A, Serena J, Vivancos J, Nombela F, Lorenzo P, Lizasoain I, Moro MA. Ischemic preconditioning reveals that GLT1/EAAT2 glutamate transporter is a novel PPARgamma target gene involved in neuroprotection. J Cereb Blood Flow Metab. 2007;27:1327–1338. doi: 10.1038/sj.jcbfm.9600438. [DOI] [PubMed] [Google Scholar]
- Rossi D, Volterrab A. Astrocytic dysfunction: Insights on the role in neurodegeneration. Brain Research Bulletin. 2009;80:224–232. doi: 10.1016/j.brainresbull.2009.07.012. [DOI] [PubMed] [Google Scholar]
- Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor-still lethal after eight years. Trends Neurosci. 1995;18:57–58. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggell C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Frujin LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Sari Y, Smith KD, Ali PK, Rebec GV. Upregulation of Glt1 attenuates cue-induced reinstatement of cocaine-seeking behavior in rats. J Neurosci. 2009;29:9239–9243. doi: 10.1523/JNEUROSCI.1746-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlag BD, Vondrasek JR, Munir M, Kalandadze A, Zelenaia OA, Rothstein JD, Robinson MB. Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Mol Pharmacol. 1998;53:355–369. doi: 10.1124/mol.53.3.355. [DOI] [PubMed] [Google Scholar]
- Schmitt A, Asan E, Lesch KP, Kugler P. A splice variant of glutamate transporter GLT1/EAAT2 expressed in neurons: cloning and localization in rat nervous system. Neuroscience. 2002;109:45–61. doi: 10.1016/s0306-4522(01)00451-1. [DOI] [PubMed] [Google Scholar]
- Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–266. doi: 10.1002/tera.1420070306. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitcheran R, Gupta P, Fisher PB, Baldwin AS. Positive and negative regulation of EAAT2 by NF-kappaB: a role for N-myc in TNFalpha-controlled repression. EMBO J. 2005;24:510–520. doi: 10.1038/sj.emboj.7600555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnewald U, Qu H, Aschner M. Pharmacology and toxicology of astrocyte- neuron glutamate transport and cycling. J Pharmacol Exp Ther. 2002;301:1–6. doi: 10.1124/jpet.301.1.1. [DOI] [PubMed] [Google Scholar]
- Storck T, Schulte S, Hofmann K, Stoffel W. Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci U S A. 1992;89:10955–10959. doi: 10.1073/pnas.89.22.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su ZZ, Lebedeva IV, Sarkar D, Gopalkrishnan RV, Sauane M, Sigmon C, Yacoub A, Valerie K, Dent P, Fisher PB. Melanoma differentiation associated gene-7, mda-7/IL-24, selectively induces growth suppression, apoptosis and radiosensitization in malignant gliomas in a p53-independent manner. Oncogene. 2003a;22:1164–1180. doi: 10.1038/sj.onc.1206062. [DOI] [PubMed] [Google Scholar]
- Su ZZ, Leszczyniecka M, Kang DC, Sarkar D, Chao W, Volsky DJ, Fisher PB. Insights into glutamate transport regulation in human astrocytes: cloning of the promoter for excitatory amino acid transporter 2 (EAAT2) Proc Natl Acad Sci U S A. 2003b;100:1955–1960. doi: 10.1073/pnas.0136555100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan R, Rauen T, Fischer F, Wiessner M, Grewer C, Bicho A, Pow DV. Cloning, transport properties, and differential localization of two splice variants of GLT-1 in the rat CNS: implications for CNS glutamate homeostasis. Glia. 2004;45:155–169. doi: 10.1002/glia.10317. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC. Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci. 1997;17:932–940. doi: 10.1523/JNEUROSCI.17-03-00932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Nogawa S, Nagata E, Ito D, Suzuki S, Dembo T, Kosakai A, Fukuuchi Y. Persistent CREB phosphorylation with protection of hippocampal CA1 pyramidal neurons following temporary occlusion of the middle cerebral artery in the rat. Exp Neurol. 2000;161:462–471. doi: 10.1006/exnr.1999.7313. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- Torp R, Lekieffre D, Levy LM, Haug FM, Danbolt NC, Meldrum BS, Ottersen OP. Reduced postischemic expression of a glial glutamate transporter, GLT1, in the rat hippocampus. Exp Brain Res. 1995;103:51–58. doi: 10.1007/BF00241964. [DOI] [PubMed] [Google Scholar]
- Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci. 2003;4:13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- Tower DB, Young OM. The activities of butyrylcholinesterase and carbonic anhydrase, the rate of anaerobic glycolysis, and the question of a constant density of glial cells in cerebral cortices of various mammalian species from mouse to whale. J Neurochem. 1973;20:269–278. doi: 10.1111/j.1471-4159.1973.tb12126.x. [DOI] [PubMed] [Google Scholar]
- Trotti D, Rolfs A, Danbolt NC, Brown RH, Jr, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2:848. doi: 10.1038/12227. [DOI] [PubMed] [Google Scholar]
- Trotti D, Aoki M, Pasinelli P, Berger UV, Danbolt NC, Brown RH, Jr, Hediger MA. Amyotrophic lateral sclerosis-linked glutamate transporter mutant has impaired glutamate clearance capacity. J Biol Chem. 2001;276:576–582. doi: 10.1074/jbc.M003779200. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Bogaert E, Dewil M, Hersmus N, Kiraly D, Scheveneels W, Bockx I, Braeken D, Verpoorten N, Verhoeven K, Timmerman V, Herijgers P, Callewaert G, Carmeliet P, Van Den Bosch L, Robberecht W. Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity. Proc Natl Acad Sci USA. 2007;104:14825–14830. doi: 10.1073/pnas.0705046104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg RJ, Mitrovic AD, Chebib M, Balcar VJ, Johnston GA. Contrasting modes of action of methylglutamate derivatives on the excitatory amino acid transporters, EAAT1 and EAAT2. Mol Pharmacol. 1997;51:809–815. doi: 10.1124/mol.51.5.809. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Wang Y, Qin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010;15:1382–402. doi: 10.1007/s10495-010-0481-0. [DOI] [PubMed] [Google Scholar]
- Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, Rothstein JD, Volsky DJ. Reduced expression of glutamate transporter EAAT2 and impaired transport in human primary astrocytes exposed to HIV-1 or gp120. Virology. 2003;312:60–73. doi: 10.1016/s0042-6822(03)00181-8. [DOI] [PubMed] [Google Scholar]
- Wu X, Kihara T, Akaike A, Niidome T, Sugimoto H. PI3K/Akt/mTOR signaling regulates glutamate transporter 1 in astrocytes. Biochem Biophys Res Commun. 2010;393:514–518. doi: 10.1016/j.bbrc.2010.02.038. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Yamada K, Watanabe M, Shibata T, Tanaka K, Wada K, Inoue Y. EAAT4 is a post-synaptic glutamate transporter at Purkinje cell synapses. Neuroreport. 1996;7:2013–2017. doi: 10.1097/00001756-199608120-00032. [DOI] [PubMed] [Google Scholar]
- Yamauchi M, Omote K, Ninomiya T. Direct evidence for the role of nitric oxide on the glutamate-induced neuronal death in cultured cortical neurons. Brain Res. 1998;780:253–259. doi: 10.1016/s0006-8993(97)01201-8. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gozen O, Watkins A, Lorenzini I, Lepore A, Gao Y, Vidensky S, Brennan J, Poulsen D, Won Park J, Li Jeon N, Robinson MB, Rothstein JD. Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron. 2009;61:880–894. doi: 10.1016/j.neuron.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasojima K, Schwab C, McGeer EG, McGeer PL. Distribution of cyclooxygenase-1 and cyclooxygenase-2 mRNAs and proteins in human brain and peripheral organs. Brain Res. 1999;830:226–236. doi: 10.1016/s0006-8993(99)01389-x. [DOI] [PubMed] [Google Scholar]
- Yokota O, Terada S, Ishizu H, Ishihara T, Nakashima H, Kugo A, Tsuchiya K, Ikeda K, Hayabara T, Saito Y, Murayama S, Ue′da K, Checler F, Kuroda S. Increased expression of neuronal cyclooxygenase-2 in the hippocampus in amyotrophic lateral sclerosis both with and without dementia. Acta Neuropathol. 2004;107:399–405. doi: 10.1007/s00401-004-0826-2. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Caspase activity sows the seeds of neuronal death. Nature Cell Biol. 1999;1:E44–E45. doi: 10.1038/10037. [DOI] [PubMed] [Google Scholar]
- Zelenaia O, Schlag BD, Gochenauer GE, Ganel R, Song W, Beesley JS, Grinspan JB, Rothstein JD, Robinson MB. Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-kappaB. Mol Pharmacol. 2000;57:667–678. doi: 10.1124/mol.57.4.667. [DOI] [PubMed] [Google Scholar]