

Abstract
Cancer pain is generally treated with pharmacological measures, relying on using opioids alone or in combination with adjuvant analgesics. Weak opioids are used for mild-to-moderate pain as monotherapy or in a combination with nonopioids. For patients with moderate-to-severe pain, strong opioids are recommended as initial therapy rather than beginning treatment with weak opioids. Adjunctive therapy plays an important role in the treatment of cancer pain not fully responsive to opioids administered alone (ie, neuropathic, bone, and visceral colicky pain). Supportive drugs should be used wisely to prevent and treat opioids’ adverse effects. Understanding the pharmacokinetics, pharmacodynamics, interactions, and cautions with commonly used opioids can help determine appropriate opioid selection for individual cancer patients.
Keywords: Cancer pain, World Health Organization, Analgesic ladder, Tramadol, Dihydrocodeine, CYP2D6, Codeine, Morphine, Fentanyl, Oxycodone, Buprenorphine, Hydromorphone, Methadone, Tapentadol, Interactions, Opioid, Pharmacokinetic, Pharmacodynamic
Introduction
Cancer pain treatment is based on the analgesic ladder, established in 1986 by the World Health Organization (WHO; see Fig. 1) [1]. Cancer pain–management guidelines in Europe are based on recommendations by the European Association for Palliative Care (EAPC). Morphine use is recommended by the Expert Working Group of the EAPC at the third step of the WHO analgesic ladder, which comprises additional opioids (ie, oxycodone, fentanyl, buprenorphine, methadone, and hydromorphone) for the treatment of moderate-to-severe pain intensity [2]. The use of an analgesic ladder should be individualized with an appropriate application of supportive drugs (laxatives and antiemetics) for the prevention and treatment of opioid adverse effects [3] and nonpharmacological measures, such as radiotherapy and invasive procedures (nerve blockades and neurolytic blocks) [4].
Fig. 1.
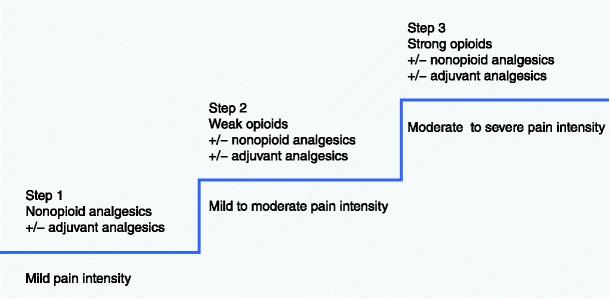
World Health Organization three-step analgesic ladder
Each step of the WHO analgesic ladder (ie, nonopioids, weak opioids [analgesics for mild-to-moderate pain], and strong opioids [opioids for moderate-to-severe pain intensity]) may be accompanied with adjuvant analgesics (coanalgesics), which can enhance opioid analgesia (Table 1). In patients with bone pain, opioids may be combined with NSAIDs, glucocorticoids, and bisphosphonates along with local or systemic radiotherapy [5]. In patients with very severe neuropathic pain, a combination of opioids and N-methyl D-aspartate (NMDA)–receptor antagonists (eg, ketamine) is recommended [6]. Opioid analgesics should be supplemented with spasmolytics in patients with visceral colicky pain, especially in the course of bowel obstruction [7].
Table 1.
Common adjuvant analgesics used in different pain types
| Bone pain |
| NSAIDs, paracetamol |
| Glucocorticoids (dexamethasone) |
| Bisphosphonates (pamidronate, zoledronate) |
| Local radiotherapy |
| Radioisotopes (strontium, samarium) |
| Neuropathic pain |
| Antidepressants (amitryptyline, nortriptyline, venlafaxine) |
| Anticonvulsants (gabapentin, pregabalin, carbamazepine) |
| Local anesthetics (lignocaine, bupivacaine) |
| NMDA-receptor antagonists (ketamine) |
| Visceral colicky pain: |
| Spasmolytics (hyoscine butylbromide, hyoscine hydrobromide, glycopyrrolate) |
NMDA N-methyl-d-aspartate; NSAIDS Nonsteroidal anti-inflammatory drugs
Opioids for Mild-to-Moderate Pain (Weak Opioids)
Tramadol
Tramadol displays opioid properties and acts on neurotransmission of noradrenalin and serotonin. Both enantiomers act synergistically and improve analgesia without increasing adverse effects. Tramadol is metabolized in the liver and excreted by the kidneys. The main metabolite is O-desmethyltramadol (M1), which displays analgesic activity with a higher affinity to μ-opioid receptors than the parent compound; (+)-M1 has 300 to 400 times greater affinity to μ-opioid receptors than tramadol and (−)-M1 mainly inhibits noradrenalin reuptake. Apart from O,N-didesmethyltramadol (M5, which has weak analgesic activity) and M1, other metabolites are inactive [8]. The elimination half-life of tramadol is 5 to 6 h and that of M1 is 8 h. During oral administration, 90% of tramadol is excreted by the kidneys and 10% in feces. Patients with renal impairment show a decreased excretion of tramadol and M1. In patients with advanced cirrhosis, there is a decrease in tramadol metabolism with decrease of hepatic clearance and increase in blood serum levels. In these patients, elimination half-life is increased 2.5-fold. The starting dose of immediate-release (IR) tramadol is 25 to 50 mg every 4 to 6 h and that of controlled-release (CR) tablets or capsules is 50 to 100 mg twice daily; the daily dose should not exceed 400 mg [9].
Patients devoid of cytochrome P450 2D6 (CYP2D6) activity (poor metabolizers) need a tramadol dose higher by 30% than those with normal CYP2D6 activity (extensive metabolizers) [10]. Tramadol analgesia depends on CYP2D6 genotype, with less analgesia in poor metabolizers being associated with lack of (+)-M1 formation [11]. Genotyping is helpful in patients with duplication of CYP2D6 gene (ultrarapid metabolizers [UM]) who are at greater risk to develop tramadol adverse effects [12•]. Tramadol metabolism through CYP2D6 may cause interactions with drugs inhibiting this enzyme (eg, cimetidine and ranitidine).
Serotonin syndrome has been reported in patients taking selective serotonin reuptake inhibitors (SSRIs) in conjunction with tramadol or opioids (see Table 2) [13]. SSRIs (eg, fluoxetine, paroxetine, and, to less extent, sertraline) used in conjunction with tramadol may cause serotonin syndrome because SSRIs inhibit tramadol metabolism and increase serotonin level; generally, they should not be coadministered with tramadol. Serotonin syndrome may appear with monoamine oxidase (MAO) inhibitors, olanzapine, risperidone, and venlafaxine. However, mianserin and mirtazapine do not influence serotonin levels and do not inhibit CYP2D6, but they are substrates of this enzyme [14].
Table 2.
Symptoms of serotonin syndrome
| Agitation |
| Restlessness |
| Headache |
| Diarrhea |
| Confusion |
| Increased heart rate and blood pressure |
| Muscle twitching |
| Shivering |
| Fever |
| Seizure |
| Loss of consciousness |
The inhibition of tramadol metabolism may attenuate analgesia due to (+)-M1 opioid analgesic activity. For example, coadministration of ondansetron (a selective 5- hydroxytryptamine receptor antagonist) blocks spinal 5-HT3 receptors and competitively inhibits CYP2D6. Tramadol analgesia also may be impaired by coadministration of carbamazepine, which accelerates tramadol and M1 metabolism. Concomitant administration of tricyclic antidepressants increases the risk of seizures. Tramadol should be avoided in patients with history of epilepsy. In rats and mice, concomitant administration of tramadol and β-blocker and the 5-HT1A/1B antagonist pindolol enhances analgesia [15].
Respiratory depression is rare in the chronic use of tramadol. When it does occur, respiratory depression is connected with the opioid mode of tramadol action, so naloxone should be administered. For example, respiratory depression was reported in a cancer patient with renal impairment (creatinine clearance 30 mL/min) and with UM genotype after renal carcinoma resection [12•]. As respiratory symptoms appeared more than 10 h after the first tramadol dose, the accumulation of M1 was the cause. The patient recovered after intravenous (IV) naloxone bolus administration (0.4 mg). This case highlights that tramadol should not be prescribed in patients with UM genotype and renal impairment [12•].
Dihydrocodeine
Dihydrocodeine (DHC) is a semisynthetic analogue of codeine. Apart from analgesic and antitussive activity, DHC also is used in the treatment of opioid addiction. After subcutaneous (SC) administration of DHC, 30 mg, analgesia is similar to that induced by 10 mg of morphine. After parenteral administration, DHC is twice as potent as codeine. Bioavailability of DHC after oral administration is 20%, which indicates that its analgesia after oral administration is slightly stronger than that of codeine (bioavailability after oral administration equals 30%–40%). After oral administration of DHC, the maximal serum concentration appears after 1.7 h, plasma half-life varies from 3.5 to 5.5 h, and analgesia lasts 4 h. Ammon et al. [16] assessed DHC pharmacokinetics in 12 extensive metabolizers of CYP2D6. They received a single oral DHC dose of 60 mg, then after 60 h, they were treated for 3 days with 60 mg dosed twice daily; for the next 3 days with 90 mg twice daily; and for 3 subsequent days with 120 mg twice daily. In the 60 to 120 mg DHC dose range, pharmacokinetics of DHC and dihydromorphine (DHM) displayed linear characteristics; area under the curve (AUC), maximum serum concentration (Cmax), and minimum serum concentration at steady state (Cssmin) for both compounds increased depending on the drug dose [17, 18]. Even though DHM displays higher affinity (about 100-fold) to the μ-opioid receptors and exhibits higher analgesic activity in comparison to the parent compound, the role of DHM and its glucuronides in DHC analgesia has not been unequivocally established. The starting dose of IR DHC is usually 30 mg every 4 to 6 h, and that of CR tablets is 60 mg twice daily [17].
Renal clearance and the clearance to DHC metabolites, glucuronidation, and O-demethylation to DHC-6-glucuronide (DHC-6-G) and DHM, respectively, are not dose dependent, which indicates that metabolism and excretion of DHC and its metabolites are not dose dependent. Moreover, the ratio of DHC to DHM for AUC does not change depending on the dose, which suggests a lack of saturation effect of the O-demethylation process of DHC to DHM depending on CYP2D6 in patients normally metabolizing the substrates of this enzyme. Pharmacokinetic parameters were similar after single and multiple doses of 60 mg of DHC [16]. Single-dose and multiple-dose pharmacokinetics of IR and CR DHC formulations provide support for a twice-daily dosage schedule of CR DHC. DHC is metabolized in the liver to its main metabolites: DHM, DHC-6-G, and nordihydrocodeine (NORDHC). NORDHC is further glucuronidated to NORDHC-6-glucuronide and O-demethylated to nordihydromorphine (NDHM). DHM undergoes glucuronidation to DHM-3-glucuronide (DHM-3-G) and DHM-6-glucuronide (DHM-6-G) and N-demethylation to NDHM. It may be concluded that DHC undergoes the first pass effect after oral administration, which is connected with the formation of significantly higher amount of metabolites after oral than after parenteral administration [18]. Studies performed to date [19, 20] indicate that DHC analgesia is independent of CYP2D6 activity [21].
Codeine
Codeine is a methylated morphine derivative that is found naturally, along with morphine, in the poppy seed. Codeine displays analgesic and antitussive activity. Codeine is available as IR and CR formulations but also in the form of paracetamol-combined preparations. IR codeine is administered every 4 to 6 h in chronic pain with a starting single dose of about 30 mg. The daily doses of DHC and codeine usually do not exceed 240 mg and 300 mg, respectively; when these analgesics are ineffective, opioids for moderate-to-severe pain (strong opioids) are introduced.
Codeine is metabolized in the liver and its bioavailability is 30% to 40% after oral administration. After oral administration of codeine, maximal plasma concentration is attained within 1 to 2 h with plasma half-life of 2.5 to 3.5 h and analgesia maintained for 4 to 6 h (IR formulations). Codeine is partially metabolized to morphine and its metabolites and to codeine metabolites norcodeine (NORC) and codeine-6-glucuronide (C-6-G) [22]. The analgesic effect of codeine is about equal to 1/10th of morphine analgesia. Polymorphism of CYP2D6 is responsible for the formation of morphine, and its metabolites may affect codeine analgesia. Other codeine metabolites, C-6-G predominantly, also display analgesic activity and contribute to codeine analgesia [23]. In healthy volunteers, codeine is metabolized to C-6-G (81.0% ± 9.3%), NORC (2.16% ± 1.44%), morphine (0.50% ± 0.39%), morphine-3-glucuronide (M-3-G; 2.10% ± 1.24%), morphine-6-glucuronide (M-6-G; 0.80% ± 0.63%), and normorphine (NORM; 2.44% ± 2.42%). The half-life of codeine is 1.47 h ± 0.32 h, and that of C-6-G is 2.75 h ± 0.79 h. The plasma AUC of C-6-G is about tenfold higher than that of codeine. Protein binding of codeine and C-6-G in vivo is 56.1% ± 2.5% and 34.0% ± 3.6%, respectively [24].
Lötsch et al. [22] explored the contributions from codeine and its metabolites to central nervous analgesic effects independent from O-demethylation of codeine to morphine. A pharmacokinetic/pharmacodynamic fit of the miotic effects by use of morphine as the only active compound was most significantly (P < 0.0001) improved when C-6-G as a second active moiety was added. CYP2D6-dependent formation of morphine does not explain exclusively the central nervous effects of codeine, and C-6-G is the most likely additional active moiety with possible contribution of NORC and the parent compound [22].
Gasche et al. [25] depicted a patient who received oral codeine in a daily dose of 75 mg (25 mg three times a day) and who, after 4 days of treatment, experienced respiratory depression. The patient recovered after IV administration of naloxone (0.4 mg). The cause of the symptoms was CYP2D6 UM phenotype. The patient was concomitantly treated with clarithromycin and voriconazole, both known inhibitors of CYP3A4. This together with CYP2D6 gene duplication led to the reduced clearance of codeine. Blood concentrations of M-3-G and M-6-G were substantially elevated, also due to renal failure [25]. Recent reports [26, 27] indicate that there is a significant risk of respiratory depression in infants whose mothers with CYP2D6 UM and UGT2B7•2/•2 genotypes taking codeine during breastfeeding [28•]. Guidelines for maternal codeine use during breastfeeding were issued in Canada [29], but it seems safer to not use codeine and substitute it with other analgesics in this patient group. Apart from morphine glucuronides, codeine and its metabolites (C-6-G and NORC) also contribute to analgesic effects [22, 23].
Opioids for Moderate-to-Severe Pain (Strong Opioids)
Morphine
Morphine still is the standard drug for the treatment of severe cancer pain and is a comparator for other strong opioids [30••]. This is predominantly due to large clinical experience and different routes of morphine administration (eg, oral, SC, IV, intrathecal, and topical). Morphine is a hydrophilic opioid and a pure opioid agonist that acts predominantly through the activation of μ-opioid receptors. Plasma half-life of IR formulations equals 2 to 3 h and the bioavailability after oral morphine administration equals about 30% to 40%. Morphine undergoes glucuronidation; thus, there is little risk of pharmacokinetic interactions with other drugs.
The active metabolite responsible for analgesia is morphine-6-glucuronide (M-6-G). The accumulation of morphine and M-6-G may cause nausea and vomiting, sedation, and finally, respiratory depression. Morphine-3-glucuronide (M-3-G) is devoid of analgesic properties but may be responsible for neurotoxic effects and opioid-induced hyperalgesia (paradoxical pain) [31]. The main drawback of morphine is the fact that M-3-G and M-6-G may accumulate especially in patients with renal impairment and renal failure, leading to possible intense adverse effects associated with accumulation of metabolites. In severe pain syndromes, a change from oral to parenteral or intrathecal route of morphine administration may be beneficial. In case of renal problems, a switch from morphine to other opioids, such as fentanyl, methadone, or buprenorphine, is recommended. Similar to other opioids, morphine often causes constipation; therefore, the use of laxative prophylaxis is recommended.
Numerous oral CR formulations of morphine, designed for 12-hour and 24-hour administration, were developed [32]. Local administration of morphine prevents systemic adverse effects. The starting daily dose of oral morphine is usually 20 to 30 mg (for opioid-naïve patients) or 40 to 60 mg (for patients unsuccessfully treated with weak opioids) [33•]. The dose of parenteral (SC or IV) morphine is one third of the morphine oral dose [34].
Fentanyl
Fentanyl is a lipophilic opioid, a μ-opioid receptor agonist, with analgesic effect about 100 times more potent than that of morphine. In chronic pain treatment, transdermal fentanyl (TF) patches are applied, usually on the upper trunk. There are five types of patches that release 12, 25, 50, 75, and 100 μg/h equal to 2.1-, 4.2-, 8.4-, 12.6-, and 16.8-mg fentanyl dose per day, respectively. Patches are changed every 72 h. Patients need access to short-acting opioid preparations (ie, oral or parenteral morphine, buccal fentanyl tablets, oral transmucosal fentanyl citrate [OTFC], or fentanyl spray) during TF therapy to effectively manage breakthrough-pain episodes. Fentanyl is metabolized mainly to inactive norfentanyl; thus, it may be used in patients with renal impairment. Because the fentanyl metabolic pathway is through CYP3A4, the drugs inhibiting or inducing this enzyme should be avoided. Caution is recommended when using drugs metabolized via CYP3A4. In comparison to morphine, the advantages of TF include milder constipation, nausea, and drowsiness [35].
When starting TF in opioid-naïve or strong opioid–naïve patients, one patch at a dose of 12 μg/h or 25 μg/h, respectively, is recommended. TF also may be used in opioid switch, especially in patients treated with morphine who suffer from intractable constipation. In an open-label study of 16 patients with cancer pain unable to take oral opioids, TF was effective and well tolerated [36]. A good analgesic effect was achieved in 11 patients, with a partial effect in an additional 2 patients. TF was effective and well tolerated in patients formerly treated with weak opioids that did not provide satisfactory analgesia [37]. The indications for TF include patients’ preferences, intense constipation during morphine treatment, morphine intolerance, nausea, and vomiting. TF should not be used in patients with unstable pain syndromes, especially with neuropathic pain component due to the long plasma half-life (20 h) of the drug, which hinders quick and effective dose titration. Fentanyl may be successfully used by other routes (eg, SC, IV, inhaled, buccal) in the treatment of breakthrough pain [38].
Oxycodone
Oxycodone is a semisynthetic thebaine derivative, a strong opioid that displays a significant affinity to κ-opioid receptors along with agonistic effect mediated by μ-opioid receptors. Limited cross-tolerance is observed between oxycodone and morphine in rats and in clinical studies [39]. In comparison to morphine, oxycodone possesses lower affinity to μ-opioid receptors and similar lipid solubility. Oxycodone permeates the blood–brain barrier very quickly, which may explain its stronger analgesic effect in comparison to other opioids. Oxycodone does not display immunosuppressive effects in experimental studies. It has high oral bioavailability (60%–87%); the plasma half-life is 2 to 3 h after IV administration, 3 h after treatment with IR oral solution, and 8 h after CR tablets. The bioavailability of rectal administration is similar to oral route (61%), but it displays greater variability.
Oxycodone is metabolized in the liver primarily to noroxycodone through CYP3A4 and, to a much less extent, to oxymorphone via CYP2D6. Noroxycodone is metabolized to noroxymorphone through CYP2D6, and oxymorphone is metabolized to noroxymorphone by CYP3A4. However, analgesia observed after oxycodone administration relies primarily on the parent compound. Noroxycodone has 17% of the potency of oxycodone. Oxymorphone, in spite of high affinity for μ-opioid receptors, is produced in very small amounts. Noroxymorphone is produced in a significant amount and displays significant affinity for opioid receptors. However, the blood–brain barrier is extremely impermeable to noroxymorphone; thus, its role in analgesia is negligible. Low blood–brain barrier permeability is also characteristic of noroxycodone and oxymorphone [40].
In patients with liver cirrhosis and hepatic diseases, the oxycodone dose should be reduced by half. Oxycodone is excreted through the kidneys. In patients with renal insufficiency, the oxycodone dose also should be reduced. In patients with renal failure, the oxycodone half-life is prolonged and ranges from 1.8 to 26 h. The elimination of noroxycodone and oxymorphone also is impaired in patients with renal failure. CYP2D6 polymorphism probably does not influence oxycodone analgesia and adverse effects. Sertraline minimally inhibits CYP2D6 and intensifies adverse effects of oxycodone (eg, hallucinations, tremors), whereas fluoxetine and quinidine (significant CYP2D6 inhibitors) do not intensify oxycodone adverse effects. Oxycodone reduces oral bioavailability of cyclosporine by half. In healthy patients, rifampin, a CYP3A4 inducer, greatly decreased oral and IV oxycodone AUC by 86% and 53%, respectively (P < 0.001), and modestly reduced analgesia and increased plasma metabolite-to–parent compound ratios for noroxycodone and noroxymorphone (P < 0.001) [41]. A pharmacodynamic interaction of oxycodone with other drugs acting on the central nervous system, such as benzodiazepines, neuroleptics, and antidepressants, may intensify oxycodone adverse effects, especially sedation, and respiratory depression may be intensified in the case of patients who are more sensitive to opioids.
Buprenorphine
Buprenorphine is a partial μ-opioid–receptor agonist and κ-receptor antagonist. A ceiling analgesic effect may be obtained at high doses (ie, 15 mg); however, such high doses are not used in clinical practice. The analgesic potency of buprenorphine is about 100 times greater than oral morphine [42]. Buprenorphine may be administered sublingually due to low oral bioavailability at doses of 0.2 to 0.8 mg, usually 3 times daily. It also may be administered by parenteral route (SC or IV).
Buprenorphine is metabolized to the active metabolite norbuprenorphine via CYP3A4. The parent compound and norbuprenorphine undergo glucuronidation; thus, the risk of pharmacokinetic interactions with other drugs is low. Compared with morphine, buprenorphine less frequently induces constipation, nausea, and vomiting, which is probably associated with higher lipophilicity. Buprenorphine is mainly excreted with feces (2/3 of the drug is excreted with feces); therefore, it may be used in patients with renal failure. Respiratory depression is rare; however, when the symptom appears, naloxone injection should be administered at a dose of 2 mg, followed by continuous infusion (4 mg/h). Buprenorphine displays antihyperalgesic activity and may be used successfully in the treatment of neuropathic pain [43].
Buprenorphine is administered in transdermal patches (TB) releasing 35, 52.5, and 70 μg/h, which correspond to 0.8, 1.2, and 1.6 mg/d, respectively. The patches are changed every 84 to 96 h. In some countries, patches releasing 5 and 10 μg/h, changed weekly, are available. The starting dose for strong opioid–naïve patients is usually one patch of 35 μg/h. However, opioid-naive patients and those with renal or hepatic impairment may start with a dose of 17.5 μg/h. The treatment is usually well-tolerated. At doses up to 140 μg/h, TB does not display ceiling analgesia [44•]. Breakthrough pain may be treated with sublingual buprenorphine tablets or with IR morphine administered by oral or parenteral route.
Hydromorphone
Hydromorphone has about 5 to 10 times more potent analgesic effect than morphine and similar pharmacodynamic properties. Hydromorphone analgesia is due to μ-opioid–receptor agonist effects. After hydromorphone administration, analgesia lasts for about 4 to 6 h and the plasma half-life is about 2.5 h. The drug is metabolized mainly to hydromorphone-3-glucuronide that may accumulate in patients with renal failure and induce neurotoxic adverse effects. Hydromorphone in small amount also is metabolized to 6-hydroxy-hydromorphone, but its role is unknown. Due to glucuronidation, the risk of hydromorphone pharmacokinetic interactions with other drugs seems to be low [45]. Adverse effects are similar to those of morphine; however, hydromorphone less frequently induces nausea and vomiting, constipation, itching, and probably more slowly develops tolerance to analgesia [46]. Hydromorphone is especially useful for patients requiring high opioid doses via parenteral route due to strong analgesic effects and the possibility of administering small volumes of the drug in SC injections.
Methadone
Methadone is a synthetic opioid and a racemate of dextrorotatory (S-methadone) and levorotatory (D-methadone) isomers. Methadone activates μ-, κ-, and Δ-receptors (D-methadone); it displays moderate antagonistic effect to NMDA receptors (both enantiomers) and strongly inhibits the reuptake of serotonin and noradrenalin in the central nervous system (S-methadone). In high doses, methadone blocks potassium channels required for rapid cardiac muscle repolarization, which may explain the risk of developing ventricular arrhythmia.
Methadone is administered mostly to patients with cancer pain who undergo opioid switch; usually, methadone is given every 8 h. In comparison to morphine, 10 times less demand for laxatives and 2 times less nausea and vomiting were observed. Methadone may be administered as the first strong opioid to patients who have been treated with opioids for moderate pain or to opioid-naïve patients (the starting dose is usually 3–5 mg every 8 h) [47]. Methadone can be administered to patients with renal impairment. It has weak immunosuppressive effect and does not suppress the functioning of natural killer cells. Methadone is tenfold less expensive than the CR morphine and 25-fold cheaper than TF.
Methadone is a highly lipophilic and basic drug with a high distribution volume (4.1 L/kg ± 0.65 L/kg) and a high affinity to tissues, where it accumulates after multiple administrations (in brain, lung, liver, gut, kidney, and muscles). The high affinity to tissues, together with a gradual and retarded release to plasma, is the cause of a prolonged half-life. The bioavailability of the drug after oral administration oscillates between 70% and 90%. The half-life is about 24 h, but it occurs in the range of 8 to 120 h. Analgesia lasts for 6 to 12 h. A stable level is reached within 2 to 4 days. Methadone is metabolized mostly via liver enzymes, but also in the intestine wall via N-demethylation to inactive metabolites. The main enzyme responsible for methadone N-demethylation is CYP3A4 with a lesser CYP1A2 and CYP2D6 involvement and a significant CYP2B6 role. The drug is excreted mainly via the alimentary tract, but also through kidneys (depending on the urine pH). In chronic renal disease, methadone does not accumulate; in severe renal failure, a dose reduction may be considered. Methadone is not eliminated in the process of hemodialysis. Methadone is more difficult to use than other opioids due to complicated pharmacokinetics, numerous drug interactions, and possible QT prolongation; therefore, it should be used by physicians experienced in management of chronic pain [48].
Tapentadol
Tapentadol chloride ([−]-[1R,2R]-3-[3-Dimethylamino-1-ethyl-2-methyl-propyl]-phenol hydrochloride) is an opioid with two analgesic mechanisms: 1) agonist of μ-opioid receptors with 50 times less affinity than morphine, and 2) inhibition of norepinephrine reuptake [49]. Bioavailability after oral administration is over 30%, the drug is metabolized to inactive metabolites through glucuronidation and excreted via kidneys [50]. In experimental studies tapentadol is effective in the treatment of neuropathic pain and in inflammatory pain. In clinical studies conducted in patients with low back pain, those with postoperative pain, and those with osteoarthritis, IR tapentadol at doses of 50, 75, and 100 mg had more favorable adverse-effects profiles with less intense gastrointestinal adverse effects (ie, nausea, vomiting, constipation) in comparison to IR oxycodone at doses of 10 and 15 mg. Clinical studies on tapentadol use in patients with cancer pain have not been published.
Conclusions
Opioids are usually effective when administered alone or with adjuvant analgesics. The traditional WHO step-by-step approach should be used individually, based on the clinical assessment of pain type and intensity. Patients with severe pain intensity should use strong opioids (opioids for moderate-to-severe pain) without climbing up the analgesic ladder. Opioids may be combined with nonopioid analgesics and adjuvant analgesics appropriate for a given pain type. Understanding important attributes of commonly used opioids can help assist selection (Tables 3 and 4).
Table 3.
Comparison among weak opioids for mild-to-moderate severity pain
| Opioida | Main mode of action | Attributes | Precaution | Typical starting dose |
|---|---|---|---|---|
| Tramadol | μ-Opioid receptor agonist, 5HT- and NOR-reuptake blocker | Less constipation than other opioids | Nausea should be prevented by antiemetics; analgesia impaired in CYP2D6 poor metabolizers | 25–50 mg q 4–6 h (IR); 50–100 mg q 12 h (CR) |
| Dihydrocodeine | μ-Opioid receptor agonist | Useful for patients with moderate pain, cough, and dyspnea | Constipation should be prevented by laxatives | 30 mg q 4–6 h (IR); 60 mg q 12 h (CR) |
| Codeine | μ-Opioid receptor agonist | Useful for patients with moderate pain, cough, and dyspnea; combined formulations with paracetamol | Constipation should be prevented by laxatives; should not be administered in CYP2D6 ultrarapid metabolizers | 30 mg q 4–6 h (IR); 60 mg q 12 h (CR) |
aTaken orally
5HT Serotonin; CR Controlled-release formulations; CYP2D6 Cytochrome P450 2D6; IR Immediate-release formulations; NOR Noradrenaline; q Every
Table 4.
Comparison among strong opioids for moderate-to-severe severity pain
| Opioida | Main mode of action | Attributes | Precaution | Typical starting dose |
|---|---|---|---|---|
| Morphine | μ-Opioid receptor agonist | May be administered by different routes: oral, SC, IV, IT, local | Active metabolites may accumulate and cause adverse effects in renal failure | 5–10 mg q 4 h (IR); 20–30 mg q 12 h (CR) |
| Fentanyl | μ-Opioid receptor agonist | Less constipation than morphine; safe in patients with renal impairment | Fever may increase absorption; should not be used for quick dose titration (unstable pain) | One patch 25 μg/h q 72 h; 12.5 μg/h q 72 h for older patients with liver or hepatic impairment |
| Oxycodone | μ- and κ-Opioid receptor agonist | Less CNS adverse effects than morphine | May accumulate in renal failure | 5 mg q 4–6 h (IR); 10–20 mg 12 h (CR) |
| Buprenorphine | Partial μ-Opioid receptor agonist, weak κ-opioid receptor antagonist | Less constipation than morphine; safe in patients with renal impairment | Fever may increase absorption; should not be used for quick dose titration (unstable pain) | One patch 35 μg/h q 84 h; 17.5 μg/h q 84–96 h for older patients with liver or hepatic impairment |
| Hydromorphone | μ-Opioid receptor agonist | Useful for patients requiring high opioid doses; less pruritus, nausea/vomiting, and sedation than morphine | Parent compound and metabolites may accumulate in renal failure | 1–2 mg q 4 h (IR); 2–4 mg q 12 h (CR) |
| Methadone | μ - and δ-Opioid receptor agonist, NMDA-receptor antagonist, NOR- and 5HT-reuptake blocker | Useful for patients with severe neuropathic pain and renal failure | Possible QT interval prolongation; numerous drug interactions; long plasma half-life | 3–5 mg q 8 h |
| Tapentadol | μ-Opioid receptor agonist and NOR-reuptake blocker | Less adverse effects from GI tract than oxycodone | May accumulate in renal failure | 50 mg q 4–6 h (IR); 100 mg q 12 h (CR) |
aTaken orally
5HT Serotonin; CNS central nervous system; CR controlled-release formulation; GI gastrointestinal; IR Immediate-release formulation; IT Intrathecal; IV Intravenous; NMDA N-methyl-D-aspartate receptors; NOR Noradrenaline; SC Subcutaneous
In case of lack of efficacy of orally or transdermally administered opioids, it may be beneficial to change the route of administration to parenteral or intrathecal. Another possibility is opioid switch that may improve analgesia and reduce adverse effects. A good example may be patients suffering from severe constipation who may benefit when switching from morphine to TF and from codeine or DHC to tramadol. A newer approach is the concomitant use of two opioids, although little evidence supports such procedure. Future studies may address genetic disposition responsible for individual patients’ response to opioid analgesics [51••].
Acknowledgments
Disclosure
No potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
An erratum to this article can be found at http://dx.doi.org/10.1007/s11916-011-0224-0
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
- 1.World Health Organization . Cancer pain relief and palliative care. Geneva: World Health Organization; 1996. [Google Scholar]
- 2.Hanks GW, de Conno F, Cherny N, Hanna M, Kalso E, McQuay HJ, et al. Expert working group of the research network of the European association for palliative care: morphine and alternative opioids in cancer pain: the EAPC recommendations. Br J Cancer. 2001;84:587–93. doi: 10.1054/bjoc.2001.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leppert W. Progress in pharmacological pain treatment with opioid analgesics (Polish) Wspolcz Onkol. 2009;13:66–73. [Google Scholar]
- 4.Eidelman A, White T, Swarm RA. Interventional therapies for cancer pain management: important adjuvants to systemic analgesics. J Natl Compr Canc Netw. 2007;5:753–60. doi: 10.6004/jnccn.2007.0075. [DOI] [PubMed] [Google Scholar]
- 5.Lussier D, Huskey AG, Portenoy RK. Adjuvant analgesics in cancer pain management. Oncologist. 2004;9:571–91. doi: 10.1634/theoncologist.9-5-571. [DOI] [PubMed] [Google Scholar]
- 6.Leppert W. The role of ketamine in the management of neuropathic cancer pain—a polish experience. Proceedings of the 3rd International Congress on Neuropathic pain, NeuPSIG, Athens (Greece), May 27—30, 2010, Ed. Christopher D. Wells. Medimond International Proceedings 2010, pp. 199–203.
- 7.Ripamonti CI, Easson AM, Gerdes H. Management of malignant bowel obstruction. Eur J Cancer. 2008;44:1105–15. doi: 10.1016/j.ejca.2008.02.028. [DOI] [PubMed] [Google Scholar]
- 8.Leppert W, Mikolajczak P. Analgesic effects and assays of controlled-release tramadol and o-desmethyltramadol in cancer patients with pain. Curr Pharmaceut Biotechnol. 2011;12:306–12. doi: 10.2174/138920111794295738. [DOI] [PubMed] [Google Scholar]
- 9.Dickman A. Tramadol: a review of this atypical opioid. Eur J Palliat Care. 2007;14:181–5. [Google Scholar]
- 10.Stamer UM, Lehnen K, Höthker F, Bayerer B, Wolf S, Hoeft A, et al. Impact of CYP2D6 genotype on postoperative tramadol analgesia. Pain. 2003;105:231–8. doi: 10.1016/S0304-3959(03)00212-4. [DOI] [PubMed] [Google Scholar]
- 11.Stamer U, Musshoff F, Kobilay M, Madea B, Hoeft A, Stuber F. Concentrations of tramadol and o-desmethyltramadol enantiomers in different CY2D6 genotypes. Clin Pharmacol Ther. 2007;82:41–7. doi: 10.1038/sj.clpt.6100152. [DOI] [PubMed] [Google Scholar]
- 12.Stamer U, Stuber F, Muders T, Musshoff F. Respiratory depression with tramadol in a patient with renal impairment and CYP2D6 gene duplication. Anesth Analg. 2008;107:926–9. doi: 10.1213/ane.0b013e31817b796e. [DOI] [PubMed] [Google Scholar]
- 13.Gnanadesigan N, Espinoza RT, Smith R, Israel M, Reuben DB. Interaction of serotonergic antidepressants and opioid analgesics: is serotonin syndrome going undetected? J Am Med Dir Associ. 2005;6:265–9. doi: 10.1016/j.jamda.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 14.Davies MP. Tramadol. In: Davies MP, Glare P, Hardy J, editors. Opioids in cancer pain. Oxford: Oxford University Press; 2005. pp. 69–82. [Google Scholar]
- 15.Leppert W. Tramadol as an analgesic for mild to moderate cancer pain. Pharmacol Rep. 2009;61:978–92. doi: 10.1016/s1734-1140(09)70159-8. [DOI] [PubMed] [Google Scholar]
- 16.Ammon S, Hofmann U, Griese EU, Gugeler N, Mikus G. Pharmacokinetics of dihydrocodeine and its active metabolite after single and multiple oral dosing. Br J Clin Pharmacol. 1999;48:317–22. doi: 10.1046/j.1365-2125.1999.00042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leppert W. Dihydrocodeine as an analgesic for the treatment of moderate to severe chronic pain. Curr Drug Metab. 2010;11:494–506. doi: 10.2174/138920010791636211. [DOI] [PubMed] [Google Scholar]
- 18.Rowell FJ, Seymour RA, Rawlins MD. Pharmacokinetics of intravenous and oral dihydrocodeine and its acid metabolites. Eur J Clin Pharmacol. 1983;25:419–24. doi: 10.1007/BF01037958. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt H, Vormfelde SV, Walchner-Bonjean M, et al. The role of active metabolites in dihydrocodeine effects. Int J Clin Pharmacol Ther. 2003;41:95–106. doi: 10.5414/cpp41095. [DOI] [PubMed] [Google Scholar]
- 20.Webb JA, Rostami-Hodjegan A, Abdul-Manap R, Hofmann U, Mikus G, Kamali F. Contribution of dihydrocodeine and dihydromorphine to analgesia following dihydrocodeine administration in man: a PK-PD modelling analysis. Br J Clin Pharmacol. 2001;52:35–43. doi: 10.1046/j.0306-5251.2001.01414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leppert W, Majkowicz M. The impact of tramadol and dihydrocodeine treatment on quality of life of patients with cancer pain. Int J Clin Pract. 2010;64:1681–7. doi: 10.1111/j.1742-1241.2010.02422.x. [DOI] [PubMed] [Google Scholar]
- 22.Lötsch J, Skarke C, Schmidt H, Rohrbacher M, Hofmann U, Schwab M, et al. Evidence for morphine-independent central nervous opioid effects after administration of codeine: contribution of other codeine metabolites. Clin Pharmacol Ther. 2006;79:35–48. doi: 10.1016/j.clpt.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Vree TB, van Dongen RTM, Koopman-Kimenai PM. Codeine analgesia is due to codeine-6-glucuronide, not morphine. Int J Clin Pract. 2000;54:395–8. [PubMed] [Google Scholar]
- 24.Vree TB, Verwey-van Wissen CP. Pharmacokinetics and metabolism of codeine in humans. Biopharm Drug Dispos. 1992;13:445–60. doi: 10.1002/bdd.2510130607. [DOI] [PubMed] [Google Scholar]
- 25.Gasche Y, Daali Y, Fathi M, et al. Codeine intoxication associated with ultrarapid CYP2D6 metabolism. N Engl J Med. 2004;351:2827–31. doi: 10.1056/NEJMoa041888. [DOI] [PubMed] [Google Scholar]
- 26.Kirchheiner J, Schmidt H, Tzetkov M, Keulen J-T, Lötsch J, Roots I, et al. Pharmacokinetics of codeine and its metabolite morphine in ultra-rapid metabolizers due to CYP2D6 duplication. Pharmacogenomics J. 2007;7:257–65. doi: 10.1038/sj.tpj.6500406. [DOI] [PubMed] [Google Scholar]
- 27.Voronov P, Przybylo HJ, Jagannathan N. Apnea in a child after oral codeine: a genetic variant—an ultra-rapid metabolizer. Pediatr Anesth. 2007;17:684–7. doi: 10.1111/j.1460-9592.2006.02182.x. [DOI] [PubMed] [Google Scholar]
- 28.Madadi P, Ross CJD, Hayden MR, Carleton BC, Gaedigk A, Leeder JS, et al. Pharmacogenetics of neonatal opioid toxicity following maternal use of codeine during breastfeeding: a case-control study. Clin Pharmacol Ther. 2009;85:31–5. doi: 10.1038/clpt.2008.157. [DOI] [PubMed] [Google Scholar]
- 29.Madadi P, Moretti M, Djokanovic N, Bozzo P, Nulman I, Ito S, et al. Guidelines for maternal codeine use during breastfeeding. Can Fam Phys. 2009;55:1077–8. [PMC free article] [PubMed] [Google Scholar]
- 30.Flemming K. The use of morphine to treat cancer-related pain: a synthesis of quantitative and qualitative research. J Pain Symptom Manage. 2010;39:139–54. doi: 10.1016/j.jpainsymman.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 31.Gretton S, Riley J. Morphine metabolites: a review of their clinical effects. Eur J Palliat Care. 2008;15:110–4. [Google Scholar]
- 32.Ridgway D, Sopata M, Burneckis A, Jespersen L, Andersen C. Clinical efficacy and safety of once-daily dosing of a novel, prolonged-release oral morphine tablet compared with twice-daily dosing of a standard controlled-release morphine tablet in patients with cancer pain: a randomized, double-blind, exploratory crossover study. J Pain Symptom Manage. 2010;39:712–20. doi: 10.1016/j.jpainsymman.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 33.Ripamonti CI, Campa T, Fagnoni E, Brunelli C, Luzzani M, Maltoni M, et al. Normal-release oral morphine starting dose in cancer patients with pain. Clin J Pain. 2009;25:386–90. doi: 10.1097/AJP.0b013e3181929b4f. [DOI] [PubMed] [Google Scholar]
- 34.Donnelly S, Davis MP, Walsh D, Naughton M. Morphine in cancer pain management: a practical guide. Support Care Cancer. 2002;10:13–25. doi: 10.1007/s005200100274. [DOI] [PubMed] [Google Scholar]
- 35.Ahmedzai S, Brooks D. Transdermal fentanyl versus sustained-release oral morphine in cancer pain: preference, efficacy and quality of life. J Pain Symptom Manage. 1997;13:254–61. doi: 10.1016/S0885-3924(97)00082-1. [DOI] [PubMed] [Google Scholar]
- 36.Leppert W, Luczak J, Gorzelinska L, Kozikowska J. Research from the palliative care department in Poznań on treatment of neoplasm pain with durogesic (transdermal fentanyl) (Polish) Przegl Lek. 2000;57:59–64. [PubMed] [Google Scholar]
- 37.Vielvoye-Kerkmeer APE, Mattern C, Uitendaal MP. Transdermal fentanyl in opioid-naive cancer pain patients: an open trial using transdermal fentanyl for the treatment of chronic cancer pain in opioid-naive patients and a group using codeine. J Pain Symptom Manage. 2000;19:185–92. doi: 10.1016/S0885-3924(99)00152-9. [DOI] [PubMed] [Google Scholar]
- 38.Slatkin NE, Xie F, Messina J, Segal TJ. Fentanyl buccal tablet for relief of breakthrough pain in opioid-tolerant patients with cancer-related chronic pain. J Support Oncol. 2008;5:327–34. [PubMed] [Google Scholar]
- 39.Maddocks I, Somogyi A, Abbott F, Hayball P, Parker D. Attenuation of morphine-induced delirium in palliative care by substitution with infusion of oxycodone. J Pain Symptom Manage. 1996;12:182–9. doi: 10.1016/0885-3924(96)00050-4. [DOI] [PubMed] [Google Scholar]
- 40.Leppert W. Role of oxycodone and oxycodone/naloxone in cancer pain management. Pharmacol Rep. 2010;62:578–91. doi: 10.1016/s1734-1140(10)70316-9. [DOI] [PubMed] [Google Scholar]
- 41.Nieminen TH, Hagelberg NM, Saari TI, Pertovaara A, Neuvonen M, Laine K, et al. Rifampin greatly reduces the plasma concentrations of intravenous and oral oxycodone. Anesthesiology. 2009;110:1371–8. doi: 10.1097/ALN.0b013e31819faa54. [DOI] [PubMed] [Google Scholar]
- 42.Likar R, Krainer B, Sittl R. Challenging the equipotency calculation for transdermal buprenorphine: four case studies. Int J Clin Pract. 2008;62:152–6. doi: 10.1111/j.1742-1241.2007.01531.x. [DOI] [PubMed] [Google Scholar]
- 43.Mercadante S, Ferrera P, Villari P. Is there a ceiling effect of transdermal buprenorphine? Preliminary data in cancer patients. Support Care Cancer. 2007;15:441–4. doi: 10.1007/s00520-006-0169-8. [DOI] [PubMed] [Google Scholar]
- 44.Kress HG. Clinical update on the pharmacology, efficacy and safety of transdermal buprenorphine. Eur J Pain. 2009;13:219–30. doi: 10.1016/j.ejpain.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 45.Sarhill N, Walsh D, Nelson KA. Hydromorphone: pharmacology and clinical applications in cancer patients. Support Care Cancer. 2001;9:84–96. doi: 10.1007/s005200000183. [DOI] [PubMed] [Google Scholar]
- 46.Wirz S, Wartenberg HC, Nadstawek J. Less nausea, emesis, and constipation comparing hydromorphone and morphine? A prospective open-labeled investigation on cancer pain. Support Care Cancer. 2008;16:999–1009. doi: 10.1007/s00520-007-0368-y. [DOI] [PubMed] [Google Scholar]
- 47.Ripamonti C, Groff L, Brunelli C, Polastri D, Stavrakis A, De Conno F. Switching from morphine to oral methadone in treating cancer pain: what is the equianalgesic dose ratio? J Clin Oncol. 1998;16:3216–21. doi: 10.1200/JCO.1998.16.10.3216. [DOI] [PubMed] [Google Scholar]
- 48.Leppert W. The role of methadone in cancer pain treatment—a review. Int J Clin Pract. 2009;63:1095–109. doi: 10.1111/j.1742-1241.2008.01990.x. [DOI] [PubMed] [Google Scholar]
- 49.Tzschentke TM, Christoph T, Kögel B, et al. (−)-(1R,2R)-3-(3-Dimethylamino-1-ethyl-2-methyl-propyl)-phenol hydrochloride (Tapentadol HCl): a novel μ-opioid receptor agonist/norepinephrine reuptake inhibitor with broad-spectrum analgesic properties. J Pharmacol Exp Ther. 2007;323:265–76. doi: 10.1124/jpet.107.126052. [DOI] [PubMed] [Google Scholar]
- 50.Kneip C, Terlinden R, Beier H, Chen G. Investigations into the drug-drug interaction potential of tapentadol in human liver microsomes and fresh human hepatocytes. Drug Metabol Lett. 2008;2:67–75. doi: 10.2174/187231208783478434. [DOI] [PubMed] [Google Scholar]
- 51.Lötsch J, Geisslinger G, Tegeder I. Genetic modulation of the pharmacological treatment of pain. Pharmacol Ther. 2009;124:168–84. doi: 10.1016/j.pharmthera.2009.06.010. [DOI] [PubMed] [Google Scholar]