

Abstract
SP/KLF transcription factors comprise an emerging group of proteins that may behave as tumor suppressors. Incidentally, many cancers displaying alterations in certain KLF proteins are also associated with a high incidence of KRAS mutations. Therefore, we here investigate whether SP/KLF proteins suppress KRAS-mediated cell growth, and more importantly, the potential mechanisms underlying these effects. Using a comprehensive, family-wide screening of the 24 SP/KLF members, we discover that SP5, SP8, KLF2, KLF3, KLF4, KLF11, KLF13, KLF14, KLF15 and KLF16 inhibit cellular growth and suppress transformation mediated by oncogenic KRAS. Each protein in this subset of SP/KLF members individually inhibits BrdU incorporation in KRAS oncogenic mutant cancer cells. SP5, KLF3, KLF11, KLF13, KLF14 and KLF16 also increase apoptosis in these cells. Using KLF11 as a representative model for mechanistic studies, we demonstrate that this protein inhibits the ability of cancer cells to form colonies in soft agar and tumor growth in vivo. Molecular studies demonstrate that these effects of KLF11 are mediated, at least in part, through silencing cyclin A via binding to its promoter and leading to cell cycle arrest in S phase. Interestingly, similar to KLF11, KLF14 and KLF16 mechanistically share the ability to modulate the expression of cyclin A. Collectively, this study stringently defines a distinct subset of SP/KLF proteins that impairs KRAS-mediated cell growth, and that mechanistically, some members of this subset accomplish this, at least in part, through regulation of the cyclin A promoter.
Keywords: KLF proteins, KLF11, KRAS, tumor suppression, pancreatic cancer, cyclin A
INTRODUCTION
The SP/KLF family constitutes a group of transcription factors that are present in organisms ranging from yeast to vertebrates [1]. Their structure is defined by the presence of three highly conserved DNA-binding zinc finger domains and a variant N-terminal domain that contains transcriptional regulatory motifs [2, 3]. Identification of an entire repertoire of SP/KLF proteins, along with characterization of their biochemical properties, has been the focus of intensive investigations. These studies have revealed that KLF proteins bind to similar, yet distinct, GC-rich target sequences, and they function either as activators or repressors in cell- and promoter-dependent manners [2–4] by interacting with co-regulator molecules via different types of regulatory domains [2, 4–6]. Interestingly, however, expression of KLF proteins is altered in many types of cancer, including breast, head and neck, colon, esophagus, pancreas, liver, lymphomas, and leukemias. Thus, in spite of extensive biochemical characterizations performed on KLF proteins, their biological functions remain a matter of intense investigation, in particular, as they relate to carcinogenesis. Moreover, whether their biochemical properties, such as targeting of specific genes promoters, have a clear impact on their cell biological function remain unclear. Thus, investigations that enhance our understanding of the role of SP/KLF proteins at the mechanistic level is of paramount importance for more complete characterization of this important, evolutionarily conserved family. Consistent with this goal, the current study focuses on the functional characterization of known members of the SP/KLF family of transcription factors as it relates to their ability to suppress oncogene-mediated cell growth. Our results, for the first time, characterize a novel subset of SP/KLF proteins that antagonize KRAS-mediated cell growth via the inhibition of proliferation and induction of apoptosis. Mechanistically, the growth suppressive function of some members of this subset of SP/KLF proteins is achieved, at least in part, through downregulation of the promoter of a key cell cycle regulator, cyclin A. Since KRAS is mutated in 30% of all tumors [7] and taking into consideration the increasing evidence supporting the involvement of KLF proteins in different types of cancer, the novel data reported here is of significant relevance to tumor biology.
EXPERIMENTAL PROCEDURES
Cell Lines, Reagents and plasmids
Unless specified, all reagents were from Sigma (St. Louis, MO). Plasmids containing SP1-8 and KLF1-16 cDNAs along with Cyclin A2 (CCNA2) promoter (containing a region of the CCNA2 promoter corresponding to -468bp to +298bp upstream of the luciferase gene) and KLF11 deletion constructs were cloned as previously described [8–11]. Oncogenic human KRASV12 plasmid was a kind gift from Dr. E. Santos (National Institutes of Health, Bethesda, MD). To obtain the −89 to −91 CGC>TTT mutant cyclin A2 promoter pGL3 reporter construct, site-directed mutagenesis was performed by the QuickChange® Site-Directed Mutagenesis kit as suggested by the manufacturer (Agilent Technologies, Inc, Santa Clara, CA). The cyclin A2 expression construct was a generous gift from Dr. Robert Sheaff (University of Minnesota, Minneapolis, MN). KLF10, KLF11 and empty vector (Ad5CMV) carrying recombinant adenoviruses were generated in collaboration with the Gene Transfer Vector Core at the University of Iowa. Human pancreatic cancer cell lines and mouse fibroblast (NIH/3T3) cells used in this study were obtained directly from the American Type Culture Collection (ATCC, Rockville, MD) and maintained according to their recommendations. Mouse embryonic fibroblasts (MEF) were freshly isolated and cultured from homogenized E13.5 embryos arising from timed mating between wild-type C57BL6 mice using standard methods [12]. The normal human pancreatic ductal epithelial cell line, HPDE6 (immortalized but not transformed), was cultured as previously described [13].
Tumor xenografts and Transformation assays
2.5×106 CAPAN2 or L3.6 cancer cells were injected subcutaneously into the hind leg of athymic nude mice and measured weekly. Nine mice were injected for each experimental condition. We estimated tumor volume (V) from the length (l) and width (w) of the tumor using the formula: 4/3 × π× [(l + w) / 4] 3 [14]. Animal experiments were approved by the Animal Facility at Mayo Clinic College of Medicine. Transformation assays were performed as described previously [15].
Proliferation and apoptosis assays
Pancreatic cell lines were electroporated using a BTX square-wave electroporator (Holliston, MA; 10msec, 360 volts). Cells were co-transfected with a GFP vector (5μg) and the indicated SP/KLF plasmid (25 μg) or control pcDNA3 vector (25μg). Forty-eight hrs post-plating, cells were pulsed with 100μM BrdU for 1hr, fixed in 3.7% formaldehyde for 10min at room temperature, and permeabilized with 70% EtOH and DNase I at 30°C for 20min. After blocking with 5% goat serum for 30min at 37°C, coverslips were incubated for 60min with an anti-BrdU mAb (Roche Applied Science, Indianapolis, IN), and then a rhodamine-conjugated anti-mouse secondary antibody (Molecular Probes, Eugene, OR) and 1μg/ml DAPI. For quantitation, 500 GFP positive cells in four different 63X fields were counted using a Zeiss confocal microscope. Western blot was performed as control of tagged-SP/KLF expression. For cyclin A2 rescue experiments, MTS assay was performed as previously described [16]. For MEF experiments, cells were plated at 6,000–8,000 cells per well in a 96-well plate. Twenty-four hours later, the media was replaced with serum free media and cells were infected with EV- or KLF11-carrying adenovirus at a MOI of 200. Media was supplemented with either 5 or 10% FBS after 3 hours. Forty-eight hours later media was aspirated, wells were washed with PBS. Subsequently, fluorescent LIVE/DEAD® Viability/Cytotoxicity assay was performed according to manufacturer’s protocol (Invitrogen, Carlsbad, CA). For apoptosis, cells undergoing death were identified by Hoechst 33860 as previously described [17]. Cells undergoing apoptosis were identified by light microscopy by their morphological criteria of nuclear fragmentation, nuclear margination, cytoplasmic blebbing, and organelle disorganization. Results were validated using Annexin V staining. For quantitation, 300 GFP positive cells were counted using a Zeiss confocal microscope.
Soft agar growth assay
1×107 cells were infected with control or KLF11 adenovirus allowed to recover 18hrs and mixed with RPMI containing 10% serum and 0.4% low melting point (LMP) agarose. This mixture was placed over hardened RPMI containing 10% BCS and 1% LMP and allowed to harden. Cells were grown for 2 weeks and visible colonies, stained with 0.4% crystal violet and containing greater than 50 cells, were counted.
RT-PCR analysis, Luciferase and Chromatin immunoprecipitation assays
Reverse transcription and luciferase assays were performed as previously described [8, 9, 11]. ChIP assay was performed as previously described [17]. A 200-bp region of the cyclin A2 promoter was amplified by PCR, using specific primers (5′-GCTCACTAGGTGGCTCAGCTTAAA-3′, 5′-TTGACGTCATTCAAGGCGACAGGG-3′).
Cell Cycle FACS Analysis
Adherent cells were harvested, filtered through a 30μm nylon mesh filter (Miltenyi Biotec, Auburn, CA) and fixed for 1h at 4°C with 100% ethanol. Fixed cells were incubated with Propidium Iodide (PI) Staining Solution (Biosure, Grass Valley, CA) and 100 μg/mL RNAse A at room temperature for 40 min. Flow cytometry was performed on a BD LSR II instrument equipped with a Coherent® Sapphire™ 20mW laser (excitation wavelength: 488nm). PI fluorescence was detected by a photomultiplier tube, a 595nm long-pass dichroic mirror and 610±20-nm bandpass emission filter. 110,000–170,000 events were collected using appropriate stop gates set in the PI fluorescence area vs. width projection to exclude cell aggregates. Acquisition settings were verified by adding internal control cells (Chick Erythrocyte Nuclei; Biosure) to an aliquot of each sample. Data files were analyzed with FlowJo software (Treestar, Asland, OR) and Dean-Jett-Fox algorithm.
Electrophoretic mobility shift assays (EMSA)
Gel shift assays were performed essentially as described [16]. Annealed, double-stranded oligonucleotides corresponding to potential KLF binding sites in the human CCNA2 promoter were end labeled with [γ-32P] ATP using T4 polynucleotide kinase (site #1: 5′-GTTTCTCCCTCCTGCCCCGCCCCTGCTCAGTTTCC-3′; site #2: 5′-GCAGGCGTTTTCTCCCGCCCCAGCCAGTTTGTTTC-3′; site #3: 5′-CTAAATCCTACCTCTCCCCGCCCCGCGCAGGCGTTTTC-3′; site #4: 5′-CGGAAGCGTCGGGCCCTAAATCCTAC-3′; and a site #1 mutant probe (5′-GTTTCTCCCTCCTGCCCTTTCCCTGCTCAGTTTCC-3′) as indicated by the manufacturer (Promega Corp., Madison, WI). A glutathione-S-transferase (GST) fusion protein carrying the zinc finger region of KLF11 was utilized for these studies. GST alone (control) and GST fusion protein expression was induced in BL21 cells (Agilent Technologies, Inc) by the addition of 2 mM isopropyl-b-D-thiogalactopyranoside and incubation for 2 h at 37 °C. Cells were lysed and subsequently purified by using glutathione Sepharose 4B affinity chromatography in accordance with the manufacturer’s instructions (GE Healthcare BioSciences Corp., Piscataway, NJ). Subsequently, gel shift assays were performed using 1 μg of purified GST or GST-KLF11 recombinant fusion proteins incubated in a buffer containing 20 mM HEPES (pH 7.5), 50 mM KCl, 5 mM MgCl2, 10 mM ZnCl2, 6% glycerol, 200 mg of bovine serum albumin per ml, and 50 mg of poly(dI-dC)•poly(dI-dC) (Sigma) per ml for 7 min at room temperature. Approximately 0.3 ng of the end-labeled probe was then added to each reaction for an additional 20 min, with or without 125X cold competitor, anti-GST antibody, or anti-mIgG antibody as indicated, and loaded immediately onto a 4% non-denaturing polyacrylamide gel. Samples were run for 2 h at 200 V at room temperature, vacuum-dried, and exposed to HyBlot CL™ autoradiography film (Denville Scientific Inc., Metuchen, NJ).
RESULTS
Systematic Screening Reveals a Role for Distinct SP/KLF Proteins in Blocking KRAS-Mediated Cell Growth
To test the effect SP/KLF in transformation, we initially performed foci formation assays to assess anchorage-dependent cell growth using the NIH-3T3 transformation model, utilizing well-established oncogenes to induce transformation and subsequently, observe any reversal of this effect with each of the 24 SP/KLF family members. NIH-3T3 cells were cotransfected with the potent oncogene, KRASV12, along with a full-length construct of each member of the SP/KLF family of transcription factors or parental vector (control). Interestingly, only SP5, SP8, KLF2, KLF3, KLF4, KLF11, KLF13, KLF14, KLF15 and KLF16 were able to suppress more than 50% of the transformation mediated by the oncogene KRAS (Fig. 1). Similar results were obtained using HRAS (data not shown). Notably, none of the SP/KLF genes induced foci formation alone or increased the transformation mediated by KRAS (Fig. 1). This is congruent with using the NIH3T3 assay where only one oncogene is needed to achieve maximal transformation, which is different than using MEF-mediated assays, which are better for studying oncogenes due to their requirement of two hits to be fully transformed. Therefore, a distinct subset of KLF proteins, namely SP5, SP8, KLF2, KLF3, KLF4, KLF11, KLF13, KLF14, KLF15 and KLF16, are able to significantly suppress K-ras mediated foci formation.
Figure 1. Inhibition of KRAS-mediated transformation is a defined feature of a subgroup of SP/KLF transcription factors.

The effect of SP/KLF family members in transformation was assessed via foci formation assay. NIH 3T3 cells were co-transfected with the indicated SP/KLF proteins and control or constitutively active KRAS. Of the 24 members, only SP5, SP8, KLF2, KLF3, KLF4, KLF11, KLF13, KLF14, KLF15 and KLF16 were able to suppress more than 50% of the KRAS-mediated transformation (p<0.05). HIS- and FLAG-tagged SP/KLF protein expression was confirmed by western blots. KRAS and housekeeping GAPDH expression was determined by RT-PCR. None of the SP/KLF genes induced foci formation alone or increased the transformation mediated by KRAS.
Suppressor-of-KRAS KLF Proteins Inhibit Anchorage-Independent Cell Growth and In Vivo Tumorigenesis
To gain an in depth insight into the molecular mechanisms underlying the suppressive function of this subgroup of SP/KLF proteins, we used a two-tiered experimental strategy. First, we chose one representative member of the subgroup of KLF proteins that suppress KRAS-mediated transformation as a model to search for novel mechanisms that may account for its suppressive function, and, subsequently, investigated whether these mechanisms were utilized by the rest of the subset. For this purpose, we performed these investigations using KLF11, a gene previously identified by our laboratory [16]. To facilitate these studies, we generated adenovirus expressing KLF11 cDNA to infect several KRAS oncogenic mutant pancreatic cancer cell lines (AsPC1, Capan2, CFPAC1, L3.6, MiaPaCa2) to determine the rate of BrdU incorporation at different time points. As shown in Figure 2a, growth in vitro was significantly inhibited in KLF11-infected cells compared with control-infected cells. In addition, we determined the ability of KLF11 to block colony formation in soft agar by these pancreatic tumor cells (Fig. 2b). KLF11 infection of Capan2, L3.6, MiaPaCa2, and PANC1 cell lines resulted in a substantial reduction of colony formation in soft agar compared to the control-infected populations. As an in vivo validation of this effect of KLF11 on tumor growth in vivo, we injected KLF11- or control-infected Capan2 and L3.6 cells subcutaneously into the flanks of irradiated male athymic nude mice and monitored the increase in tumor volume over a six-week period. We found that in vivo tumor formation was significantly delayed in the mice with injected tumor cells expressing exogenous KLF11 as compared to control mice (Fig. 2c–d). We confirmed expression of KLF11 in the KLF11-infected cells by western blotting (Fig. 2c). These data indicate that KLF11 inhibits the transformed phenotype in vitro and in vivo to support its role as a tumor suppressor in KRAS mutant tumors.
Figure 2. KLF11 inhibits proliferation, anchorage-independent growth and tumor formation in vivo in pancreatic cancer cells.

a. KLF11 inhibits cell proliferation in pancreatic cell lines. As a measurement of proliferation, the rate of BrdU incorporation was assessed in AsPC1, Capan2, CFPAC, L3.6 and MiaPaCa2 with a control or KLF11- adenovirus. Cell proliferation was significantly inhibited in KLF11-infected cells compared with control-infected cells (p<0.05). b. KLF11 decreases anchorage-independent growth. KLF11 infection of Capan2, L3.6, MiaPaCa2 and PANC1 cell lines resulted in substantial reduction of colony formation in soft agar compared to control (p<0.05). c. KLF11 inhibits pancreatic tumorigenesis in vivo. KLF11- or control-infected Capan2 and L3.6 cells were injected subcutaneously into the flanks of athymic nude mice and tumor volume was monitored. In vivo tumor formation was significantly reduced in mice injected with tumor cells expressing KLF11 as compared to control mice (p<0.05). A representative example of tumor mass is shown. Bottom panel shows western blot results of lysates from the pool of injected cells to confirm KLF11 expression. d. Graphical representation of the mean tumor volume after 6 weeks is shown, indicating that KLF11 inhibits the transformed phenotype of pancreatic cells in vivo (p<0.05) to support its role as a tumor suppressor in pancreatic cancer.
Suppressor-of-KRAS KLF Proteins Inhibit Cell Proliferation and Induce Apoptosis
Subsequently, we investigated the cellular mechanisms underlying the effect of the additional KRAS suppressor KLF proteins by examining if the suppression of neoplastic transformation by SP/KLFs was, similar to KLF11, due at least in part to a change in cell proliferation. For this purpose, we tested the ability of these proteins to regulate BrdU incorporation at various time points first, in PANC1, which are cancer cells containing endogenous oncogenic mutant KRAS. As shown in Figure 3a, transfection with SP5, SP8, KLF2, KLF3, KLF4, KLF11, KLF13, KLF14, KLF15 and KLF16, the same subset that suppressed KRAS-mediated transformation, resulted in decreased proliferation in PANC1. Subsequently, we repeated this experiment in two additional cell lines, BxPC3 and NIH3T3, both cell lines that contain wild type endogenous KRAS, which were then transfected with the oncogenic variant of KRAS. BXPC3 (Fig. 3b), and NIH3T3 (Fig. 3c) confirm that the same subset, with the exception of KLF13, decrease proliferation of these cells in the presence of oncogenic KRAS, as determined by the reduction in the number of BrdU positive cells. Next, we examined the effect of these SP/KLFs on cell death via morphological criteria of nuclear fragmentation, nuclear margination, cytoplasmic blebbing, and organelle disorganization as visualized by Hoechst DNA staining. Interestingly, only SP5, KLF3, KLF11, KLF13, KLF14 and KLF16 increased the number of apoptotic cells by more than 2-fold in PANC1 cells (Fig. 3d). Thus, together, these data identify a subgroup of SP/KLF transcription factors that suppress oncogenesis and suggest that suppression of KRAS neoplastic transformation is explained, in part, by the ability of these molecules to inhibit cell proliferation and for some of this subset, to also induce apoptosis. Conceptually, these results identify two important cellular mechanisms underlying the suppression of neoplastic transformation by these proteins, namely an increase in apoptosis and concurrent decrease in cellular proliferation. More importantly, these results demonstrate that KLF11 is a reliable suppressor protein to utilize as a robust model in gathering more mechanistic data.
Figure 3. Suppressor-of-KRAS KLF proteins inhibit cell proliferation and induce apoptosis.

a. The same subset of SP/KLF proteins that suppress KRAS-mediated transformation, namely SP5, SP8, KLF2, KLF3, KLF4, KLF11, KLF13, KLF14, KLF15 and KLF16, decrease cell proliferation in PANC1 via BrdU incorporation (p<0.05). Two additional cell lines, BxPC3 and NIH3T3, both cell lines that contain wild type endogenous KRAS, were transfected with the oncogenic variant of KRAS. b and c. BXPC3 (b) cells and NIH 3T3 (c) confirm that this subset, with the exception of KLF13, decreases cell proliferation in the presence of oncogenic KRAS, as determined by the reduction in BrdU positive cells (p<0.05). d. The effect of the entire family of SP/KLF proteins on apoptosis was evaluated by microscopy for morphological criteria of nuclear fragmentation, nuclear margination, cytoplasmic blebbing, and organelle disorganization via Hoechst DNA staining in PANC1 cells. Interestingly, only SP5, KLF3, KLF11, KLF13, KLF14 and KLF16 increased the number of apoptotic cells by more than 2-fold (p<0.05).
Distinct Suppressor-of-KRAS KLF Proteins Share a Common Mechanism of Silencing Cyclin A
Since inhibition of cellular proliferation was common among all members of the SP/KLF family that suppressed KRAS-mediated transformation, we investigated the molecular mechanisms underlying this effect of the suppressor-of-KRAS KLF proteins. Consequently, we performed RT-PCR for genes involved in regulating the cell cycle, including cyclin D1, cyclin A2 and cyclin B1. Notably, messenger RNA levels of cyclin A2 were significantly decreased in three SP/KLF transcription factors with suppressor-of-KRAS growth functions, specifically KLF11, KLF14, and KLF16 (60.8%±15%, 47.0%±7.4%, and 70.2%±0.1% of control levels, respectively) (Fig. 4a). These KLF proteins were also able to repress the promoter activity of cyclin A2 (CCNA2) via luciferase-based reporter assay (KLF11, 39.4%±12.1%; KLF14, 11.4%±3.0%; and KLF16, 33.4%±6.1% compared to control) (Fig. 4b). Again, using KLF11 as a model, we analyzed the cell cycle profile via flow cytometry in KLF11-overexpressing cells, which resulted in a lower percentage of the cell population in G0/G1 with a mean change of 12.025% ±1.4% compared to control with a concordant increase of cells in S phase with a mean difference of 11.26%±2.1% compared to the control (Fig. 4c). There was no change in cells in G2/M between the parental vector (EV) control and KLF11 groups (meanΔ 0.475%±2.95%). These results, along with decreased BrdU incorporation mediated by KLF11 (Fig. 2a and 3a–c), suggest that this increased population of cells in S phase represent an arrest at this stage of the cell cycle. We find that this effect of KLF11 on the CCNA2 promoter is not only functional in cancer cells, as similar results on cell growth via fluorescent LIVE/DEAD® Viability/Cytotoxicity assay (approximately 30% reduction in cell survival compared with control-infected cells, p<0.05) and CCNA2 promoter activity (30.9% ± 18.4% relative to control, p<0.05) were obtained in normal fibroblasts and normal pancreatic ductal cells, respectively (Fig. 4d–e).
Figure 4. KLF11 Represses the Cyclin A Promoter and Causes Cell Cycle Arrest.

a. Of the SP/KLF transcription factors with suppressor-of-KRAS activity, KLF11, KLF14, and KLF16 demonstrate a significant decrease in cyclin A2 expression (60.8%±15%, 47.0%±7.4%, and 70.2%±0.1%, respectively, p<0.05). The results of qRT-PCR are shown relative to control in PANC1 cells. b. This same subset, consisting of KLF11, KLF14, and KLF16, was able to significantly repress cyclin A2 promoter activity as determined by luciferase assay (p<0.05). PANC1 were transfected with a cyclin A2 promoter-luciferase reporter construct and either a control or the indicated SP/KLF expression vector. Luciferase activity was measured and the mean and standard deviation were determined for each experimental condition from at least three independent experiments. c. FACS-based cell cycle analysis demonstrates that KLF11 causes an increase in the percentages of cells in the S phase of the cell cycle (mean increase of 11.26%±2.1%) with a concomitant decrease in G0/G1 (mean decrease of 12.025% ±1.4%) compared to control cells (p<0.05). No change was observed in the percentage of the KLF11-expressing cell population in G2/M compared to control cells (mean Δ 0.475%±2.95%). Graph represents the percentage of cells from the empty vector (control) and KLF11 groups in various phases of the cell cycle from four independent experiments. A representative figure of DNA content measured by FACS analysis is shown on the right. d. KLF11-mediated inhibition of cell growth is operational in normal primary fibroblasts. Freshly isolated wild type MEF were infected with a control empty vector (EV) or KLF11 carrying adenovirus. After 48 hours, KLF11-infected MEF had approximately 30% reduction in cell survival compared with control-infected cells (1.13±0.1 vs. 0.81±0.09, p<0.05). e. KLF11-mediated repression of the CCNA2 promoter is functional in normal pancreatic ductal cells. HPDE6 cells were transfected with the cyclin A2 promoter-luciferase reporter construct and either control or KLF11 expression vector. KLF11 significantly repressed cyclin A2 promoter activity (30.9% ± 18.4% normalized to EV; p<0.05) Luciferase activity was measured and the mean and standard deviation were determined from three independent experiments done in triplicate.
To determine whether the effect of KLF11 on cyclin A2 is due to a direct interaction with its promoter, we performed chromatin immunoprecipitation (ChIP) assay. Indeed, KLF11 was found on the endogenous CCNA2 promoter, while KLF10, a protein that does not regulate the promoter in our experiments, is not detected on the promoter in the same ChIP experiments (Fig. 5a). KLF10, the SP/KLF family member with highest similarity to KLF11, is part of the same TIEG (TGFβ-Inducible Early Gene) subfamily [5] but does not alter KRAS-mediated transformation or proliferation in this study, therefore supporting the specificity of binding to this promoter. Subsequently, we performed genetic rescue experiments in which cyclin A2 was overexpressed from a heterologous promoter along with KLF11 in PANC1 cells. Under these circumstances, KLF11 arrests cell growth (22% ± 12.9% normalized to EV), however, the expression of exogenous Cyclin A2 rescues the KLF11 effect (88% ± 11.3% normalized to EV), indicating that the cell cycle regulatory function of this protein depends on its repression of the CCNA2 promoter (Fig. 5b). KLF11 contains three distinct repressor domains, R1, R2, and R3, respectively located in the N-terminal domain of the protein [8, 11]. Consequently, using deletion mutants, we investigated which of these domains is involved in CCNA2 promoter repression. KLF11 transcriptional repression depends on the specific interaction of corepressor molecules with each of these domains [9, 10]. Specifically, the interaction of the R1 domain with the Sin3A-HDAC corepressor complex is required for the transcriptional regulatory activity of certain genes. Regarding how these gene regulatory pathways impact on cell biology, our laboratory has previously described that many KLF11 biological functions require its gene silencing activity through this corepressor complex [9, 18]. Interestingly, however, in the context of cyclin A2 expression, as studied here, further characterization of the mechanism revealed that KLF11 requires the intact full length of the protein in order to repress this promoter activity in cancer cells (Fig. 5c). In fact, none of the deletion mutants had any statistically significant difference with the control values. Thus, this result suggests that KLF11 uses a novel mechanism to achieve this function, which involves all of its transcriptional repressor domains. Since other co-repressors of KLF11 function currently remain unknown, the Sin3-independent regulation of this promoter by KLF11 raises an interesting area of future investigation, focused on how distinct chromatin-mediated events triggered by this KLF protein results in the repression of the cyclin A2 promoter and suppression of KRAS-mediated transformation.
Figure 5. KLF11 inhibits cell proliferation via direct binding and repression of the CCNA2 promoter in a Sin3/HDAC independent manner.
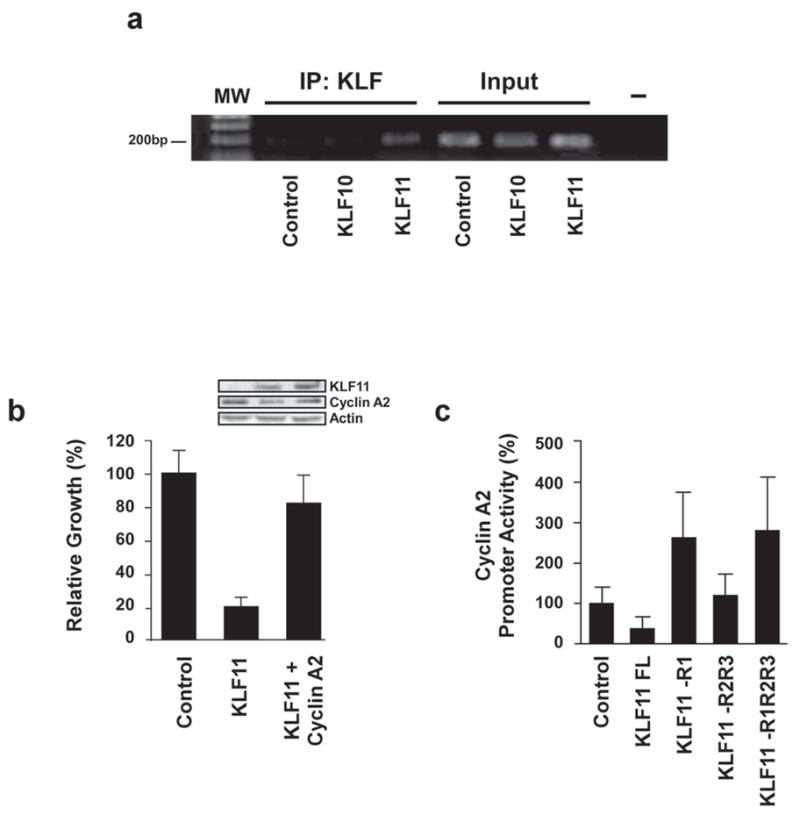
a. KLF11 occupies the cyclin A2 promoter in vivo. PANC1 cells were infected with His-tagged KLF10, KLF11 or empty vector control and chromatin immunoprecipitation was performed. Utilizing an antibody against the His-tag to immunoprecipitate protein/DNA complexes demonstrates that only the KLF11-infected cells amplify a band to indicate its presence on the CCNA2 promoter, whereas KLF10 and empty vector serve as negative controls. KLF10, the SP/KLF family member with highest similarity to KLF11, is part of the same TIEG subfamily but does not alter KRAS-mediated transformation or proliferation. Positive amplification of PCR products is shown in the input DNA lanes demonstrating that this region of the CCNA2 promoter is present in all samples before immunoprecipitation. b. KLF11-mediated regulation of the CCNA2 promoter plays a direct role in cell growth arrest. PANC1 cells were transfected with control parental vector, KLF11 alone or the combination of KLF11and cyclin A2 expression construct. Forty-eight hours post-transfection, cell growth was measured by MTS assay, which demonstrates that the KLF11-mediated inhibition of cell proliferation is rescued by cyclin A2 overexpression (22% ± 12.9% normalized to EV for KLF11 alone versus 88% ± 11.3% normalized to EV for KLF11 + cyclin A2). Control western blot shows expression of exogenous KLF11 and cyclin A proteins. c. KLF11 represses the cyclin A2 promoter in a Sin3/HDAC-independent manner. PANC1 cells were transfected with various KLF11 deletion mutants lacking the R1 (Sin3-interacting domain), R2 and R3 domains along with cyclin A2 promoter-luciferase reporter construct. Luciferase activity demonstrates that full length KLF11 is required for CCNA2 promoter repression, indicating a Sin3-independent mechanism. Statistical analysis determined that only full length KLF11 had any significant difference with the control (35.2% ± 13.7%, p<0.05). The mean and standard deviation are shown from at least three independent experiments.
To specifically characterize the KLF11-mediated regulation of cyclin A2, we first analyzed the CCNA2 promoter for KLF binding sites via bioinformatics using pairwise comparison with KLF consensus sites defined according to rules derived from crystal structures [16, 19], as well as empirical DNA binding matrices developed by our laboratory [18, 20] to find 4 potential sites (Fig. 6a). To determine which of these candidate sites KLF11 indeed binds, we performed electromobility shift assay (EMSA). Interestingly, the specific binding of recombinant KLF11 was observed only for site #1, corresponding to −87 through −92 of the CCNA2 promoter (Fig. 6b, lane 1 and 6c, lane 4). The binding of recombinant KLF11 to this sequence was found to be specific by super-shift assays (Fig. 6c, lanes 5–6). Furthermore, competition was performed with cold wild type but not mutant probe, confirming high specificity in binding (Fig. 6c, lanes 7 and a). Binding, however, was not observed with a mutant probe to this site (Fig. 6c, lanes b–d, nucleotides corresponding to −89 to −91 CGC>TTT). We also performed site-directed mutagenesis on the CCNA2 promoter to change the corresponding KLF11 binding site from CCCCGCCCC to CCCTTTCCC. Indeed, upon mutagenesis of this site, KLF11 was no longer able to repress promoter activity and was similar to the EV control (Fig. 6d; 41.4% ± 3.7% normalized to EV for WT promoter vs. 116% ± 8.8% normalized to EV for the −89 to −91 CGC>TTT mutant promoter), confirming that this is indeed the site of KLF11 binding to the CCNA2 promoter and this site is necessary for KLF11-mediated repression of this promoter. Taken together, these data indicate that KLF11 can inhibit tumor cell proliferation and therefore, transformation, at least in part, through the modulation of cyclin A2, and that this mechanism requires full length KLF11 protein, which directly binds to −87 through −92 of the CCNA2 promoter.
Figure 6. KLF11-mediated repression requires an SP/KLF site at −87 through −92 of the CCNA2 promoter.

a. The cyclin A2 promoter has four putative KLF binding sites. The diagram represents a schematic of the human CCNA2 proximal promoter (−85 to −180 relative to TSS) containing four GC-rich KLF/Sp-like elements (underlined, labeled #1–4 relative to TSS). b. KLF11 binds to putative KLF site #1 of the CCNA2 promoter in vitro. Gel shift assays with probes containing each of the four putative binding sites demonstrate binding of KLF11 to site #1 (lane 1), but not sites #2–4 (lanes 3, 5, 7). EMSA was performed using recombinant KLF11 GST-fusion protein (lanes 1, 3, 5, 7) or GST protein alone (lanes 2, 4, 6, 8) with radiolabeled oligonucleotides for each of the four putative KLF binding sites, as indicated. Each of the probes were also loaded without protein (Probe alone lanes, a–d). A KLF11/CCNA2 oligo complex was only detected with site #1, as indicated by the arrow. c. Full characterization of KLF11 binding to site #1 demonstrates binding specificity in vitro. EMSA was performed with the wild-type human CCNA2 KLF site #1 (WT site #1; lanes 1, 3–8) with GST protein (lane 3), recombinant KLF11 (KLF11; lanes 2, 4–8) or probe alone (lane 1). The specific complexes that form between KLF11 and the probe, as well as the free probe, are indicated by the arrows on the left. Although the anti-GST antibody shifted the KLF11/CCNA2 WT site #1 complex (lane 5), the same amount of an anti-mIgG antibody did not (lane 6), indicating specificity. Furthermore, the addition of an excess of unlabeled WT site #1 probe robustly competed for the binding (125X; lane 7), whereas the addition of the mutant probe did not (125X; lane a). Notably, KLF11 did not shift the mutant probe corresponding to a −89 to −91 CGC>TTT substitution (mut site #1; lane d). The mutant probe was utilized for gel shift assays (lanes b–d) with GST protein (lane c), recombinant KLF11 (lane d) or probe alone (lane b). Distinct lanes of the same EMSA gel are separated by a line to indicate irrelevant lanes removed for presentation purposes. Non-specific radioactive background from lanes 1–3 was removed using Image J 1.44 software (NIH). d. KLF11 requires an intact site #1 to repress the CCNA2 promoter. WT and site #1 mutant CCNA2 promoter reporter constructs were utilized in luciferase reporter assays. Activity of the WT promoter was significantly repressed by KLF11 (41.4% ± 3.7% normalized to EV; *p<0.05), however the site #1 mutant promoter (−89 to −91 CGC>TTT), which is unable to bind KLF11 via EMSA, loses KLF11-mediated repression (116% ± 8.8% normalized to EV; #p<0.05). Graphical depiction of the results is the mean and standard error from three independent experiments performed in triplicate.
DISCUSSION
KLF proteins are eliciting significant attention because of their mis-expression in different cancers, as well as their role in cell proliferation, apoptosis, senescence, migration, adhesion, and angiogenesis [2, 21]. Many of these tumors and the cell processes mediated by KLF proteins share the activation of the KRAS pathway in common either by signaling or mutations. However, the role of KLF proteins in KRAS-mediated transformation has remained poorly understood. The current study was designed to fill this existing gap in knowledge and outlines novel mechanistic information on how KLF-mediated transcriptional pathways can regulate cancer-associated processes. Using well-established assays, we have performed the first family-wide functional screening for KLF proteins, identifying a subset that inhibits the ability of KRAS to mediate neoplastic transformation. Our functional screening yielded SP5, SP8, KLF2, KLF3, KLF11, KLF13, KLF14, KLF15 and KLF16 as novel SP/KLF molecules with growth suppressor properties in the context of the an oncogenic KRAS pathway. This SP/KLF subgroup has suppressive activity on KRAS transformation similar to our positive control, KLF4 [22]. KLF5, which exhibits well-characterized opposite effects to KLF4 [23], did not suppress K-RAS transformation thereby serving as an internal negative control for the assay. Thus, these experiments reveal that mechanistically at the cellular level, the suppression activities of these KLF proteins shared at least two functional mechanisms (Fig. 3), namely decreased cell proliferation and enhanced apoptosis. Collectively, these experiments identify a subset of KLF proteins that antagonize KRAS-mediated transformation and outline two cellular mechanisms that primarily account for this effect.
Insights into molecular mechanisms shared by suppressor KLF genes was derived from a more focused functional characterization of KLF11, chosen as a model for this group of proteins. We selected this gene for further study because of its growing medical relevance, due to recent reports describing its methylation-dependent inactivation and downregulation in several malignancies including leukemia, myeloproliferative disorders, esophageal adenocarcinoma, pancreatic cancer, germ cell tumors, as well as head and neck cancer, supporting its candidacy as an actual tumor suppressor in humans [17, 24–28]. Congruent with this idea, our investigations at the cellular level show that KLF11 not only inhibits the KRAS-mediated transformed phenotype in foci formation and agar colony formation in vitro but also the in vivo growth of KRAS mutant xenografts (Fig. 2). At the molecular level, we find that KLF11 and two other KLF suppressor proteins, KLF14 and KLF16, share the ability to downregulate cyclin A2 at the transcriptional level. This downregulation of cyclin A2 is congruent with the existence of multiple potential cis-regulatory SP/KLF sites in the promoter of this gene as defined by bioinformatics and based upon DNA binding matrices with stringent theoretical and empirical support [18, 20]. As demonstrated for KLF11, downregulation of cyclin A2 occurs via binding of this SP/KLF transcription factor to the CCNA2 promoter to silence its expression. Similar to what happens with other tumor suppressor proteins, downregulation of cyclin A2 triggers cell cycle arrest in S phase [29]. A rescue experiment, performed by the concomitant overexpression of cyclin A2, abolishes this effect, further supporting the veracity of this phenomenon (Fig. 5B). Therefore, together with our cell biological data, this molecular information demonstrates that some of these KLF proteins have the ability to antagonize KRAS-mediated transformation, at least in part, by inhibiting cell growth at S phase via a shared biochemical mechanism, namely the downregulation of cyclin A.
Careful molecular dissection of CCNA2 promoter repression revealed that the full length KLF11 protein was required for its repressive effect and specific binding of KLF11 occurs at site #1, corresponding to −87 through −92 of the CCNA2 promoter (Fig. 6). In particular, the requirement for the full length KLF11 protein, unlike previously characterized gene targets for KLF11-mediated repression [17, 27], indicates that cyclin A2 repression is Sin3a/HDAC-independent, a complex that interacts with a defined 20 amino acid peptide located within the R1/SID region of KLF11. Notably, since other co-repressors of KLF11 function currently remain unknown, the Sin3-independent regulation of this promoter by KLF11 suggests a yet-undiscovered mechanism of repression among the three KLF proteins observed to have a significant effect on cyclin A2 expression, namely KLF11, KLF14 and KLF16. Rationally, the best shared characteristic of these proteins, outside of Sin3 binding, is that all possess a similar SP1-like DNA binding domain. Consequently, the ability of KLF proteins to compete with each other through their zinc finger domains for binding to the same promoter target may explain some measure of redundancy currently assumed in this field of research [3, 5]. In this theoretical context, our results suggest that different KLF proteins might downregulate cyclin A2 in different cells or their specificity, in this regard, might come from their relative abundance in distinct cell types. Moreover, since many KLF proteins are regulated at the transcriptional and posttranslational level, some of these proteins may not be functional for regulating cyclin A2 in particular cells where these mechanisms are operational in a context dependent manner. Nevertheless, the biochemical ability of these proteins to silence cyclin A2 expression, as described here, should fuel future studies that will define the cell-specific and the biological context, based on expression and signaling, in which each suppressor KLF protein performs this function.
In conclusion, the current study identifies a subgroup of SP/KLF proteins that blocks KRAS-mediated neoplastic transformation, thus significantly expanding our understanding of the repertoire of proteins that can antagonize the function of the KRAS oncogene. Regarding SP/KLF proteins, this study identifies not only new modulators of the neoplastic phenotype, but also provides novel mechanisms underlying the function of these proteins. At the cellular level, there appear to be a network of SP/KLF proteins with the ability to negatively regulate growth by inhibition of cell proliferation and the induction of apoptosis. At the molecular level, the results described here advance the field of SP/KLF proteins and their role in the control of cancer-associated processes, since a regulatory role for any SP/KLF on cyclin A2 expression has not previously been determined. Therefore, collectively, both the phenomenological and mechanistic discoveries of this study further our understanding of the role of KLF proteins in cell cycle control, tumor suppression and KRAS antagonism.
Acknowledgments
FUNDING
This work was supported by National Institutes of Health Grants DK 52913 (to R. U.) and Mayo Clinic Pancreatic SPORE P50 CA102701, NCI CA136526, Mayo Clinic Cancer Center, Division of Oncology Research (to M. E. F.-Z.) and DK 76845 (to N.S.B.), as well as theMayo Clinic Center for Cell Signaling in Gastroenterology NIDDK P30DK084567.
Abbreviations
- BrdU
5-bromo-2-deoxyuridine
- ChIP
chromatin immunoprecipitation
- EV
empty vector
- FACS
fluorescence-activated cell sorting
- HDAC
histone deacetylase
- KLF
Kruppel-Like Factor
- KRAS
V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
- MEF
mouse embryonic fibroblast
- PI
propidium iodide
- RT-PCR
Reverse transcription polymerase chain reaction
- TIEG
TGFβ-Inducible Early Gene
Footnotes
AUTHOR CONTRIBUTION
Martin E. Fernandez-Zapico, Gwen A. Lomberk and Raul Urrutia designed all of the experimental work. Martin E. Fernandez-Zapico, Gwen A. Lomberk, Shoichiro Tsuji, Cathrine J. DeMars, Michael R. Bardsley, Yi-Hui Lin, Luciana L. Almada, Jing-Jing Han, and Navtej S. Buttar participated in the experiments. Martin E. Fernandez-Zapico, Gwen A. Lomberk, and Raul Urrutia analyzed and interpreted the results, as well as directed and supervised the research. Michael R. Bardsley and Tamas Ordog performed and interpreted results of cell cycle analysis. The paper was written by Gwen A. Lomberk and Raul Urrutia and revised before submission by Martin E. Fernandez-Zapico, Navtej S. Buttar, Debabrata Mukhopadhyay and Tamas Ordog.
The authors declare no financial interests.
References
- 1.Zhao C, Meng A. Sp1-like transcription factors are regulators of embryonic development in vertebrates. Dev Growth Differ. 2005;47:201–211. doi: 10.1111/j.1440-169X.2005.00797.x. [DOI] [PubMed] [Google Scholar]
- 2.Black A, Black J, Azizkhan-Clifford J. Sp1 and krüppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol. 2001;188:143–160. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- 3.Kaczynski J, Cook T, Urrutia R. Sp1- and Kruppel-like transcription factors. Genome Biol. 2003;4:206. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Safe S, Abdelrahim M. Sp transcription factor family and its role in cancer. Eur J Cancer (Oxford, England : 1990) 2005;41:2438–2448. doi: 10.1016/j.ejca.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Lomberk G, Urrutia R. The family feud: turning off Sp1 by Sp1-like KLF proteins. Biochem J. 2005;392:1–11. doi: 10.1042/BJ20051234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsumura T, Suzuki T, Aizawa K, Munemasa Y, Muto S, Horikoshi M, Nagai R. The Deacetylase HDAC1 Negatively Regulates the Cardiovascular Transcription Factor Kruppel-like Factor 5 through Direct Interaction. J Biol Chem. 2005;280:12123–12129. doi: 10.1074/jbc.M410578200. [DOI] [PubMed] [Google Scholar]
- 7.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 8.Cook T, Gebelein B, Belal M, Mesa K, Urrutia R. Three Conserved Transcriptional Repressor Domains Are a Defining Feature of the TIEG Subfamily of Sp1-like Zinc Finger Proteins. J Biol Chem. 1999;274:29500–29504. doi: 10.1074/jbc.274.41.29500. [DOI] [PubMed] [Google Scholar]
- 9.Ellenrieder V, Buck A, Harth A, Jungert K, Buchholz M, Adler G, Urrutia R, Gress T. KLF11 mediates a critical mechanism in TGF-β signaling that is inactivated by Erk-MAPK in pancreatic cancer cells. Gastroenterology. 2004;127:607–620. doi: 10.1053/j.gastro.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 10.Ellenrieder V, Zhang J, Kaczynski J, Urrutia R. Signaling disrupts mSin3A binding to the Mad1-like Sin3-interacting domain of TIEG2, an Sp1-like repressor. EMBO J. 2002;21:2451–2460. doi: 10.1093/emboj/21.10.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Moncrieffe M, Kaczynski J, Ellenrieder V, Prendergast F, Urrutia R. A Conserved α-Helical Motif Mediates the Interaction of Sp1-Like Transcriptional Repressors with the Corepressor mSin3A. Mol Cell Biol. 2001;21:5041–5049. doi: 10.1128/MCB.21.15.5041-5049.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huser M, Luckett J, Chiloeches A, Mercer K, Iwobi M, Giblett S, Sun X-M, Brown J, Marais R, Pritchard C. MEK kinase activity is not necessary for Raf-1 function. EMBO J. 2001;20:1940–1951. doi: 10.1093/emboj/20.8.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furukawa T, Duguid WP, Rosenberg L, Viallet J, Galloway DA, Tsao MS. Long-term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am J Pathol. 1996;148:1763–1770. [PMC free article] [PubMed] [Google Scholar]
- 14.Lee MD, She Y, Soskis MJ, Borella CP, Gardner JR, Hayes PA, Dy BM, Heaney ML, Philips MR, Bornmann WG, Sirotnak FM, Scheinberg DA. Human mitochondrial peptide deformylase, a new anticancer target of actinonin-based antibiotics. J Clin Invest. 2004;114:1107–1116. doi: 10.1172/JCI22269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gebelein B, Fernandez-Zapico M, Imoto M, Urrutia R. KRAB-independent suppression of neoplastic cell growth by the novel zinc finger transcription factor KS1. J Clin Invest. 1998;102:1911–1919. doi: 10.1172/JCI1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cook T, Gebelein B, Mesa K, Mladek A, Urrutia R. Molecular Cloning and Characterization of TIEG2 Reveals a New Subfamily of Transforming Growth Factor-β-inducible Sp1-like Zinc Finger-encoding Genes Involved in the Regulation of Cell Growth. J Biol Chem. 1998;273:25929–25936. doi: 10.1074/jbc.273.40.25929. [DOI] [PubMed] [Google Scholar]
- 17.Fernandez-Zapico ME, Mladek A, Ellenrieder V, Folch-Puy E, Miller L, Urrutia R. An mSin3A interaction domain links the transcriptional activity of KLF11 with its role in growth regulation. EMBO J. 2003;22:4748–4758. doi: 10.1093/emboj/cdg470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neve B, Fernandez-Zapico M, Ashkenazi-Katalan V, Dina C, Hamid Y, Joly E, Vaillant E, Benmezroua Y, Durand E, Bakaher N, Delannoy V, Vaxillaire M, Cook T, Dallinga-Thie G, Jansen H, Charles M, Clement K, Galan P, Hercberg S, Helbecque N, Charpentier G, Prentki M, Hansen T, Pedersen O, Urrutia R, Melloul D, Froguel P. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci U S A. 2005;102:4807–4812. doi: 10.1073/pnas.0409177102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gebelein B, Urrutia R. Sequence-Specific Transcriptional Repression by KS1, a Multiple-Zinc-Finger-Kruppel-Associated Box Protein. Mol Cell Biol. 2001;21:928–939. doi: 10.1128/MCB.21.3.928-939.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez-Zapico ME, van Velkinburgh JC, Gutierrez-Aguilar R, Neve B, Froguel P, Urrutia R, Stein R. MODY7 gene, KLF11, is a novel p300-dependent regulator of Pdx-1 (MODY4) transcription in pancreatic islet beta cells. J Biol Chem. 2009;284:36482–36490. doi: 10.1074/jbc.M109.028852. Epub 32009 Oct 36420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bureau C, Hanoun N, Torrisani J, Vinel JP, Buscail L, Cordelier P. Expression and Function of Kruppel Like-Factors (KLF) in Carcinogenesis. Curr Genomics. 2009;10:353–360. doi: 10.2174/138920209788921010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei D, Kanai M, Jia Z, Le X, Xie K. Kruppel-like Factor 4 Induces p27Kip1 Expression in and Suppresses the Growth and Metastasis of Human Pancreatic Cancer Cells. Cancer Res. 2008;68:4631–4639. doi: 10.1158/0008-5472.CAN-07-5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghaleb AM, Nandan MO, Chanchevalap S, Dalton WB, Hisamuddin IM, Yang VW. Kruppel-like factors 4 and 5: the yin and yang regulators of cellular proliferation. Cell Res. 2005;15:92–96. doi: 10.1038/sj.cr.7290271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Potapova A, Hasemeier B, Römermann D, Metzig K, Göhring G, Schlegelberger B, Länger F, Kreipe H, Lehmann U. Epigenetic inactivation of tumour suppressor gene KLF11 in myelodysplastic syndromes*. Eur J Haematol. 2010;84:298–303. doi: 10.1111/j.1600-0609.2009.01389.x. [DOI] [PubMed] [Google Scholar]
- 25.Wermann H, Stoop H, Gillis AJ, Honecker F, van Gurp RJ, Ammerpohl O, Richter J, Oosterhuis JW, Bokemeyer C, Looijenga LH. Global DNA methylation in fetal human germ cells and germ cell tumours: association with differentiation and cisplatin resistance. J Pathol. 2010;221:433–442. doi: 10.1002/path.2725. [DOI] [PubMed] [Google Scholar]
- 26.Gebhard C, Schwarzfischer L, Pham T-H, Schilling E, Klug M, Andreesen R, Rehli M. Genome-Wide Profiling of CpG Methylation Identifies Novel Targets of Aberrant Hypermethylation in Myeloid Leukemia. Cancer Res. 2006;66:6118–6128. doi: 10.1158/0008-5472.CAN-06-0376. [DOI] [PubMed] [Google Scholar]
- 27.Buttar NS, DeMars CJ, Lomberk G, Rizvi S, Bonilla-Velez J, Achra S, Rashtak S, Wang KK, Fernandez-Zapico ME, Urrutia R. Distinct Role of Kruppel-like Factor 11 in the Regulation of Prostaglandin E2 Biosynthesis. J Biol Chem. 2010;285:11433–11444. doi: 10.1074/jbc.M109.077065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belbin TJ, Singh B, Barber I, Socci N, Wenig B, Smith R, Prystowsky MB, Childs G. Molecular classification of head and neck squamous cell carcinoma using cDNA microarrays. Cancer Res. 2002;62:1184–1190. [PubMed] [Google Scholar]
- 29.Blanchard J-M. Cyclin A2 transcriptional regulation: modulation of cell cycle control at the G1/S transition by peripheral cues. Biochem Pharmacol. 2000;60:1179–1184. doi: 10.1016/s0006-2952(00)00384-1. [DOI] [PubMed] [Google Scholar]