

Abstract
Excessive signalling by excitatory neurotransmitters like glutamate and ATP can be deleterious to neurons and oligodendroglia, and cause disease. In particular, sustained activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), kainate and N-methyl-d-aspartate (NMDA) receptors damages oligodendrocytes, a feature that depends entirely on Ca2+ overload of the cytoplasm and that can be initiated by disruption of glutamate homeostasis. Thus, inhibition of glutamate uptake by activated microglia can compromise glutamate homeostasis and induce oligodendrocyte excitotoxicity. Moreover, non-lethal, brief activation of kainate receptors in oligodendrocytes rapidly sensitizes these cells to complement attack as a consequence of oxidative stress. In addition to glutamate, ATP signalling can directly trigger oligodendrocyte excitotoxicity via activation of Ca2+-permeable P2X7 purinergic receptors, which mediates ischaemic damage to white matter (WM) and causes lesions that are reminiscent of multiple sclerosis (MS) plaques. Conversely, blockade of P2X7 receptors attenuates post-ischaemic injury to WM and ameliorates chronic experimental autoimmune encephalomyelitis, a model of MS. Importantly, P2X7 expression is elevated in normal-appearing WM in patients with MS, suggesting that signalling through this receptor in oligodendrocytes may be enhanced in this disease. Altogether, these observations reveal novel mechanisms by which altered glutamate and ATP homeostasis can trigger oligodendrocyte death. This review aims at summarizing current knowledge about the mechanisms leading to WM damage as a consequence of altered neurotransmitter signalling, and their relevance to disease. This knowledge will generate new therapeutic avenues to treat more efficiently acute and chronic WM pathology.
Keywords: ATP, demyelination, excitotoxicity, glutamate, human brain, ischaemia, oligodendrocyte death
Introduction
White matter is unique as opposed to grey matter in that it mostly contains axons and glial cells supporting their correct functioning. Macroglial cells in white matter (WM) are also diverse as astrocytes have a fibrous morphological phenotype and it bears the majority of oligodendrocytes. In humans, WM comprises about half of the CNS volume, which is about a three–fourfold greater proportion than in other mammals, including those typically used for animal experiments (Zhang & Sejnowski, 2000). This feature may have misrepresented the importance of WM damage to the outcome of CNS diseases in humans.
White matter damage typically involves primary or secondary disruption of axons, which leads to the impairment of motor and sensory functions, behaviour and cognition (Filley, 2001; Desmond, 2002). Intriguingly, WM cells and axons possess the molecular machinery to signal by means of neurotransmitters (Stys, 2005; Butt, 2006; Constantinou & Fern, 2009; Matute, 2010), a property whose functional significance remains elusive. Yet, it is now well established that excessive neurotransmitter signalling is deleterious to WM and that it can trigger disease and/or contribute to its progression. In this review, I summarize progress in the understanding of the mechanisms by which neurotransmitters cause structural and functional damage to WM, and emphasize the case of glutamate and ATP, two major excitatory transmitters that can act as potent neurotoxins.
Glutamate signalling in WM glia and axons
The major intermediaries of glutamate signalling are glutamate receptors (GluRs) and glutamate transporters (GluTs). Glutamate activates ionotropic and metabotropic receptors (for reviews, see Dingledine et al. 1999; Cull-Candy & Leszkiewicz, 2004; Swanson et al. 2005), which are expressed in glial cells in grey (for reviews, see Belachew & Gallo, 2004; Kettenmann & Steinhäuser, 2005; Matute et al. 2006; Verkhratsky & Kirchhoff, 2007; Bakiri et al. 2009) and in WM (reviewed in Matute, 2010). In particular, cells of the oligodendrocyte lineage express functional α-amino-3-hydroxy-5-methyl4-isoxazolepropionic acid (AMPA) and kainate type receptors throughout a wide range of developmental stages and species, including humans (Matute et al. 2007a). In addition, immature and mature oligodendrocytes express N-methyl-d-aspartate (NMDA) receptors, which can be activated during injury (Káradóttir et al. 2005; Salter & Fern, 2005; Micu et al. 2006; reviewed in Matute, 2006). Moreover, oligodendrocytes also express receptors of all three groups of metabotropic GluRs, but their levels are developmentally regulated and are very low in mature cells of this lineage (Deng et al. 2004).
Glutamate uptake from the extracellular space by specific GluTs is essential for the shaping of excitatory postsynaptic currents and for the prevention of excitotoxic death due to overstimulation of GluRs (Rothstein et al. 1996). At least five GluTs have been cloned (Danbolt, 2001; Huang & Bergles, 2004). Of these, glutamate transporter 1 (GLT-1, also named as EAAT2) exhibits the highest level of expression, and it is responsible for most glutamate transport (Danbolt, 2001). GluTs are expressed by astrocytes and oligodendrocytes. The main transporter expressed by oligodendrocytes is glutamate aspartate transporter (GLAST; also named as EAAT1). The neuronal transporter, termed excitatory amino acid carrier 1 (EAAC1 or EAAT3), is present in a subpopulation of adult oligodendrocyte progenitor cells (Domercq et al. 1999). It thus appears that all macroglial cells differentially express the three major GluTs present in the CNS. These transporters maintain basal levels of extracellular glutamate in the range of 1–2 μm, and thus prevent overactivation of GluRs under physiological conditions. In turn, GluTs can contribute to glutamate release in WM by reversal of Na+-dependent glutamate transport during depolarization (Domercq et al. 1999; Li et al. 1999).
Ionotropic GluRs expressed in glial cells have similar properties to their neuronal counterparts. However, the fact that these receptors are edited to a lesser extent in the WM, and that AMPA receptors in oligodendrocytes do not have GluR2 subunits, suggest that they have a higher Ca2+ permeability than those present in grey matter (Matute et al. 2006). In turn, NMDA receptors are expressed in WM oligodendrocytes at all developmental stages, and their activation generates a membrane depolarization and a rise in cytosolic Ca2+ (Bakiri et al. 2009). Interestingly, NMDA receptors are expressed in clusters on oligodendrocyte processes and myelin, whereas AMPA and kainate receptors are diffusely located on oligodendrocyte somata (Káradóttir et al. 2005; Salter & Fern, 2005; Micu et al. 2006). In addition, oligodendrocytes also express all three subtypes of mGluRs, but their levels are developmentally regulated and are very low in mature cells of this lineage (Deng et al. 2004). However, little is still known about the specific properties of ionotropic and metabotropic GluRs expressed on WM astrocytes and microglia (Matute et al. 2006).
In addition to glial cells, axons are also endowed with GluRs and GluTs. Thus, axons in the dorsal column of the spinal cord are depolarized via activation of AMPA receptors (Ouardouz et al. 2006). In turn, they can be damaged by overactivation of AMPA/kainate receptors (Matute, 1998; Fowler et al. 2003) and protected by blockers of these receptors in models of WM injury (Pitt et al. 2000; Tekkök & Goldberg, 2001). In axons, native AMPA receptors are formed by the GluR4 subunit, and kainate receptors are composed of at least GluR5 and GluR6 subunits, which in all instances are located in the internodes (Ouardouz et al. 2009a,b;). Moreover, axon demise may also be secondary to oligodendrocyte loss by excitotoxicity and the ensuing demyelination rather than by activation per se of GluRs in axons.
Axons release glutamate during electrical activity and in pathological conditions by reversal of Na+-dependent glutamate transport (Li et al. 1999; Bakiri et al. 2009). The major GluT expressed by axons is GLT-1, though some significant levels of GLAST are also present (Li et al. 1999). Other factors that contribute to glutamate homeostasis include the glutamate-producing enzyme glutaminase, which is present in oligodendrocytes and microglia (Domercq et al. 1999; Werner et al. 2001), as well as the glutamate-cystine exchanger xCT− expressed in microglia (Domercq et al. 2007). The function and levels of glutaminase and xCT− exchanger is altered in activated microglia during inflammation, which in turn alters GluT function, all resulting in disrupted glutamate homeostasis and excitotoxicity (Matute, 2010).
Axonal AMPA receptors are weakly permeable to Ca2+, the entry of which in turn releases further Ca2+ from the axoplasmic reticulum by opening intracellular Ca2+ channels known as ryanodine receptors (Ouardouz et al. 2009a). In contrast, axonal kainate receptors with the GluR5 subunit are coupled to phospholipase C activation (Ouardouz et al. 2009a). In addition, activation of kainate receptors with the GluR6 subunit induces a small amount of Ca2+ entry that stimulates nitric oxide synthase, as well as a local depolarization which activates L-type Ca2+ channels and subsequently ryanodine receptors in the axoplasmic reticulum (Ouardouz et al. 2009b). The functional significance of these signalling mechanisms by GluRs in axons is unknown, but they may serve to amplify axonal Ca2+ signals that seem to be weak because of the limited quantity of cation available in the narrow space (Ouardouz et al. 2009b). In turn, high local concentrations of Ca2+ generated by these receptors may result in focal swellings and irreversible axonal transections (Ouardouz et al. 2009b).
Finally, axons are competent sources of neurotransmission within WM as they form functional synapses with NG2 glial progenitors (Kuckley et al. 2007; Ziskin et al. 2007). Thus, action potentials induce vesicular release of glutamate from unmyelinated axons of the corpus callosum, which activate AMPA receptors during development and in the mature brain. Consequently, axonal transmitter release represents a widespread mechanism for activity-dependent signalling between axons and glial precursors in WM (Kuckley et al. 2007; Ziskin et al. 2007). Notably, NG2-expressing glia also is endowed with the synaptic protein synaptophysin, which suggests that they may also be capable of vesicular release and bidirectional communication with axons via their cellular contacts (Bakiri et al. 2009). To add to this complex mosaic of glutamate signals, axonal–glial synapses may also be modulated by vesicular release of glutamate from astrocytes, as observed in classical synapses between neurons (Volterra & Meldolesi, 2005).
Glutamate signalling in WM pathology
White matter, like grey matter, is vulnerable to glutamate insults resulting from deregulation of glutamate homeostasis. I summarize below current evidence regarding the specific mechanisms linking glutamate dyshomeostasis to glial cell death and axonal damage, and its relevance to human diseases involving WM (Table 1).
Table 1.
Neurotransmitter receptors mediating WM pathology
| Disease | Experimental paradigm | Receptor types | References |
|---|---|---|---|
| Stroke | Cultured oligodendrocytes | AMPA/kainate | Fern & Möller (2000) |
| Isolated optic nerve | NMDA | Káradóttir et al. (2005), Salter & Fern (2005), Micu et al. (2006) | |
| Older optic nerve | AMPA/kainate | Baltan et al. (2008), Baltan (2009) | |
| Oligodendrocytes and optic nerve | P2X7 | Domercq et al. (2010) | |
| Transient focal ischaemia | NMDA | Schäbitz et al. (2000) | |
| Transient focal ischaemia | AMPA | McCracken et al. (2002) | |
| Perinatal ischaemia | Rat model | AMPA and NMDA | Follett et al. (2004), Manning et al. (2008) |
| Immature optic nerve | AMPA and NMDA | Alix & Fern (2009) | |
| Rat model | P2X7 | Wang et al. (2009) | |
| MS | Acute and chronic EAE | AMPA | Pitt et al. (2000), Matute et al. (2001), Smith et al. (2000), Kanwar et al. (2004) |
| Microglia activation in optic nerve | AMPA/kainate | Domercq et al. (2007) | |
| Oligodendrocytes and optic nerve | Kainate | Alberdi et al. (2006) | |
| Chronic EAE | GluK2 | Pérez-Samartín et al. (2009) | |
| Chronic EAE | P2X7 | Matute et al. (2007b) | |
| Traumatic injury | Spinal cord | AMPA/kainate | Li & Stys (2000) |
| Toxicity | Optic nerve | Adenosine | González-Fernández et al. (2010) |
| Optic nerve | Nicotinic and adrenergic | Constantinou & Fern (2009) |
AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; EAE, experimental autoimmune encephalomyelitis; GluK2, kainate receptor subunit 2; MS, multiple sclerosis; NMDA, N-methyl-d-aspartate.
Glutamate excitotoxicity
Prolonged activation of AMPA, kainate and NMDA receptors causes oligodendrocyte death and primary and/or secondary myelin destruction. A central event to this process is Ca2+ influx upon receptor activation and the ensuing accumulation of this cation within mitochondria, which leads to depolarization of this organelle, increased production of radical oxygen species, and release of proapoptotic factors that activate caspases (Verkhratsky et al. 1998; Matute et al. 2006). The types of oligodendrocyte death induced by activation of AMPA and kainate receptors depend on the intensity and duration of the excitotoxic insult. Notably, the molecular cascades initiated by AMPA and kainate receptors are not identical, indicating that different intracellular domains are involved in executing the death program triggered by these receptors (Matute et al. 2007a). In particular, insults channelled through kainate receptors activate caspases 9 and 3 leading to apoptosis. In contrast, those activating AMPA receptors induce apoptosis by recruiting caspase 8, which leads to the truncation of the Bid protein. This in turn activates caspase 3 and PARP-1 polymerase, or cause necrosis (Sánchez-Gómez et al. 2003; Matute et al. 2006). The mechanisms triggered by NMDA receptor-mediated insults to oligodendrocytes have not been studied in detail yet.
Glutamate uptake is crucial to terminate glutamate signalling in WM and to prevent excitotoxicity. Thus, inhibition of glutamate uptake in pure cultures of oligodendrocyte increases glutamate levels and causes excitotoxicity, which is prevented by AMPA and kainate receptor antagonists (Domercq et al. 2005). Furthermore, inhibition of GluT function and/or expression in axonal tracts in vivo leads to oligodendroglial loss, massive demyelination and severe axonal damage (Fig. 1; Domercq et al. 2005). Therefore, the integrity of WM depends on proper GluT function and, conversely, its deregulation may contribute to acute and chronic WM damage.
Fig. 1.
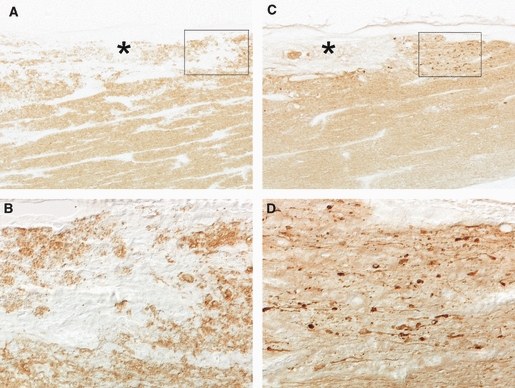
Oligodendroglial and axonal damage occurs after inhibition of glutamate transport in optic nerve. (A, B) Glutamate transporter blocker dihydrokainate induces local demyelination (*) as revealed by immunostaining with antibodies to myelin basic protein. (B) is a higher-magnification of the inset in (A). (C, D) Immunohistochemistry with antibodies to dephosphorylated neurofilaments shows axonal loss in demyelinated areas (*) and severe axonal dystrophy in nearby regions after glutamate transport inhibition. (D) Higher-magnification of inset in (C), illustrating the swelling characteristics of axonal damage. Modified from Domercq et al. (2005).
Other factors that may contribute to perturbing glutamate homeostasis include altered activity of the glutamate-producing enzyme glutaminase in activated macrophages/microglia in close proximity to dystrophic axons (Werner et al. 2001) and reduced expression of the glutamate transporters EAAT-1 and EAAT-2 in oligodendrocytes as a consequence of enhanced exposure to the proinflammatory cytokine TNF-α (Pitt et al. 2003). Overall, these alterations likely lead to high extracellular glutamate levels and an increased risk of oligodendrocyte excitotoxicity in multiple sclerosis (MS).
Stroke and perinatal ischaemia
Injury of central WM is a major cause of functional disability in cerebrovascular disease (Goldberg & Ransom, 2003). Damage to WM as a consequence of hypoxic–ischaemic injury occurs in periventricular leukomalacia (PVL) in the neonatal period, stroke and cardiac arrest in adults, as well as in vascular dementia in the aging brain. Indeed, the metabolic rate of WM is only modestly lower than that of grey matter, and animal studies suggest that WM can be damaged even by brief ischaemia (Pantoni et al. 1996). Energy supply failure during ischaemia results in ion gradient break down, membrane depolarization and ultimately leads to Ca2+ overload of the cytosol, which activates Ca2+-dependent enzymes such as calpains, phospholipases and other enzymes resulting in irreversible damage of WM glia and axons (Stys, 2004).
Immature and differentiated oligodendrocytes in vitro are the most sensitive cells of the WM to transient oxygen and glucose deprivation (Fern & Möller, 2000; reviewed in Matute et al. 2006). In both instances, cell death is prevented in the absence of Ca2+ or by AMPA/kainate receptor antagonists, but not by the blockade of other potential sources of Ca2+ influx, which suggests that Ca2+ entry through the receptor channel is sufficient to initiate cell demise. Notably, simulated ischaemia induces an inward current in oligodendrocytes in situ, which is partly mediated by NMDA and AMPA/kainate receptors (Káradóttir et al. 2005). In addition, Ca2+ levels also increase in myelin itself during ischaemia, an effect that is abolished by broad-spectrum NMDA receptor antagonists, causing ultrastructural damage to both axon cylinders and myelin (Micu et al. 2006). Taken together, these results using in vitro models simulating ischaemia confirm earlier findings suggesting that AMPA and NMDA receptor blockade protects from post-ischaemic damage to both grey matter and WM following temporary focal occlusion (Schäbitz et al. 2000; McCracken et al. 2002).
White matter becomes intrinsically more vulnerable to ischaemia in older animals as the mechanisms of injury change as a function of age (Baltan et al. 2008). Thus, removal of extracellular Ca2+, or blockade of Ca2+ entry via reversal of the Na+–Ca2+ exchanger, improves WM function in young but not in older animals (Baltan et al. 2008). Indeed, Ca2+-free conditions worsen the recovery from the ischaemic insult in the latter, suggesting that Ca2+ release from intracellular Ca2+ stores may become more critical during ischaemia in aging WM (Baltan, 2009). In turn, ischaemic WM injury in older mice is predominately mediated by glutamate release via reverse glutamate transport and the ensuing activation of AMPA/kainate-type GluRs (Baltan et al. 2008). Intriguingly, blockade of NMDA receptors aggravates the outcome of ischaemia in older animals (Baltan et al. 2008).
Together, these findings clearly indicate that the mechanisms of ischaemic damage to WM involve Ca2+ dyshomeostasis induced by excessive glutamate signaling, which is age-dependent. This important finding has profound consequences for the development of optimized age-specific therapies for the treatment of brain damage after stroke. Furthermore, novel approaches using conditional knockout mice (e.g. lacking NMDA receptors in oligodendrocytes) are required to clarify the apparent opposing roles of these receptors in ischaemia-mediated WM damage in young and old brains.
Periventricular leukomalacia is the major neuropathological lesion in premature infants, and involves focal WM necrosis and subsequent hypomyelination. Its pathophysiology is multifactorial, and includes hypoxia–ischaemia-induced glutamate excitotoxicity, oxidative stress and inflammation (Volpe, 2009). Injury to oligodendrocyte progenitors caused in part by glutamate contributes to the pathogenesis of myelination disturbances in this illness (Back & Rivkees, 2004). In the immature human brain, the susceptibility of developing oligodendrocytes to hypoxia–ischaemia correlates with their expression of GluRs of the AMPA receptor subtypes on those cells in the immature human brain (Talos et al. 2006), and systemic administration of AMPA receptor antagonists attenuates injury in a rat model of PVL (Follett et al. 2004). In addition, developing oligodendrocytes also express NMDA receptors, and their blockade with memantine attenuates oligodendrocyte loss and prevents the long-term reduction in cerebral mantle thickness in experimental PVL (Manning et al. 2008).
Finally, ischaemic injury to axons is also a feature of PVL, and it occurs early in local and diffuse damage associated with this pathology (Haynes et al. 2008). Interestingly, experimental ischaemia in immature axons produces action potential failure and focal breakdown of the axolemma of small premyelinated axons at sites of contact with oligodendrocytes processes, which are also disrupted (Alix & Fern, 2009). Axon damage is prevented by NMDA and AMPA/kainate receptor blockers, suggesting that glutamate receptor-mediated injury to oligodendrocyte processes in contact with premyelinated axons precedes disruption of the underlying axon (Alix & Fern, 2009).
MS
The major demyelinating disease of the CNS is MS, which is the foremost disabling pathology among young adults. MS is a chronic, degenerative disease of the CNS, which is characterized by focal lesions with inflammation, demyelination, infiltration of immune cells, oligodendroglial death and axonal degeneration (Prineas et al. 2002). These cellular alterations are accompanied by neurological deficits, such as sensory disturbances, lack of motor coordination and visual impairment. It is widely accepted that the aetiology of this illness has autoimmune and inflammatory grounds, and that a derailment of the immune system leads to cell- and antibody-mediated attacks on myelin.
Both genetic and environmental factors contribute to MS susceptibility (Zamvil & Steinman, 2003). Among them, primary and/or secondary alterations in glutamate signalling cause excitotoxicity that contribute to MS pathology. Thus, numerous studies carried out in cellular and animal models of MS as well as in post mortem brain and in patients indicate that excitotoxicity mediated by Ca2+-permeable GluRs contributes to oligodendrocyte death, demyelination and tissue damage in MS (Matute et al. 2001; Srinivasan et al. 2005; Vallejo-Illarramendi et al. 2006). In particular, experimental autoimmune encephalomyelitis (EAE), an animal model that exhibits the clinical and pathological features of MS, is alleviated by AMPA and kainate receptor antagonists (Pitt et al. 2000; Smith et al. 2000). Indeed, mice deficient in the kainate receptor subunit GluK2 are less susceptible to EAE (Pérez-Samartín et al. 2009). Remarkably, blockade of these receptors in combination with anti-inflammatory agents is effective even at an advanced stage of unremitting EAE, as assessed by increased oligodendrocyte survival and remyelination, and corresponding decreased paralysis, inflammation, CNS apoptosis and axonal damage (Kanwar et al. 2004). Importantly, a recent genome-wide association screening study identified associated alleles in AMPA receptor genes in patients with MS showing highest levels of glutamate and brain volume loss (Baranzini et al. 2010). These findings provided a novel quantitative endophenotype that may contribute to clarify the pathophysiology of the heterogeneity of clinical expression in MS.
In contrast, blockade of NMDA receptors with MK-801 does not attenuate EAE symptoms (Matute, 2010), an event that calls into question the proposed relevance of NMDA receptors in demyelinating diseases (Bakiri et al. 2009). EAE experiments carried out in genetically modified mice lacking NMDA receptors specifically in oligodendrocytes may help clarify this issue.
Glutamate levels are increased in the human brain (Srinivasan et al. 2005) as a consequence of altered glutamate homeostasis (Vallejo-Illarramendi et al. 2006) and, thus, trigger excitotoxic destruction of oligodendrocytes and myelin as well as of axons (Domercq et al. 2005). Glutamate dyshomeostasis results from primary and/or secondary inflammation as a consequence of the autoimmune attack to the CNS and/or resulting from ongoing cell damage within the brain and spinal cord. Thus, activated microglia releases cytokines and free radicals that diminish glutamate uptake. This in turn elevates the extracellular levels of this transmitter, resulting in overactivation of Ca2+-permeable GluRs, which leads to oligodendrocyte excitotoxicity (Domercq et al. 2007). Moreover, activated microglia increases their expression of the glutamate-cystine exchanger, which contributes further to raising the levels of glutamate and its toxicity (Domercq et al. 2007). Other mechanisms accounting for glutamate dyshomeostasis include genetic variability in the promoter of the major glutamate transporter, EAAT2, which results in lower transporter expression (Pampliega et al. 2008). Finally, an additional component of the genetic background linking MS and deregulation of glutamate signalling may lie in a polymorphism in the Ca2+-permeable AMPA receptor subunit GluR3, an abundantly expressed subunit in oligodendrocytes, which is associated with a subgroup of patients responding to interferon beta therapy in MS (Comabella et al. 2009).
Glutamate at non-toxic concentrations also contributes to demyelinating pathology by inducing oligodendrocyte death via sensitization of these cells to complement attack (Alberdi et al. 2006). Intriguingly, complement toxicity is induced by activation of kainate, but not of AMPA, NMDA or metabotropic GluRs. Oligodendrocyte death by complement requires the formation of the membrane attack complex, which in turn increased membrane conductance, induced Ca2+ overload and mitochondrial depolarization as well as a rise in the level of reactive oxygen species (Alberdi et al. 2006). Sensitization by glutamate to complement attack may initiate MS lesions with massive oligodendrocytes apoptosis, as described earlier (Barnett & Prineas, 2004).
Spinal cord injury (SCI)
Traumatic injury to the CNS inevitably involves damage to WM, and causes primary mechanical destruction of glia and axons. In addition, secondary impairment of tissue occurs as a consequence of a prolonged pathological response involving chronic inflammation, microglial activation and astroglial scar formation, which can ultimately result in the development of a large cavity at the site of the lesion and persistent functional deficits (Dumont et al. 2001). In particular, acute traumatic SCI results in a devastating loss of neurological function below the level of injury. The pathobiology of SCI involves a primary mechanical insult to the spinal cord and activation of a delayed secondary cascade of events, which ultimately causes progressive degeneration of the spinal cord. Whereas cell death from the mechanical injury is predominated by necrosis, secondary injury events trigger a continuum of necrotic and apoptotic cell death mechanisms, which include glutamate excitotoxicity (Park et al. 2004; Baptiste & Fehlings, 2006). Thus, tissue destruction after SCI leads to the release of high levels of glutamate, which cause Ca2+-dependent excitotoxic damage to WM astrocytes, oligodendrocytes and myelin, but not to axons (Li & Stys, 2000). In particular, the density of oligodendrocytes declines in the next few hours after injury, and oligodendroglia show caspase-3 activation and other apoptotic features (Xu et al. 2008).
ATP signalling in WM glia and axons
Glial cells also express a heterogeneous repertoire of ATP receptor, including an ample variety of ionotropic (P2X) and metabotropic (P2Y) purinergic receptor subtypes (Table 1; Butt, 2006; Verkhratsky et al. 2009). ATP-gated P2X channels are formed by P2X1–P2X7 subunits and have marked Ca2+ permeability. Activation of P2X1 and P2X3 results in fast, rapidly desensitizing currents. In contrast, P2X7, and also P2X2 and P2X4, are capable of a conformational change, which results in larger pore diameter following prolonged exposure to ATP.
Astrocytes express most of the P2X and P2Y receptor subtypes whose activation mediates signalling through the astrocyte syncytium (James & Butt, 2002; Fields & Burnstock, 2006). In particular, activation of P2X7 receptors in astrocytes increases [Ca2+]i and causes the release of purines. Optic nerve astrocytes also express a variety of P2X receptors, which are highly permeable to Ca2+, and of P2Y receptors, which mobilize this cation from intracellular stores (James & Butt, 2002), as reported in grey matter astrocytes.
Cells of the oligodendrocyte lineage are endowed with P2X (Table 2) and P2Y receptors, which can act as mediators of axo-oligodendroglial communication related in myelination control. In particular, ATP induces a rise in cytosolic Ca2+ in oligodendrocytes by activating ionotropic P2X7 receptors (James & Butt, 2002; Matute et al. 2007b) and metabotropic P2Y receptors (Kirischuk et al. 1995; James & Butt, 2002). Moreover, mature oligodendrocytes of the optic nerve express most of the P2X receptor subtypes, with the P2X7 subtype being the most predominant; this is located in the oligodendrocyte soma and in the myelin sheath (James & Butt, 2002; Matute et al. 2007b). P2X receptors with higher affinity may be activated by ATP released during axonal electrical activity and from astrocytes (Butt, 2006). In contrast, the functional significance of lower affinity P2X7 receptors in oligodendrocytes is not known, as unusual high concentrations of ATP in the extracellular space are needed to activate them. However, in pathological conditions, ATP levels may rise sufficiently upon tissue damage to stimulate P2X7 receptors and therefore they may be relevant to acute and chronic injury to WM (Matute et al. 2007b). Indeed, sustained activation of P2X7 receptors in oligodendrocytes in vitro and in vivo results in overload of the cytosol with Ca2+, caspase-3 activation and chromatin condensation and cell death (Matute et al. 2007b).
Table 2.
Expression of P2X receptor subunit in oligodendrocytes*
| Subunit | P2X1 | P2X2 | P2X3 | P2X4 | P2X5 | P2X6 | P2X7 |
|---|---|---|---|---|---|---|---|
| Optic nerve cultures | Few cells | High | None | High | Few cells | Few cells | High |
| Optic nerve | None | Moderate | None | High | Low | None | High |
| Spinal cord | None | High | n.d. | High | n.d. | n.d. | High |
According to Matute et al. (2007b).
Microglia expresses several P2X and P2Y receptors, which act as sensors of astrocyte activity and trigger cytokine release (Färber & Kettenmann, 2006; Fields & Burnstock, 2006). In particular, microglial P2X7 receptors drive microglial activation and proliferation (Monif et al. 2009), and are functionally linked to the release of several substances including pro-inflammatory cytokines, such as interleukin-1β, which influence pathological processes and promote neurodegeneration (Färber & Kettenmann, 2006). Moreover, ATP is a potent immunomodulator controlling microglial recruitment and activation (Davalos et al. 2005; Nimmerjahn et al. 2005) by acting at P2Y12 receptors to induce microglial chemotaxis at early stages of the response to local CNS injury (Haynes et al. 2006).
Consequently, activation of purinoceptors and in particular P2X7 receptors by ATP can cause primary and/or secondary damage to WM, and is an important component of the glial response to injury in the CNS.
ATP signalling in WM pathology
Purinergic signalling is relevant to neuroinflammation, which arises in the CNS from a number of neurodegenerative diseases, as well as from ischaemic and traumatic brain injuries (Friedle et al. 2010). These pathologies give rise to increased levels of extracellular adenine nucleotides, which, via activation of a variety of cell surface P2 purinergic receptors, influence the inflammatory activities of responding immune cells. In particular, the P2X7 receptor potentiates the release of pro-inflammatory cytokines, such as interleukin (IL)-1β from microglia and induces cell death. Accordingly, neuroprotective properties of classical and novel selective and non-selective antagonists of P2X7 receptors have been observed in various cellular and animal models of CNS disorders to which excessive inflammatory activities contribute (reviewed in Friedle et al. 2010).
On the other hand, little is known about the relevance of ATP damage as a primary event in the pathophysiology of acute and chronic diseases. However, the release of ATP from dying cells may well contribute as a secondary mechanism to aggravate the extent of ongoing damage in numerous pathological conditions (Matute et al. 2007b).
White matter, like grey matter, is vulnerable to excessive ATP signalling. This section summarizes specific mechanisms by which ATP damages WM glia and axons, and its relevance to human diseases involving WM (Table 1).
Mechanisms of P2X7 receptor-mediated cell death
ATP, when in excess, is a potent endogenous toxin that can directly kill oligodendrocytes via activation of P2X7 receptors (Matute et al. 2007a,b;). ATP excitotoxicity in oligodendrocytes is Ca2+-dependent, and induces cell death by apoptosis or necrosis depending on the intensity of the insult. In addition, P2X7 receptor engagement activates several second messenger and enzyme cascades. In macrophages/monocytes and microglia, P2X7 receptor stimulation rapidly activates c-Jun N-terminal kinases 1 and 2, extracellular signal-regulated kinases, and p38 MAPK (reviewed in Skaper et al. 2010). Transcription factors such as nuclear factor-κB, nuclear factor of activated T-cells, cyclic AMP response element-binding protein and activator protein 1, whose activation and nuclear translocation are associated with the expression of inflammatory genes, are also activated by P2X7 receptors in microglia. Moreover, stimulation of P2X7 receptors involves Ca2+ signalling and increases protein tyrosine phosphorylation, ultimately leading to MAPK pathway activation (reviewed in Skaper et al. 2010).
Stroke and perinatal ischaemia
Earlier findings in experimental transient cerebral ischaemia suggested that P2X7 receptors are not primary mediators of experimentally induced neuronal death, while antagonists of the IL-1 receptor proved to be beneficial (Le Feuvre et al. 2003). However, P2X7 upregulation appears to be an event associated to ischaemia in experimental models (e.g. Cavaliere et al. 2004; reviewed in Sperlágh et al. 2006). In turn, brief oxygen deprivation causes a decline on brain intracellular ATP levels with a concomitant efflux of ATP into the extracellular space (Volonté et al. 2003), which can activate microglia in the vicinity of the site of injury (Honda et al. 2001). Consistent with that idea, ATP acting at P2X7 receptors in microglia is deleterious to cultured neurons as a consequence of oxidative stress, an effect that is absent in microglia lacking those receptors (Skaper et al. 2010). These observations suggest that P2X7 receptor activation on microglia is critical for microglia-mediated damage.
Perinatal ischaemia triggers, in addition to glutamate excitotoxicity (see above), lethal activation of P2X7 receptors in oligodendrocyte precursors (Wang et al. 2009). Thus, oligodendrocytes precursors express P2X7 receptors whose levels are reduced after oxygen–glucose deprivation in vitro and neonatal hypoxic–ischaemic injury, which suggested a role for this receptor in the pathophysiology of hypoxic–ischaemic brain injury (Wang et al. 2009). Indeed, ischaemia triggers in oligodendrocytes an inward current and cytosolic Ca2+ overload, which is partially mediated by P2X7 receptors (Domercq et al. 2010). P2X7 receptors in oligodendrocytes are activated, at least in part, by ATP released through pannexin channels opening during oxygen and glucose deprivation. This leads to mitochondrial depolarization as well as oxidative stress culminating in oligodendrocyte death, which is attenuated by P2X7 receptor antagonists, by the ATP-degrading enzyme apyrase and by blockers of pannexin hemichannels (Domercq et al. 2010). Likewise, these drugs ameliorate structural and functional damage to axonal tracts (Fig. 2; Domercq et al. 2010).
Fig. 2.
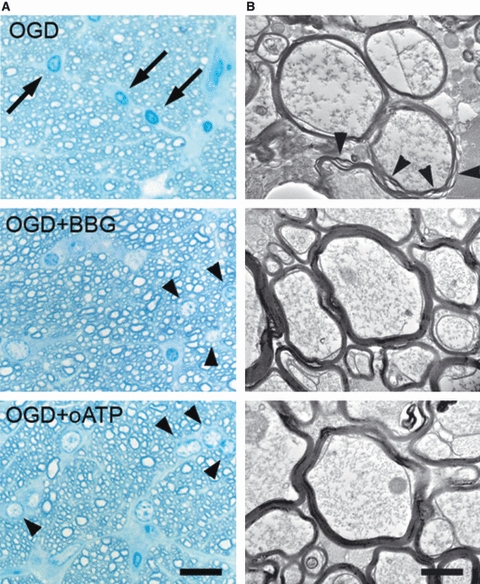
P2X7 inhibition protects myelin against ischaemic damage. (A) High-resolution analysis after 1 h oxygen-glucose deprivation (OGD) showed the presence of numerous pyknotic oligodendrocytes (arrows in left) and severe damage to myelin, with separation of the compact lamellae (arrowheads in right). In the presence of Brilliant Blue G (BBG; 50 nm) and oATP (1 mm), the number of pyknotic oligodendrocytes and the extent of myelin damage alter OGD was greatly diminished. Scale bar: 20 and 1 μm in left and right, respectively. (B) Graph summarizing the effect of BBG and oATP in oligodendroglial pyknosis and myelin damage. *P < 0.05; ***P < 0.001. Modified from Domercq et al. (2010).
These data indicate that ATP is released during ischaemia, and that the subsequent activation of P2X7 receptor appears to be relevant to WM demise during stroke.
MS
White matter damage, including oligodendrocyte death, demyelination and axonal damage are hallmarks of MS. As mentioned above, ATP signalling can trigger oligodendrocyte excitotoxicity via activation of Ca2+-permeable P2X7 purinergic receptors expressed by these cells. Importantly, sustained activation of P2X7 receptors in vivo causes lesions that are reminiscent of the major features of MS plaques, and treatment with P2X7 antagonists of chronic EAE reduces demyelination and ameliorates the associated neurological symptoms (Fig. 3; Matute et al. 2007b). These results are in line with data in P2X7 null mice showing that this deficiency suppresses the development of EAE (Sharp et al. 2008), and at odds with earlier observations indicating that the lack of P2X7 receptors aggravates EAE (Chen & Brosnan, 2006) and our own unpublished data. These apparent discrepancies may be caused by the different strains and knockout mice used, and to solve this issue will require the development of conditional P2X7 receptor knockout mice missing this transcript in specific glial cell populations. In turn, the availability of these technical resources would overcome the problem of interfering with the immune system whereby P2X7 receptors may be relevant intermediaries of the response to myelin antigens in EAE.
Fig. 3.
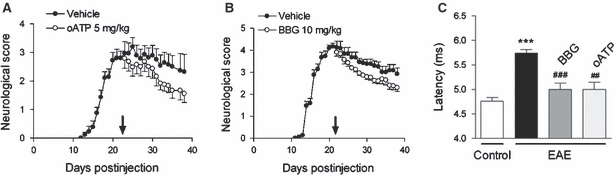
P2X7 antagonists ameliorate chronic experimental autoimmune encephalomyelitis (EAE)-associated neurological symptoms and restore normal axon conduction velocity. (A, B) Established chronic EAE induced in C57BL/6 mice by immunization with myelin oligodendrocyte glycoprotein (MOG) improves after treatment with oATP (P < 0.001) and BBG (P < 0.01) starting at 21 days after injection (arrows). (C) The increase in axon conduction latency in the corticospinal tract of mice with chronic EAE (***P < 0.001 vs. control, non-immunized animals) is greatly attenuated after treatment with oATP and Brilliant Blue G (BBG) (##P < 0.01 and ###P < 0.001, respectively, compared with MOG-immunized mice). Modified from Matute et al. (2007b).
In addition, P2X7 RNA and protein levels are elevated in normal-appearing axon tracts in patients with MS, suggesting that signalling through P2X7 receptors in oligodendroglia is enhanced in this disease, which may render this cell type more vulnerable to ATP dysregulation (Matute et al. 2007b). The increased expression of P2X7 receptors in axon tracts before lesions are formed indicates that this feature may constitute a risk factor associated with newly forming lesions in MS; this receptor subunit may thus prove to be a diagnostic and/or prognostic clinical biomarker for MS. On the other hand, blockade of ATP P2X7 receptors protects oligodendrocyte from dying; this property has therapeutic potential for halting the progression of tissue damage in MS.
SCI
ATP, which is also present at high levels in the extracellular space after traumatic CNS damage, may cause Ca2+-dependent gliotoxicity either directly by activating P2X receptors or after degradation to adenosine and subsequent activation of P1 purinergic receptors (Verkhratsky et al. 2009), but the later putative deleterious effects on WM have not yet been demonstrated to occur after SCI. Thus, SCI is associated with prolonged P2X7 receptor activation and ensuing neuronal excitotoxicity (Wang et al. 2004). Strikingly, systemic administration of a P2X7 antagonist permeable to the blood–brain barrier ameliorates the motor behaviour of animals who had been subjected to spinal cord contusion, indicating that neuroprotection after injury can preserve function (Peng et al. 2009). Oligodendrocyte preservation by P2X7 receptor blockade in those experimental conditions may also be critical to attenuate WM destruction and the ensuing motor and sensory deficits.
Concluding remarks
White matter can be damaged by aberrantly enhanced glutamate and ATP signalling in acute and chronic diseases, including MS, ischaemia and traumatic injury. Oligodendrocytes, the major cell type in WM, display great vulnerability to overactivation of ionotropic glutamate receptors and of P2X7 purinoceptors. In addition, it is conceivable that axons, which express AMPA and kainate receptors, may undergo direct glutamate excitotoxicity. The proper functioning of glutamate uptake is critical to prevent glutamate-induced damage to WM, and drugs that regulate the function and expression of GluTs have the potential to attenuate glutamate insults. On the other hand, the control of ATP release and degradation during acute and chronic inflammation and also at early stages of post-ischaemic and post-traumatic injury may prove therapeutic.
Signalling by other neurotransmitters including acetylcholine and noradrenaline is also potentially harmful to WM at least in acute experimental settings (Domingues et al. 2010). The underlying mechanisms of that damage are currently unknown, but the phenomena are consistent with the expression of nicotinic and adrenergic receptors in WM glia (Constantinou & Fern, 2009). Moreover, prolonged exposure to adenosine is also toxic to oligodendrocytes derived from optic nerve, an effect that is prevented by adenosine receptor antagonists (González-Fernández et al. 2010).
A deeper knowledge about the mechanisms leading to WM demise mediated by glutamate and ATP receptors, and by other neurotransmitters, will facilitate new pharmacological strategies for the treatment of CNS disorders in which WM is severely compromised.
Acknowledgments
Studies carried out in my laboratory were supported by the Gobierno Vasco, Universidad del País Vasco and Ministerio de Educación y Ciencia and by CIBERNED. Technical and human support provided by SGIker (UPV/EHU, MICINN, GV/EJ, ESF) is also gratefully acknowledged.
References
- Alberdi E, Sánchez-Gómez MV, Torre I, et al. Activation of kainate receptors sensitizes oligodendrocytes to complement attack. J Neurosci. 2006;26:3220–3228. doi: 10.1523/JNEUROSCI.3780-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alix JJ, Fern R. Glutamate receptor-mediated ischemic injury of premyelinated central axons. Ann Neurol. 2009;66:682–693. doi: 10.1002/ana.21767. [DOI] [PubMed] [Google Scholar]
- Back SA, Rivkees SA. Emerging concepts in periventricular white matter injury. Semin Perinatol. 2004;28:405–414. doi: 10.1053/j.semperi.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Bakiri Y, Burzomato V, Frugier G, et al. Glutamatergic signaling in the brain's white matter. Neuroscience. 2009;158:266–274. doi: 10.1016/j.neuroscience.2008.01.015. [DOI] [PubMed] [Google Scholar]
- Baltan S. Ischemic injury to white matter: an age-dependent process. Neuroscientist. 2009;15:126–133. doi: 10.1177/1073858408324788. [DOI] [PubMed] [Google Scholar]
- Baltan S, Besancon EF, Mbow B, et al. White matter vulnerability to ischemic injury increases with age because of enhanced excitotoxicity. J Neurosci. 2008;28:1479–1489. doi: 10.1523/JNEUROSCI.5137-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptiste DC, Fehlings MG. Pharmacological approaches to repair the injured spinal cord. J Neurotrauma. 2006;23:318–334. doi: 10.1089/neu.2006.23.318. [DOI] [PubMed] [Google Scholar]
- Baranzini SE, Srinivasan R, Khankhanian P, et al. Genetic variation influences glutamate concentrations in brains of patients with multiple sclerosis. Brain. 2010;133:2603–2611. doi: 10.1093/brain/awq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol. 2004;55:458–468. doi: 10.1002/ana.20016. [DOI] [PubMed] [Google Scholar]
- Belachew S, Gallo V. Synaptic and extrasynaptic neurotransmitter receptors in glial precursors’ quest for identity. Glia. 2004;48:185–196. doi: 10.1002/glia.20077. [DOI] [PubMed] [Google Scholar]
- Butt AM. Neurotransmitter-mediated calcium signalling in oligodendrocyte physiology and pathology. Glia. 2006;54:666–675. doi: 10.1002/glia.20424. [DOI] [PubMed] [Google Scholar]
- Cavaliere F, Amadio S, Sancesario G, et al. Synaptic P2X7 and oxygen/glucose deprivation in organotypic hippocampal cultures. J Cereb Blood Flow Metab. 2004;24:392–398. doi: 10.1097/00004647-200404000-00004. [DOI] [PubMed] [Google Scholar]
- Chen L, Brosnan CF. Exacerbation of experimental autoimmune encephalomyelitis in P2X7R-/- mice: evidence for loss of apoptotic activity in lymphocytes. J Immunol. 2006;176:3115–3126. doi: 10.4049/jimmunol.176.5.3115. [DOI] [PubMed] [Google Scholar]
- Comabella M, Craig DW, Morcillo-Suárez C, et al. Genome-wide scan of 500,000 single-nucleotide polymorphisms among responders and nonresponders to interferon beta therapy in multiple sclerosis. Arch Neurol. 2009;66:972–978. doi: 10.1001/archneurol.2009.150. [DOI] [PubMed] [Google Scholar]
- Constantinou S, Fern R. Conduction block and glial injury induced in developing central white matter by glycine, GABA, noradrenalin, or nicotine, studied in isolated neonatal rat optic nerve. Glia. 2009;57:1168–1177. doi: 10.1002/glia.20839. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;255:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Deng W, Wang H, Rosenberg PA, et al. Role of metabotropic glutamate receptors in oligodendrocyte excitotoxicity and oxidative stress. Proc Natl Acad Sci USA. 2004;101:7751–7756. doi: 10.1073/pnas.0307850101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmond DW. Cognition and white matter lesions. Cerebrovasc Dis. 2002;13:1353–1357. doi: 10.1159/000049151. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, et al. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Domercq M, Sánchez-Gómez MV, Areso P, et al. Expression of glutamate transporters in rat optic nerve oligodendrocytes. Eur J Neurosci. 1999;11:2226–2236. doi: 10.1046/j.1460-9568.1999.00639.x. [DOI] [PubMed] [Google Scholar]
- Domercq M, Etxebarria E, Pérez-Samartín A, et al. Excitotoxic oligodendrocyte death and axonal damage induced by glutamate transporter inhibition. Glia. 2005;52:36–46. doi: 10.1002/glia.20221. [DOI] [PubMed] [Google Scholar]
- Domercq M, Sánchez-Gómez MV, Sherwin C, et al. System xc- and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J Immunol. 2007;178:6549–6556. doi: 10.4049/jimmunol.178.10.6549. [DOI] [PubMed] [Google Scholar]
- Domercq M, Perez-Samartin A, Aparicio D, et al. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia. 2010;58:730–740. doi: 10.1002/glia.20958. [DOI] [PubMed] [Google Scholar]
- Domingues AM, Taylor M, Fern R. Glia as transmitter sources and sensors in health and disease. Neurochem Int. 2010;57:359–366. doi: 10.1016/j.neuint.2010.03.024. [DOI] [PubMed] [Google Scholar]
- Dumont RJ, Okonkwo DO, Verma S, et al. Acute spinal cord injury, part I: pathophysiological mechanisms. Clin Neuropharmacol. 2001;24:254–264. doi: 10.1097/00002826-200109000-00002. [DOI] [PubMed] [Google Scholar]
- Färber K, Kettenmann H. Purinergic signaling and microglia. Pflugers Arch. 2006;452:615–621. doi: 10.1007/s00424-006-0064-7. [DOI] [PubMed] [Google Scholar]
- Fern R, Möller MT. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci. 2000;20:34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Burnstock G. Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci. 2006;7:423–436. doi: 10.1038/nrn1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filley CM. The Behavioral Neurology of White Matter. New York: Oxford University Press; 2001. [Google Scholar]
- Follett PL, Deng W, Dai W, et al. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J Neurosci. 2004;24:4412–4420. doi: 10.1523/JNEUROSCI.0477-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler JH, McCracken E, Dewar D, et al. Intracerebral injection of AMPA causes axonal damage in vivo. Brain Res. 2003;991:104–112. doi: 10.1016/j.brainres.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Friedle SA, Curet MA, Watters JJ. Recent patents on novel P2X(7) receptor antagonists and their potential for reducing central nervous system inflammation. Recent Pat CNS Drug Discov. 2010;5:35–45. doi: 10.2174/157488910789753530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MP, Ransom BR. New light on white matter. Stroke. 2003;34:330–332. doi: 10.1161/01.str.0000054048.22626.b9. [DOI] [PubMed] [Google Scholar]
- González-Fernández E, Sánchez-Gómez MV, Matute C. Activation of adenosine receptors induces apoptosis in oligodendrocytes. Sixth Cajal Winter Conference, Role of Glia in Health and Disease. 2010:5. [Google Scholar]
- Haynes RL, Billiards SS, Borenstein NS, et al. Diffuse axonal injury in periventricular leukomalacia as determined by apoptotic marker fractin. Pediatr Res. 2008;63:656–661. doi: 10.1203/PDR.0b013e31816c825c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1463–1464. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Honda S, Sasaki Y, Ohsawa K, et al. Extracellular ATP and ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci. 2001;21:1975–1982. doi: 10.1523/JNEUROSCI.21-06-01975.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Bergles DE. Glutamate transporters bring competition to the synapse. Curr Opin Neurobiol. 2004;14:346–352. doi: 10.1016/j.conb.2004.05.007. [DOI] [PubMed] [Google Scholar]
- James G, Butt AM. P2Y and P2X purinoceptor mediated Ca2+ signalling in glial cell pathology in the central nervous system. Eur J Pharmacol. 2002;447:247–260. doi: 10.1016/s0014-2999(02)01756-9. [DOI] [PubMed] [Google Scholar]
- Kanwar JR, Kanwar RK, Krissansen GW. Simultaneous neuroprotection and blockade of inflammation reverses autoimmune encephalomyelitis. Brain. 2004;127:1313–1331. doi: 10.1093/brain/awh156. [DOI] [PubMed] [Google Scholar]
- Káradóttir R, Cavelier P, Bergersen LH, et al. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. 2005;438:1162–1166. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Steinhäuser C. Receptors for neurotransmitters and hormones. In: Kettenmann H, Ransom BR, editors. Neuroglia. 2nd edn. New York: Oxford University Press; 2005. pp. 131–145. [Google Scholar]
- Kirischuk S, Scherer J, Kettenmann H, et al. Activation of P2-purinoreceptors triggered Ca2+ release from InsP3-sensitive internal stores in mammalian oligodendrocytes. J Physiol. 1995;483:41–57. doi: 10.1113/jphysiol.1995.sp020566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuckley M, Capetillo-Zarate E, Dietrich D. Vesicular glutamate release from axons in white matter. Nat Neurosci. 2007;10:311–320. doi: 10.1038/nn1850. [DOI] [PubMed] [Google Scholar]
- Le Feuvre RA, Brough D, Touzani O, et al. Role of P2X7 receptors in ischemic and excitotoxic brain injury in vivo. J Cereb Blood Flow Metab. 2003;23:381–384. doi: 10.1097/01.WCB.0000048519.34839.97. [DOI] [PubMed] [Google Scholar]
- Li S, Stys PK. Mechanisms of ionotropic glutamate receptor-mediated excitotoxicity in isolated spinal cord white matter. J Neurosci. 2000;20:1190–1198. doi: 10.1523/JNEUROSCI.20-03-01190.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Mealing GA, Morley P, et al. Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na+-dependent glutamate transport. J Neurosci. 1999;19:RC16. doi: 10.1523/JNEUROSCI.19-14-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning SM, Talos DM, Zhou C, et al. NMDA receptor blockade with memantine attenuates white matter injury in a rat model of periventricular leukomalacia. J Neurosci. 2008;28:6670–6678. doi: 10.1523/JNEUROSCI.1702-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C. Characteristics of acute and chronic kainate excitotoxic damage to the optic nerve. Proc Natl Acad Sci USA. 1998;95:10229–10234. doi: 10.1073/pnas.95.17.10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C. Oligodendrocyte NMDA receptors: a novel therapeutic target. Trends Mol Med. 2006;12:289–292. doi: 10.1016/j.molmed.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Matute C. Calcium dyshomeostasis in white matter pathology. Cell Calcium. 2010;47:150–157. doi: 10.1016/j.ceca.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Matute C, Alberdi E, Domercq M, et al. The link between excitotoxic oligodendroglial death and demyelinating diseases. Trends Neurosci. 2001;24:224–230. doi: 10.1016/s0166-2236(00)01746-x. [DOI] [PubMed] [Google Scholar]
- Matute C, Domercq M, Sánchez-Gómez MV. Glutamate-mediated glial injury: mechanisms and clinical importance. Glia. 2006;53:212–224. doi: 10.1002/glia.20275. [DOI] [PubMed] [Google Scholar]
- Matute C, Alberdi E, Domercq M, et al. Excitotoxic damage to white matter. J Anat. 2007a;210:693–702. doi: 10.1111/j.1469-7580.2007.00733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C, Torre I, Perez-Cerdá F, et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci. 2007b;27:9525–9533. doi: 10.1523/JNEUROSCI.0579-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken E, Fowler JH, Dewar D, et al. Grey matter and white matter ischemic damage is reduced by the competitive AMPA receptor antagonist, SPD 502. J Cereb Blood Flow Metab. 2002;22:1090–1097. doi: 10.1097/00004647-200209000-00006. [DOI] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, et al. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–992. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- Monif M, Reid CA, Powell KL, et al. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci. 2009;29:3781–3791. doi: 10.1523/JNEUROSCI.5512-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Malek S, Coderre E, et al. Complex interplay between glutamate receptors and intracellular Ca2+ stores during ischaemia in rat spinal cord white matter. J Physiol. 2006;577:191–204. doi: 10.1113/jphysiol.2006.116798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouardouz M, Coderre E, Basak A, et al. Glutamate receptors on myelinated spinal cord axons: I. GluR6 kainate receptors. Ann Neurol. 2009a;65:151–159. doi: 10.1002/ana.21533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouardouz M, Coderre E, Zamponi GW, et al. Glutamate receptors on myelinated spinal cord axons: II. AMPA and GluR5 receptors. Ann Neurol. 2009b;65:160–166. doi: 10.1002/ana.21539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampliega O, Domercq M, Villoslada P, et al. Association of an EAAT2 polymorphism with higher glutamate concentration in relapsing multiple sclerosis. J Neuroimmunol. 2008;195:194–198. doi: 10.1016/j.jneuroim.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Pantoni L, Garcia JH, Gutierrez JA. Cerebral white matter is highly vulnerable to ischemia. Stroke. 1996;27:1641–1646. doi: 10.1161/01.str.27.9.1641. [DOI] [PubMed] [Google Scholar]
- Park E, Velumian AA, Fehlings MG. The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J Neurotrauma. 2004;21:754–774. doi: 10.1089/0897715041269641. [DOI] [PubMed] [Google Scholar]
- Peng W, Cotrina ML, Han X, et al. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci USA. 2009;106:12489–12493. doi: 10.1073/pnas.0902531106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Samartín A, Pérez-Cerdá F, Matute C. Methodological tools for research in multiple sclerosis. Eur J Anat. 2009;13(Suppl.1):27. [Google Scholar]
- Pitt D, Nagelmeier IE, Wilson HC, et al. Glutamate uptake by oligodendrocytes: Implications for excitotoxicity in multiple sclerosis. Neurology. 2003;61:1113–1120. doi: 10.1212/01.wnl.0000090564.88719.37. [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- Prineas JW, McDonald WI, Franklin RJM. Demyelinating diseases. In: Graham DI, Lantos PL, editors. Greenfield's Neuropathology. Vol. 2. London: Arnold; 2002. pp. 471–550. [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, et al. Knock out of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 2005;438:1167–1171. doi: 10.1038/nature04301. [DOI] [PubMed] [Google Scholar]
- Sánchez-Gómez MV, Alberdi E, Ibarretxe G, et al. Caspase-dependent and caspase-independent oligodendrocyte death mediated by AMPA and kainate receptors. J Neurosci. 2003;23:9519–9528. doi: 10.1523/JNEUROSCI.23-29-09519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäbitz WR, Li F, Fisher M. The N-methyl-D-aspartate antagonist CNS 1102 protects cerebral gray and white matter from ischemic injury following temporary focal ischemia in rats. Stroke. 2000;31:1709–1714. doi: 10.1161/01.str.31.7.1709. [DOI] [PubMed] [Google Scholar]
- Sharp AJ, Polak PE, Simonini V, et al. P2X7 deficiency suppresses development of experimental autoimmune encephalomyelitis. J Neuroinflammation. 2008;8:33. doi: 10.1186/1742-2094-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD, Debetto P, Giusti P. The P2X7 purinergic receptor: from physiology to neurological disorders. FASEB J. 2010;24:337–345. doi: 10.1096/fj.09-138883. [DOI] [PubMed] [Google Scholar]
- Smith T, Groom A, Zhu B, et al. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6:62–66. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- Sperlágh B, Vizi ES, Wirkner K, et al. P2X7 receptors in the nervous system. Prog Neurobiol. 2006;78:327–346. doi: 10.1016/j.pneurobio.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Sailasuta N, Hurd R, et al. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain. 2005;128:1016–1025. doi: 10.1093/brain/awh467. [DOI] [PubMed] [Google Scholar]
- Stys PK. White matter injury mechanisms. Curr Mol Med. 2004;4:113–130. doi: 10.2174/1566524043479220. [DOI] [PubMed] [Google Scholar]
- Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci. 2005;233:3–13. doi: 10.1016/j.jns.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Swanson CJ, Bures M, Johnson MP, et al. Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat Rev Drug Discov. 2005;4:131–144. doi: 10.1038/nrd1630. [DOI] [PubMed] [Google Scholar]
- Talos DM, Follett PL, Folkerth RD, et al. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. Human cerebral white matter and cortex. J Comp Neurol. 2006;497:61–77. doi: 10.1002/cne.20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekkök SB, Goldberg MP. AMPA/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci. 2001;21:4237–4248. doi: 10.1523/JNEUROSCI.21-12-04237.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo-Illarramendi A, Domercq M, Pérez-Cerdá F, et al. Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol Dis. 2006;21:154–164. doi: 10.1016/j.nbd.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Kirchhoff F. NMDA receptors in glia. Neuroscientist. 2007;13:28–37. doi: 10.1177/1073858406294270. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Orkand RK, Kettenmann H. Glial calcium: homeostasis and signaling function. Physiol Rev. 1998;78:99–141. doi: 10.1152/physrev.1998.78.1.99. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Krishtal OA, Burnstock G. Purinoceptors on neuroglia. Mol Neurobiol. 2009;39:190–208. doi: 10.1007/s12035-009-8063-2. [DOI] [PubMed] [Google Scholar]
- Volonté C, Amadio S, Cavaliere F, et al. Extracellular ATP and neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2003;2:403–412. doi: 10.2174/1568007033482643. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009;8:110–124. doi: 10.1016/S1474-4422(08)70294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Wang X, Arcuino G, Takano T, et al. P2X7 receptor inhibition improves recovery after spinal cord injury. Nat Med. 2004;10:821–827. doi: 10.1038/nm1082. [DOI] [PubMed] [Google Scholar]
- Wang LY, Cai WQ, Chen PH, et al. Downregulation of P2X7 receptor expression in rat oligodendrocyte precursor cells after hypoxia ischemia. Glia. 2009;57:307–319. doi: 10.1002/glia.20758. [DOI] [PubMed] [Google Scholar]
- Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. 2001;50:169–180. doi: 10.1002/ana.1077. [DOI] [PubMed] [Google Scholar]
- Xu GY, Liu S, Hughes MG, et al. Glutamate-induced losses of oligodendrocytes and neurons and activation of caspase-3 in the rat spinal cord. Neuroscience. 2008;153:1034–1047. doi: 10.1016/j.neuroscience.2008.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamvil SS, Steinman L. Diverse targets for intervention during inflammatory and neurodegenerative phases of multiple sclerosis. Neuron. 2003;38:685–688. doi: 10.1016/s0896-6273(03)00326-x. [DOI] [PubMed] [Google Scholar]
- Zhang K, Sejnowski TJ. A universal scaling law between gray matter and white matter of cerebral cortex. Proc Natl Acad Sci USA. 2000;97:5621–5626. doi: 10.1073/pnas.090504197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziskin JL, Nishiyama A, Rubio M, et al. Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci. 2007;10:321–330. doi: 10.1038/nn1854. [DOI] [PMC free article] [PubMed] [Google Scholar]