

Abstract
Aims/hypothesis
The role of TNF-α in impaired wound healing in diabetes was examined by focusing on fibroblasts.
Methods
Small excisional wounds were created in the db/db mice model of type 2 diabetes and normoglycaemic littermates, and in a streptozotocin-induced type 1 diabetes mouse model and control mice. Fibroblast apoptosis was measured by the TUNEL assay, proliferation by detection of proliferating cell nuclear antigen, and forkhead box O1 (FOXO1) activity by DNA binding and nuclear translocation. TNF-α was specifically inhibited by pegsunercept.
Results
Diabetic wounds had increased TNF-α, fibroblast apoptosis, caspase-3/7 activity and activation of the pro-apoptotic transcription factor FOXO1, and decreased proliferating cell nuclear antigen positive fibroblasts (p<0.05). TNF-α inhibition improved healing in the diabetic mice and increased fibroblast density. This may be explained by a decrease in fibroblast apoptosis and increased proliferation when TNF-α was blocked (p <0.05). Although decreased fibroblast proliferation and enhanced FOXO1 activity were investigated in type 2 diabetes, they may also be implicated in type 1 diabetes. In vitro, TNF-α enhanced mRNA levels of gene sets related to apoptosis and Akt and p53 but not mitochondrial or cell-cycle pathways. FOXO1 small interfering RNA reduced gene sets that regulate apoptosis, Akt, mitochondrial and cell-cycle pathways. TNF-α also increased genes involved in inflammation, cytokine, Toll-like receptor and nuclear factor-kB pathways, which were significantly reduced by FOXO1 knockdown.
Conclusions/interpretation
These studies indicate that TNF-α dysregulation in diabetic wounds impairs healing, which may involve enhanced fibroblast apoptosis and decreased proliferation. In vitro, TNF-α induced gene sets through FOXO1 that regulate a number of pathways that could influence inflammation and apoptosis.
Keywords: Diabetes, Fibroblast, FOXO, Nuclear translocation, PCNA, Proliferation, TNF-α
Introduction
A serious complication of diabetes is impaired healing, which can lead to diminished physical activity and in some cases chronic wounds and limb amputation [1–4]. Multiple factors are likely to contribute to deficient healing in patients with diabetes. They include an altered host response, diminished anti-bacterial defences, prolonged inflammation, altered protease activity, a tendency for vascular abnormalities, the generation of an inadequate number of cells to accomplish rapid and robust healing, decreased growth factor production, a failure to form a sufficient amount of extracellular matrix, and alterations in apoptosis that may interfere with healing by decreasing the number of cells that participate in new tissue formation [2, 5–13].
Wound healing is a complex process that involves inflammation, formation of granulation tissue, production of new structures and tissue remodelling [14, 15]. These processes are regulated by cytokines and growth factors and modulated by systemic conditions such as diabetes [4, 11]. A critical component of a vigorous healing response is the generation of a sufficient number of cells to participate in repair. Processes that interfere with the ability to produce enough cells, such as inappropriately high levels of fibroblast apoptosis, may limit healing [16, 17]. On the other hand, apoptosis of cells at later stages of healing is important in removing cells that are no longer needed [18–20].
A potential mechanism through which diabetes may increase apoptosis is the excessive production of the TNF-α pleiotropic cytokine that plays an important role in inflammation and immunity [21]. Overproduction of TNF-α is thought to contribute to a number of disease processes associated with persistent inflammation and tissue destruction [22–24]. TNF-α levels are elevated in non-healing ulcers [4] and associated with impaired healing in type 2 diabetes mouse models [25]. Chronic elevation of TNF-α has been shown to impair cutaneous wound healing and to cause a decrease in collagen production, while exogenous TNF-α results in a decrease in wound strength [26, 27]. Moreover, TNF-α is associated with the aetiological processes in both type 1 and type 2 diabetes [28, 29], as well as diabetic complications. For example, high levels of TNF-α are involved in diabetic retinopathy and nephropathy associated with both forms of diabetes [30, 31]. To investigate the contribution of TNF-α to diminished wound healing in diabetes, we created small wounds in db/db and matched normoglycaemic littermates, and in a streptozotocin-induced mouse model of type 1 diabetes and control mice.
The results indicate that diabetes enhanced TNF-α levels, decreased fibroblast density and proliferation and increased fibroblast apoptosis and activation of the pro-apoptotic transcription factor, forkhead box O1 (FOXO1). When TNF-α is blocked there is improved healing, increased fibroblast proliferation and reduced apoptosis and greater fibroblast density in the diabetic group. Fibroblast proliferation and FOXO1 activity were investigated only in a type 2 diabetes model. However, the same pathways may be involved in type 1 diabetes. In vitro studies were carried out to examine the effect of TNF-α on fibroblasts and the role of the transcription factor FOXO1 in microarray experiments in conjunction with RNA interference (RNAi). Gene set enrichment analysis (GSEA) of mRNA profiling results indicates that FOXO1 plays an important role in both pro-apoptotic and pro-inflammatory gene expression and may thereby contribute to impaired healing in diabetes. The latter represents a previously unrecognised role of FOXO1.
Methods
Preparation of the animals
Genetically diabetic C57BL/KsJ-Lepr−/− (db/db) mice and their non-diabetic littermates, C57BL/KsJ-Lepr−/+ (db/db+), were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). They exhibit many aspects of diabetes impaired wound healing seen in humans [6, 8, 10, 25, 32]. Eight-week-old male CD-1 mice (Charles River Laboratories, Wilmington, MA, USA) were rendered diabetic by multiple low dose i.p. injection of streptozotocin (40 mg/kg; Sigma, St Louis, MO, USA) in 10 mmol citrate buffer daily for 5 days, as we have previously described [33]. Control mice were treated identically with vehicle alone, 10 mmol citrate buffer. Mice were considered to be hyperglycaemic when serum glucose levels were >13.875 mmol/l. Experiments were started when mice had been hyperglycaemic for at least 3 weeks, with typically n=6 mice per group. Two excisional wounds of 1.5 mm were made in the scalp with a 1.5 mm disposable biopsy punch, as we have previously described [34, 35]. Small wounds heal by connective tissue fill and epithelial bridging rather than by contraction, do not require an occlusal dressing and rarely become infected [35, 36]. Pegsunercept, generously provided by Amgen (Thousand Oaks, CA, USA), was administered by i.p. injection (5 mg/kg). Pegsunercept is a recombinant soluble TNF receptor type 1 (TNF-R1) linked to polyethylene glycol, which has been shown to be specific, highly efficacious and minimally toxic [37]. It has been reported that a pegsunercept-related molecule, etanercept, binds to soluble TNF-α but has reduced avidity for membrane-integrated forms of TNF-α [38]. However, studies have also reported that TNF-R1-based inhibitors also block membrane-bound TNF-α [39, 40]. To avoid interfering with the early inflammatory events, pegsunercept was administered starting 2 days after wounding and on days 5 and 8 based on its half-life of approximate 4 days [37]. Mice were killed at the indicated time points by CO2 overdose and decapitation. All animal procedures were approved by the Institutional Animal Care and Use Committee, Boston University Medical Centre.
Histological analysis
The scalp and attached calvarial bone were fixed in 4% (wt/vol.) paraformaldehyde and decalcified in Immunocal (Decal Chemical Corporation, Congreve, NY, USA). Five micrometre sagittal paraffin sections were prepared. The epithelial and connective tissue gaps and the per cent connective tissue fill at the wound site were measured with computer-assisted image analysis in haematoxylin and eosin-stained sections taken at the centre of each lesion. Polymorphonuclear leucocytes (PMNs) were identified by their characteristic appearance in haematoxylin and eosin-stained sections at ×1,000 magnification. Fibroblasts were identified by immunohistochemistry using an antibody against heat-shock protein 47 (HSP47; Stressgen, Ann Arbor, MI, USA), a specific fibroblast marker [41]. There was no immunostaining with matched control IgG (data not shown). In some experiments, immunohistochemistry was combined with a TUNEL assay in situ by means of an ApopTag Peroxidase In Situ Kit (Chemicon, Temecula, CA, USA). HSP47 was localised by detection with a biotin-labelled secondary antibody and avidin–biotin–alkaline phosphatase complex with Vector Red substrate (Vector Laboratories, Burlingame, CA, USA). TUNEL-positive cells were detected by 3,3′-diaminobenzidine tetrahydrochloride-nickel (Vector Laboratories). TNF-α-positive cells were identified by immunohistochemistry using an antibody specific for TNF-α (Santa Cruz Biothechnology, Santa Cruz, CA, USA). There was no immunostaining with matched control IgG (data not shown). To analyse proliferating fibroblasts, specimens were double-immunostained for HSP47 and proliferating cell nuclear antigen (PCNA) (Santa Cruz Biotechnology) with secondary detection by avidin–biotin–peroxidase complex and avidin–biotin–alkaline phosphatase complex (Vector Laboratories). Chromogens 3,3′-diaminobenzidine and Vector Red were from Vector Laboratories. Cell counts were made at ×1,000 magnification. All counting and measurements were done under blinded conditions by one examiner and confirmed by a second independent examiner, with both examiners calibrated by a trained pathologist.
Confocal laser scanning microscopy
FOXO1 nuclear translocation of fibroblasts was detected by three-colour confocal laser scanning microscopy with antibodies specific for FOXO1 and HSP47 combined with nuclear staining with 7-aminoactinomycin D (7-AAD; Molecular Probes, Eugene, OR, USA). FOXO1 antibody was detected by biotinylated secondary antibody followed by Alexa Fluor 488-conjugated streptavidin (Molecular Probes) and HSP47 antibody was detected by Alexa Fluor 514-conjugated secondary antibody (Molecular Probes). For better colour contrast, HSP47 staining was displayed by a blue colour using Carl Zeiss LSM image software. Immunofluorescence images were captured by confocal laser scanning microscopy (Axiovert-100M; Carl Zeiss; Thornwood, NY, USA) of healing connective tissue. Fibroblasts with FOXO1 in the nuclear compartment were counted by comparing individual images with merged images (see Electronic supplementary material [ESM] Fig. 1). For each antibody, a matched control antibody was used as a negative control and no immunostaining was detected (data not shown).
Biochemical analysis of wounded tissue
Tissue at the wounded site was harvested using a 2.0 mm punch biopsy and frozen in liquid nitrogen. Specimens from a group were combined, placed in cytoplasmic lysis buffer containing protease inhibitors (Pierce, Rockford, IL, USA) and disrupted using Fast Prep (Q-Biogene, Solon, OH, USA). The nuclei were separated from cytoplasmic proteins by centrifugation. Protein in each lysate was determined using a BCA protein assay kit (Pierce). TNF-α levels were quantified using an ELISA kit (R&D Systems, Minneapolis, MN, USA). Caspase-3/7 activity was measured by a luminometric kit (Promega, Madison, WI, USA). FOXO1 activation was measured in nuclear protein extracts obtained by lysis of the nuclear pellet. DNA binding activity was measured by ELISA (Active Motif, Carlsbad, CA, USA). To quantify the mRNA levels, total RNA was extracted using an RNeasy total RNA isolation kit (Qiagen, Valencia, CA, USA) and a QIAshredder spin column (Qiagen). cDNA was prepared using a reverse transcription kit (Applied Biosystems, Foster City, CA, USA). TaqMan primer and probe sets for murine Tnf-α (also known as Tnfa) and caspase-3 (Casp-3) were purchased from Applied Biosystems. Results were normalised with respect to an 18S ribosomal primer and probe set purchased from Applied Biosystems. Each value is the mean of three independent assays ± SEM. One-way ANOVA was used to determine significance between groups at the p<0.05 level.
mRNA profiling and FOXO1 siRNA
Primary human adult dermal fibroblasts were purchased from Cambrex (Walkers-ville, MD, USA) and cultured in Dulbecco’s modified Eagle’s medium (Cambrex) supplemented with 10% (vol./vol.) FBS. All assays were performed under serum-free conditions. Fibroblasts were transfected with silencing FOXO1 small interfering RNA (siRNA) (r[GCCCUGGCUCUCACAGCAA]d[TT]) or non-silencing siRNA (r[GATGGCCTCTACTTTACCC]dTT) for 48 h followed by TNF-α (20 ng/ml) or alone for 6 h as described previously [42]. Some cells were incubated serum-free for 24 h and then stimulated with TNF-α (20 ng/ml) for 6 h. Total RNA was isolated using an RNeasy kit (Qiagen, Valencia, CA, USA). mRNA profiling was performed using a GeneChip Human Genome U133 Plus 2.0 microarray (Affymetrix, Santa Clara, CA, USA). Reverse transcription and real-time quantitative PCR (qPCR) of selected genes was performed to validate microarray result using Taqman reverse transcription reagents and primers and probe sets (Applied Biosystems). Results were normalised with respect to the value obtained for the housekeeping gene, RPL32, a ribosomal protein. GSEA was performed using GSEA software (www.broadinstitute.org/gsea, accessed 26 August 2009) as described previously [43]. GSEA determines whether an a priori defined set of genes shows statistically significant differences between two groups. GSEA was performed using gene sets downloaded from the GSEA websites. No data collapsing or filtering was performed.
Statistical analysis
For histological sections, eight to 12 fields were typically examined per section. One-way ANOVA was used to determine significance between groups. For real-time qPCR Student’s t test was used to determine significance between groups. Significance was set at the p<0.05 level.
Results
A small excisional wound was placed in the scalp of normoglycaemic and diabetic db/db mice. On day 4, a time point at which there was little wound closure in either diabetic or normoglycaemic groups, the diabetic group revealed threefold higher mRNA and protein levels of TNF-α compared with the normoglycaemic mice (p<0.05; Fig. 1a, b). We next examined the caspase-3 activity, apoptosis and FOXO1 DNA binding activity in healing wounds. The level of caspase-3/7 activity in healing wound tissue was almost fourfold higher in diabetic mice than in normoglycaemic mice during wound healing (Fig. 1c). When fibroblast apoptosis was measured there was a 2.5-fold higher level in the diabetic group compared with the normoglycaemic mice (Fig. 1d). Since TNF-α induces fibroblast pro-apoptotic activity through induction of the transcription factor FOXO1 [42], we measured FOXO1 DNA binding activity in healing tissue. Diabetes increased FOXO1 DNA binding activity twofold compared with wounds in normoglycaemic animals (Fig. 1e). Thus, diabetic healing in vivo is characterised by a number of variables that have been shown to be stimulated by TNF-α in vitro, including diabetes-enhanced fibroblast apoptosis, caspase-3/7 activity and FOXO1 activation.
Fig. 1.

Diabetes increases TNF-α mRNA and protein levels, fibroblast apoptosis, caspase-3/7 activity and FOXO1 DNA binding in healing wounds. A 1.5 mm excisional full thickness wound was created in the scalp of db/db diabetic (black bars) and normoglycaemic mice (white bars). a Total RNA was isolated and TNF-α mRNA levels were measured by real-time PCR. b TNF-α protein levels were measured in protein extracts of healing wounds by ELISA and are representative of two experiments. c Caspase activity was measured in cytoplasmic extracts with a luminometric kit. d, e The wounded tissue was recovered with a 2.0 mm dermal microtome and cytoplasmic and nuclear proteins were extracted. d Apoptotic fibroblasts were measured by the TUNEL assay 4 days after wounding (n=6 per group). Prior to wounding there was little to no apoptosis detected (data not shown). e FOXO1 activity was measured in extracted nuclear proteins obtained 4 days after wounding using a transcription factor activation ELISA. For a, c and e each value is the mean±SEM of three separate experiments. *Significant difference between normoglycaemic and diabetic mice (p<0.05)
To examine the role of TNF-α in impaired healing, mice were treated with the specific inhibitor pegsunercept or vehicle alone. On day 5, the gap between the edges of the healing connective tissue and healing epithelium was approximately 1.7-fold larger in the diabetic mice (Fig. 2a, c and ESM Fig. 2a, b). In the normoglycaemic mice, new connective tissue filled approximately 50% of the original wound site (Fig. 2e), whereas it was approximately 25% filled in the diabetic animals. For both normoglycaemic and diabetic groups, treatment with TNF-α inhibitor had no effect on healing on day 5. From day 5 to day 9 after wounding there was relatively little improvement in the diabetic wounds, while the normoglycaemic animals had healed extensively (Fig. 2 and ESM Fig. 2). Treatment with pegsunercept had a significant effect on diabetic healing at this time point. Vehicle-treated diabetic mice had gaps between epithelial and connective tissue edges of the wounds that were approximately 70% larger than in the pegsunercept-treated diabetic mice (Fig. 2b, d; p<0.05). Pegsunercept also increased the percentage of the wound filled with new connective tissue by approximately 50% in the diabetic mice and had no effect on normoglycaemic healing (Fig. 2f; p<0.05).
Fig. 2.

Healing of excisional wounds is impaired in db/db mice and improved by inhibition of TNF-α. A 1.5 mm excisional wound was placed in the scalp of db/db mice and their non-diabetic littermates. Mice were treated by i.p. injection of pegsunercept (5 mg/kg) or vehicle alone starting 2 days after wounding and killed 5 days (a, c, e) or 9 days (b, d, f) after wounding. The gap between the epithelium (a, b), connective tissue (c, d) and the per cent fill of the wound site by connective tissue (e, f) was assessed by image analysis of haematoxylin and eosin-stained sections at the centre of each lesion. Each value is the mean ± SE of n=6–7 mice. *p<0.05 between pegsunercept- and vehicle-treated groups. White bars, vehicle-injected normoglycaemic mice; diagonal hatched bars, pegsunercept-injected normoglycaemic mice; black bars, vehicle-injected db/db diabetic mice; horizontal hatched bars, pegsunercept-injected db/db mice
Experiments were performed to assess the level of fibroblast apoptosis with and without TNF-α inhibitor. Day 9 was examined in db/db mice since pegsunercept had a significant impact on healing at this time point. The percentage of apoptotic fibroblasts in diabetic healing wounds was fivefold higher compared with the normoglycaemic group (Fig. 3a and ESM Fig. 3). After inhibition of TNF-α, fibroblast apoptosis was reduced by >50% in both the diabetic and non-diabetic mice, although the absolute decrease in the diabetic mice was much greater (p<0.05; Fig. 3a). The number of fibroblasts per mm2 was almost twofold higher in the normoglycaemic compared with the diabetic (Fig. 3c, ESM Fig. 4; p<0.05). Fibroblast density in diabetic mice treated with TNF-α inhibitor increased by 78% compared with vehicle-treated mice (p<0.05).
Fig. 3.

TNF-α inhibition reduces fibroblast apoptosis and increases fibroblast numbers in diabetic mice. Excisional wounds were created in the scalp of db/db (a, c, e) and streptozotocin-induced diabetic mice (b, d) or their matched normoglycaemic controls. Mice were treated with pegsunercept or vehicle alone starting on day 2 as described in Fig. 2 and killed 9 days after wounding (a, c) or 5 days after wounding (b, d, e). The number of fibroblasts per mm2 was assessed by immunohistochemistry using an antibody specific for HSP47, a fibroblast marker (c, d). There was no immunostaining with matched control IgG (data not shown). Apoptotic fibroblasts were measured by double staining as TUNEL-positive and HSP47-positive (a, b). Proliferative fibroblasts were identified as both HSP47- and PCNA-positive by immunohistochemistry (e). Each value is the mean ± SEM for n=5–7 mice per group. *p<0.05 between pegsunercept- and vehicle-treated groups. a, c, e White bars, diagonal hatched bars, black bars and horizontal hatched bars indicate vehicle-injected normoglycaemic mice, pegsunercept-injected normoglycaemic mice, vehicle-injected db/db diabetic mice and pegsunercept-injected db/db mice, respectively. b, d White bars, black bars and horizontal hatched bars indicate normoglycaemic mice, streptzotocin-induced type 1 diabetic mice with vehicle injection, and pegsunercept-injected streptzotocin-induced diabetic mice, respectively
The impact of diabetes on fibroblast apoptosis and density was also measured in streptozotocin-induced diabetic mice. The percentage of apoptotic fibroblasts was increased fourfold in the streptozotocin diabetic compared with the matched normoglycaemic mice (Fig. 3b). Treatment with pegsunercept blocked the increase in these mice caused by diabetes. The number of fibroblasts per mm2 was almost 50% higher in the normal compared with the streptozotocin-induced diabetic animals (Fig. 3d). Treatment with the pegsunercept increased fibroblast density by 71% compared with streptozotocin-induced diabetic mice treated with vehicle alone. The increase in cell density is consistent with the large change in absolute number of apoptotic fibroblasts in the diabetic animals of both models.
The impact of diabetes and inhibition of TNF-α on proliferation of fibroblasts was measured (Fig. 3e). The number of proliferative fibroblasts was fourfold higher in the normoglycaemic than diabetic mice. Treatment with pegsunercept in diabetic mice significantly increased the number of proliferating fibroblasts. However, pegsunercept had no effect on fibroblast proliferation in normoglycaemic mice, indicating that the high levels of TNF-α in the diabetic mice were problematic whereas they were not in the normoglycaemic group.
In order to explore the effect of diabetes and TNF-α inhibition on inflammation, PMNs were counted in both diabetic and normoglycaemic groups of the two animal models. In the normoglycaemic and diabetic db/db groups the inflammatory infiltrate was relatively large on day 5 (Fig. 4a and ESM Fig. 5). However, it was significantly less on day 9 in the normoglycaemic group but remained high at this time point in the diabetic mice. When diabetic mice were treated with pegsunercept there was a significant reduction in inflammatory infiltrate on days 5 and 9, while there was an improvement in the normoglycaemic group only on day 5 (p<0.05; Fig. 4a). The inflammatory infiltrate was also assayed in the streptozotocin model (Fig. 4b). The PMN infiltrate was significantly higher in the diabetic group than control group (p<0.05) and TNF-α inhibition reduced the PMN infiltrate in the diabetic group (p<0.05), consistent with its anti-inflammatory activity.
Fig. 4.
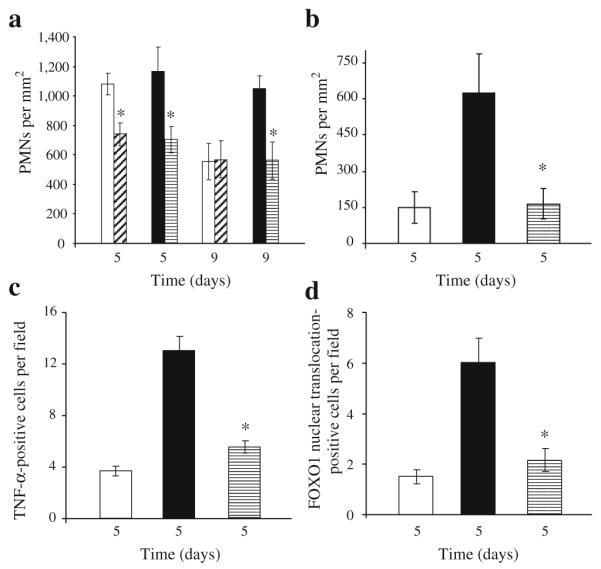
TNF-α inhibition decreases infiltration of inflammatory cells and FOXO1 activation. Excisional wounds were created in db/db (a, d) or streptozotocin-induced diabetic mice (b, c) or their matched normoglycaemic controls and treated with pegsunercept or vehicle alone starting on day 2. The number of PMNs was counted in haematoxylin and eosin-stained sections (a, b). TNF-α-immunopositive fibroblasts were identified by double immunohisto-chemistry using an antibody specific for TNF-α simultaneously with an antibody specific for HSP47 (c)(n=5 per group). FOXO1 nuclear translocation in fibroblasts was assessed in three-colour confocal laser scanning microscopy with antibodies specific for FOXO1 or HSP47. Nuclei were identified by fluorescent nuclear stain, 7-AAD. There was no immunostaining with matched control IgG (data not shown). Each value is the mean±SEM of n=5–7 mice. *p < 0.05 between pegsunercept- and vehicle-treated groups. a, d White bars, diagonal hatched bars, black bars and horizontal hatched bars indicate vehicle-injected normoglycaemic mice, pegsunercept-injected normoglycaemic mice, vehicle-injected db/db diabetic mice and pegsunercept-injected db/db mice, respectively. b, c White bars, black bars, and horizontal hatched bars indicate normoglycaemic mice, streptzotocin-induced type 1 diabetic mice with vehicle injection and pegsunercept-injected streptzotocin-induced diabetic mice, respectively
In vitro, TNF-α stimulates FOXO1 DNA binding activity in fibroblasts [42]. We assessed the number of cells that produced TNF-α and determined whether it was affected by treatment with pegsunercept (Fig. 4c). The number of TNF-α-positive cells was 3.5-fold higher in the diabetic group than the normoglycaemic and treatment with pegsunercept reduced the number of TNF-α-producing cells by 60%, both of which were significant. When FOXO1 is activated it translocates to the nucleus and exhibits increased DNA binding activity. Upon deactivation it is translocated out of the nucleus. To assess FOXO1 activation in vivo we examined the level of FOXO1 nuclear translocation in fibroblastic cells by three-colour confocal laser scanning microscopy (Fig. 4d). FOXO1 nuclear translocation in fibroblasts in type 2 diabetic mice was increased threefold compared with the normoglycaemic mice on day 5. Blocking TNF-α inhibited almost all of the increase caused by diabetes, suggesting that TNF-α played a significant role in increased FOXO1 activation in fibroblasts in diabetic wound healing.
We have previously shown that TNF-α induces fibroblast apoptosis through induction of the transcription factor, FOXO1 [42]. To better understand the impact of TNF-α on fibroblasts and the role that FOXO1 might play, GSEA was performed. We compared TNF-α-stimulated vs unstimulated fibroblasts as well as fibroblasts transfected with FOXO1 siRNA compared with scrambled siRNA prior to TNF-α stimulation. A false discovery rate was set at <5%. Compared with non-stimulated cells, TNF-α significantly increased mRNA levels of genes involved in apoptosis by increasing apoptosis, Akt and p53 gene sets but not mitochondrial or cell-cycle gene sets (Table 1). FOXO1 siRNA decreased gene expression in apoptosis, Akt, mitochondrial and cell-cycle gene sets but not p53. Individual genes identified by the GSEA software for the ‘apoptosis’ pathway are listed in Table 2. Several were upregulated by TNF-α more than 1.7-fold and their upregulation was blocked by FOXO1 siRNA (0.58 or less), demonstrating the functional role of FOXO1 in their regulation. These included TNF-α (also known as TNF), TNF superfamily member 10 (TNFSF10), baculoviral IAP repeat-containing 2 (BIRC2 ), BIRC3, interferon regulatory factor-1 (IRF1), IRF2, IRF7, v-rel reticuloendotheliosis viral oncogene homologue A (RELA), nuclear factor of kappa light polypeptide gene enhancer in B cells-1 (NFKB1), nuclear factor κB (NFKB) inhibitor (NFKBI), NFKBI-alpha (NFKBIA), NFKBI-beta (NFKBIB), NFKBI-epsilon (NFKBIE), TNF receptor-associated factor 1 (TRAF1), CASP1, CASP7, CASP10, TNF receptor super-family member 6 (FAS), TNF receptor superfamily member 1B (TNFRSF1B), TNFRSF10B, BCL2-like 11 (BCL2L11), BH3 interacting domain death agonist (BID) and Jun oncogene (JUN). Genes included in the other apoptotic pathways and their stimulation by TNF-α and effect of FOXO1 siRNA are shown in ESM Tables 1, 2, 3 and 4.
Table 1.
Gene set enrichment identifies TNF-α- and FOXO1-modulated pathways
| Apoptosis | Akt | p53 | Mitochondrial | Cell cycle | Inflammation | Cytokine | Toll | NFKB | |
|---|---|---|---|---|---|---|---|---|---|
| TNF-α stimulation | + | + | + | Nc | Nc | + | + | + | + |
| FOXO1 siRNA | − | − | Nc | − | − | − | − | − | − |
To examine TNF-α-induced gene sets, primary human fibroblasts were treated as described in the Methods. mRNA profiling was carried out using Affymetrix microarrays. Each group was examined using triplicate arrays. TNF-α-stimulated cells were compared with unstimulated cells and FOXO1 siRNA was compared with scrambled siRNA. GSEA analysis was applied to different apoptotic and inflammatory related pathways +, significantly increased; − significantly decreased; Nc, no significant change
Table 2.
Apoptotic genes induced by TNF-α and inhibited by FOXO1 siRNA
| Gene | TNF-α |
FOXO1 siRNA |
||
|---|---|---|---|---|
| Median | Mean | Median | Mean | |
| TNFSF10 a | 20.03 | 14.26 | 0.04 | 0.04 |
| BIRC3 a | 17.73 | 18.69 | 0.03 | 0.40 |
| IRF7 a | 12.94 | 9.67 | 0.11 | 0.27 |
| NFKB1E a | 7.06 | 10.90 | 0.05 | 0.25 |
| TRAF1 a | 6.97 | 28.05 | 0.04 | 0.25 |
| CASP1 a | 4.34 | 3.07 | 0.25 | 0.28 |
| NFKB1A a | 4.31 | 6.14 | 0.10 | 0.28 |
| IRF1 a | 3.98 | 13.66 | 0.04 | 0.30 |
| NFKB1 a | 3.27 | 4.19 | 0.16 | 0.35 |
| NFKB1B a | 3.20 | 3.23 | 0.28 | 0.48 |
| FAS a | 2.80 | 2.94 | 0.18 | 0.19 |
| BCL2L11 a | 2.78 | 2.10 | 0.47 | 0.53 |
| RELA a | 2.56 | 2.36 | 0.32 | 0.25 |
| CASP7 a | 2.51 | 1.99 | 0.44 | 0.43 |
| BID | 2.48 | 2.28 | 0.50 | 0.39 |
| CASP10 a | 2.44 | 2.48 | 0.40 | 0.46 |
| TNFRSF1B a | 2.32 | 2.20 | 0.52 | 0.58 |
| TNF-α a | 2.18 | 7.84 | 0.02 | 0.30 |
| IRF2 a | 2.14 | 2.09 | 0.52 | 0.42 |
| MYC | 2.07 | 1.90 | 0.55 | 0.73 |
| TNFRSF10B a | 2.04 | 2.06 | 0.43 | 0.43 |
| BIRC2 a | 1.99 | 2.22 | 0.37 | 0.48 |
| CASP3 | 1.95 | 1.66 | 0.67 | 0.68 |
| JUN a | 1.93 | 4.63 | 0.11 | 0.08 |
| TRADD | 1.80 | 1.47 | 0.59 | 0.52 |
| CASP4 | 1.77 | 1.46 | 0.66 | 0.54 |
| CYCSs | 1.63 | 1.63 | 0.44 | 0.53 |
| TNFRSF1A | 1.58 | 1.37 | 0.81 | 0.68 |
| CASP9 | 1.56 | 1.28 | 0.96 | 0.82 |
| BCL2L11 | 1.50 | 1.52 | 0.26 | 0.33 |
| IKBKB | 1.43 | 1.28 | 0.25 | 0.37 |
| RIPK1 | 1.42 | 1.34 | 0.41 | 0.51 |
| MDM2 | 1.41 | 1.32 | 0.41 | 0.33 |
| TRAF2 | 1.39 | 1.44 | 0.60 | 0.47 |
|
BIRC4 (also known as XIAP) |
1.36 | 1.36 | 0.52 | 0.40 |
| IRF5 | 1.33 | 1.24 | 0.76 | 0.75 |
| FASL | 1.31 | 1.09 | 0.69 | 0.62 |
| TRAF3 | 1.30 | 1.40 | 0.43 | 0.69 |
| IRF3 | 1.30 | 1.42 | 0.63 | 0.65 |
| IKBKG | 1.30 | 1.31 | 0.80 | 0.72 |
| PRF1 | 1.26 | 1.19 | 0.79 | 0.69 |
| CHUK | 1.25 | 1.19 | 0.81 | 0.78 |
| CASP2 | 1.19 | 1.26 | 0.46 | 0.64 |
| LTA | 1.19 | 2.95 | 1.06 | 0.77 |
| MAPK10 | 1.15 | 1.46 | 0.59 | 0.66 |
| BAD | 1.13 | 1.30 | 0.52 | 0.64 |
| BCL2 | 1.08 | 2.70 | 0.31 | 0.42 |
| BIRC5 | 1.07 | 1.09 | 0.46 | 0.51 |
| BNIP3L | 1.04 | 0.85 | 1.52 | 1.95 |
| MAP2K4 | 1.02 | 0.99 | 0.70 | 0.82 |
| DFFA | 1.02 | 0.91 | 0.96 | 1.13 |
| IRF6 | 1.00 | 1.34 | 0.59 | 0.59 |
| TNFRSF25 | 0.99 | 1.05 | 0.42 | 0.71 |
| APAF1 | 0.99 | 1.01 | 0.67 | 0.71 |
| FADD | 0.98 | 0.84 | 0.99 | 1.11 |
| CASP6 | 0.97 | 0.79 | 0.77 | 1.41 |
| TP53 | 0.95 | 1.01 | 0.58 | 0.87 |
| CASP8 | 0.94 | 1.08 | 0.15 | 0.50 |
| TP73 | 0.94 | 1.11 | 0.30 | 0.74 |
| HELLS | 0.93 | 0.97 | 0.39 | 0.32 |
| GZMB | 0.93 | 0.96 | 0.92 | 0.78 |
| BAX | 0.85 | 0.83 | 0.69 | 0.71 |
| HRK | 0.83 | 0.94 | 0.39 | 0.98 |
| TNFRSF21 | 0.79 | 0.69 | 0.47 | 0.58 |
| IRF4 | 0.74 | 1.12 | 1.05 | 0.92 |
| MAP3K1 | 0.72 | 1.06 | 0.65 | 0.47 |
Human primary fibroblasts were treated as described in the Methods. mRNA profiling was carried out. The mean and median values for TNF-α vs unstimulated and FOXO1 siRNA plus TNF-α vs scrambled siRNA plus TNF-α are shown for triplicate experiments. Genes defined in the apoptosis pathway gene set (www.broadinstitute.org/gsea) are shown
Both a 1.7-fold or more stimulation by TNF-α and a 1.7-fold decrease (0.58, which is the fold decrease equivalent to 1.7 fold stimulation) by FOXO1 siRNA for both mean and median values
Because TNF-α also induces inflammation, inflammatory pathways were examined including pathways designated by the GSEA software as ‘inflammation’, ‘cytokines’, ‘Toll’ and ‘NFKB’. GSEA indicated that that each of these gene sets had mRNA levels enhanced by TNF-α (Table 1). Unexpectedly, FOXO1 silencing significantly reduced mRNA levels for all four of these gene sets. Mediators included in the ‘inflammation’ gene set that were upregulated by TNF-α stimulation and blocked by FOXO1 silencing included TNF-α, interleukin-1 alpha (IL1α), interleukin-6 (IL6), IL8, IL15, colony-stimulating factor 1 (CSF1), CSF2 and CSF3 (Table 3). Genes included in the other inflammatory pathways and their stimulation by TNF-α and effect of FOXO1 siRNA are shown in ESM Tables 5, 6 and 7.
Table 3.
Inflammatory genes induced by TNF-α and inhibited by FOXO1 siRNA
| Gene | TNF-α |
FOXO1 siRNA |
||
|---|---|---|---|---|
| Median | Mean | Median | Mean | |
| IL8 | 298.69 | 294.75 | 0.01 | 0.26 |
| IL11 | 86.27 | 57.7 | 0.58 | 0.76 |
| IL6 a | 14.52 | 13.53 | 0.04 | 0.39 |
| IL1-α a | 10.83 | 66.63 | 0.01 | 0.17 |
| CSF2 a | 6.24 | 6.95 | 0.15 | 0.30 |
| IL15 a | 3.60 | 3.32 | 0.16 | 0.20 |
| TNF-α a | 2.18 | 7.84 | 0.02 | 0.30 |
| IL4 | 2.18 | 2.07 | 1.23 | 0.90 |
| CSF1 a | 2.16 | 2.31 | 0.07 | 0.19 |
| IL12a | 2.07 | 1.56 | 0.36 | 0.35 |
| CSF3 a | 1.91 | 2.11 | 0.10 | 0.08 |
| IFNB1 | 1.54 | 1.57 | 0.02 | 0.04 |
| TGFB2 | 1.47 | 1.53 | 0.14 | 0.11 |
| IL10 | 1.31 | 1.31 | 1.00 | 0.86 |
| PDGF-α | 1.27 | 1.26 | 0.17 | 0.18 |
| IL2 | 1.24 | 1.60 | 1.02 | 0.90 |
| TGFB3 | 1.23 | 1.08 | 0.79 | 0.78 |
| LT-α (also known as LTA) | 1.19 | 2.95 | 1.06 | 0.77 |
| IL7 | 1.17 | 1.04 | 0.21 | 0.20 |
| IL13 | 1.11 | 1.01 | 0.68 | 0.74 |
| IFNA1 | 1.08 | 1.15 | 0.83 | 0.83 |
| TGFB1 | 1.06 | 1.19 | 0.82 | 0.79 |
| IL3 | 1.04 | 1.14 | 0.48 | 1.06 |
| CD4 | 0.94 | 6.79 | 0.73 | 0.64 |
| IFNG | 0.82 | 0.97 | 0.94 | 0.87 |
| IL5 | 0.80 | 0.86 | 0.90 | 0.84 |
| IL12B | 0.74 | 0.80 | 1.23 | 0.98 |
| HLA-DRA | 0.47 | 2.00 | 0.38 | 2.72 |
| HLA-DRB1 | 0.12 | 2.06 | 0.63 | 2.64 |
Human primary fibroblasts were treated as described in the Methods. mRNA profiling was carried out. The mean and median values for TNF-α vs unstimulated and FOXO1 siRNA plus TNF-α vs scrambled siRNA plus TNF-α are shown for triplicate experiments. Genes defined in the inflammation pathway gene set (www.broadinstitute.org/gsea) are shown
Both a 1.7-fold or more stimulation by TNF-α and a 1.7-fold decrease (0.58, which is the fold decrease equivalent to 1.7 fold stimulation) by FOXO1 siRNA for both mean and median values
To validate microarray results, real-time qPCR was carried out for selected genes. When microarray results were directly compared with real-time qPCR results TNF-α was shown to stimulate a more than 1.7-fold increase in IL1α, IL6, IL8, TNF-α and FAS in both types of assays (Fig. 5a). Similarly, when cells were first transfected with FOXO1 siRNA or scrambled siRNA both microarray and real-time qPCR results indicated that FOXO1 siRNA caused a 1.7-fold or more decrease in the mRNA values for each gene tested (Fig. 5b).
Fig. 5.
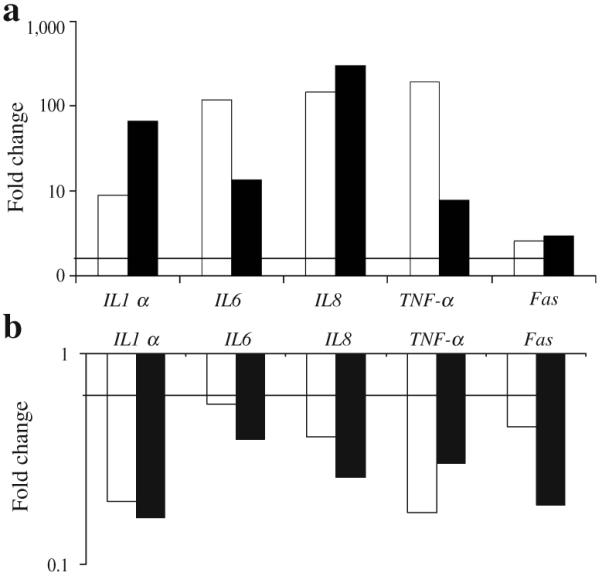
FOXO1 RNAi inhibits TNF-α upregulation of pro-inflammatory and pro-apoptotic genes in human fibroblasts. Real-time qPCR (white bars) was performed on total RNA isolated from human fibroblasts that had been stimulated with TNF-α (20 ng/ml) for 6 h. In some groups, cells were transfected with FOXO1 or scrambled siRNA and then stimulated with TNF-α. a mRNA levels for TNF-α compared with unstimulated cells. b mRNA levels for FOXO1 siRNA plus TNF-α compared with scrambled siRNA plus TNF-α. For real-time qPCR the experiment was conducted three times with similar results. A representative experiment is shown. The corresponding mean values obtained by microarray (black bars) analysis (see Tables 2 and 3) are presented. The horizontal bar represents a 1.7-fold increase (a) or a 1.7-fold decrease (b) in mRNA levels. Note that vertical axes are on logarithmic scales
Discussion
Multiple factors contribute to impaired wound healing. Results presented here indicate that TNF-α is elevated in both type 1 and type 2 diabetic wounds. When TNF-α is specifically inhibited, healing of both type 1 and type 2 diabetic wounds is improved, including the rate of epithelial coverage and formation of new connective tissue. TNF-α inhibition significantly enhanced fibroblast density. To investigate mechanisms, we further examined fibroblast proliferation and apoptosis. The effect of diabetes on fibroblast proliferation and apoptosis was reversed by blockage of TNF-α. To further investigate how TNF-α affects fibroblast apoptosis in diabetic wound healing we examined the pro-apoptotic transcription factor FOXO1. Wounds in type 2 diabetic mice had increased FOXO1 DNA binding activity and increased FOXO1 nuclear translocation in fibroblasts in vivo. Moreover, this increase was driven by TNF-α, since nuclear translocation was significantly reduced by TNF-α inhibition. In contrast, inhibiting TNF-α had relatively little effect on the normoglycaemic mice, suggesting that normal levels of TNF-α are not problematic. Increased FOXO1 activity may also be implicated in type 1 diabetic mice, since elevated TNF-α was observed in type 1 diabetic mice.
Apoptosis is rapid, and at the end-stage of apoptosis the cell breaks down into small apoptotic bodies that are rapidly removed and are not retained in the tissue. We counted only identifiable cells undergoing apoptosis, which exist transiently during a window of ~2 h. A rate of 2.5% apoptotic fibroblasts in a 2 h time frame would translate to 30% over a 24 h period, indicating that the level observed could have a physiological impact. Since the level of fibroblast apoptosis was significantly higher in diabetic wounds, the cumulative effect would be greater in diabetic mice.
It is well known that diabetes causes prolonged inflammation during wound healing [1, 4, 6]. One mechanism through which diabetes may cause enhanced inflammation during the healing process is through the activity of AGEs or elevated levels of TNF-α [25]. The latter is supported by a recent report that blocking TNF-α with an antibody improves healing, which was associated with a decrease in inflammation [44]. It has also been reported that mice with genetic ablation of TNF-R1 have accelerated wound healing with reduced leucocyte infiltration [45]. Persistently high levels of TNF-α from systemic or local application in vivo interfere with dermal wound healing [26, 46, 47], which is consistent with the effects seen in diabetic wound healing where TNF-α is elevated.
To explore pathways that may be regulated by TNF-α and to determine whether they were mediated by FOXO1 in fibroblasts, siRNA studies were carried out in conjunction with mRNA profiling and GSEA. GSEA examines an entire ‘gene set’ and can distinguish differences between groups that might be missed if each gene were examined separately, since individual genes may exhibit changes that do not meet a pre-conceived threshold, or there may be a confusing pattern of inconsistent changes [43, 48]. This analysis indicated, as expected, that mRNA levels of an ‘apoptosis’ gene set were higher in TNF-α-stimulated cells. TNF-α also increased mRNA levels of Akt and p53 apoptotic genes. Diabetic wound healing was associated with increased nuclear translocation in fibroblasts that was reversed by the inhibition of TNF-α. To determine whether FOXO1 mediated the TNF-α-induced apoptotic pathway, RNAi studies were carried out in conjunction with GSEA. FOXO1 siRNA blocked TNF-α induced upregulation of apoptosis pathway and Akt pathway gene sets, but had no effect on the p53 pathway gene set. Interestingly, FOXO1 siRNA decreased levels of the mitochondrial apoptotic and cell-cycle gene sets even though they were not modulated by TNF-α. The former is consistent with a report that FOXO1 plays an important role in apoptosis related to DNA damage that involves the activity of genes that regulate the cell cycle [49]. Surprisingly, FOXO1 siRNA also blocked each of the pro-inflammatory gene sets induced by TNF-α, indicating a previously unrecognised function of FOXO1 in mediating TNF-α-induced inflammatory gene expression.
In summary, diabetes affects wound healing by significantly increasing fibroblast apoptosis and decreasing fibroblast proliferation while decreasing fibroblast density. Each of these effects is reversed by TNF-α inhibition. The improved healing from decreased fibroblast apoptosis is consistent with a previous report that treatment with a caspase inhibitor that blocks apoptosis increases fibroblast density and improves the repair of connective tissue following a bacteria-induced wound [50]. In vitro results indicate that TNF-α enhances inflammatory and pro-apoptotic gene expression in fibroblasts through the transcription factor FOXO1. This may be important and is supported by findings that FOXO1 is elevated during healing of wounds in diabetes.
Supplementary Material
Acknowledgements
This work was supported by a grant from the NIDCR, DE017732 and DE018307. We would like to thank S. Kabani from the Department of Oral and Maxillofacial Pathology at Boston University (MA, USA) for assistance in quantitative assessment of histological sections and A. Ruff, Administrative Assistant from the Department of Oral Biology at Boston University, for administrative help in preparing this manuscript.
Abbreviations
- 7-AAD
7-Aminoactinomycin D
- FOXO1
Forkhead box O1
- GSEA
Gene set enrichment analysis
- HSP47
Heat-shock protein 47
- NFKB
Nuclear factor-κB
- PCNA
Proliferating cell nuclear antigen
- PMN
Polymorphonuclear (leucocyte)
- qPCR
Quantitative PCR
- RNAi
RNA interference
- siRNA
Small interfering RNA
- TNF-R1
TNF receptor type 1
Footnotes
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
Electronic supplementary material The online version of this article (doi:10.1007/s00125-009-1529-y) contains supplementary material, which is available to authorised users.
Contributor Information
M. F. Siqueira, Department of Periodontology and Oral Biology, Boston University School of Dental Medicine, Boston, MA, USA
J. Li, Department of Periodontics, University of Medicine and Dentistry of New Jersey, 110 Bergen Street C-781, Newark, NJ 07101, USA
L. Chehab, Department of Periodontology and Oral Biology, Boston University School of Dental Medicine, Boston, MA, USA
T. Desta, Department of Periodontics, University of Medicine and Dentistry of New Jersey, 110 Bergen Street C-781, Newark, NJ 07101, USA
T. Chino, Department of Periodontics, University of Medicine and Dentistry of New Jersey, 110 Bergen Street C-781, Newark, NJ 07101, USA
N. Krothpali, Department of Periodontology and Oral Biology, Boston University School of Dental Medicine, Boston, MA, USA
Y. Behl, Department of Periodontology and Oral Biology, Boston University School of Dental Medicine, Boston, MA, USA
M. Alikhani, Department of Orthodontics, NYU School of Dentistry, New York, NY, USA
J. Yang, Department of Periodontology and Oral Biology, Boston University School of Dental Medicine, Boston, MA, USA
C. Braasch, Department of Periodontology and Oral Biology, Boston University School of Dental Medicine, Boston, MA, USA
D. T. Graves, Department of Periodontics, University of Medicine and Dentistry of New Jersey, 110 Bergen Street C-781, Newark, NJ 07101, USA
References
- 1.Lioupis C. Effects of diabetes mellitus on wound healing: an update. J Wound Care. 2005;14:84–86. doi: 10.12968/jowc.2005.14.2.26738. [DOI] [PubMed] [Google Scholar]
- 2.Claxton MJ, Armstrong DG, Boulton AJ. Healing the diabetic wound and keeping it healed: modalities for the early 21st century. Curr Diab Rep. 2002;2:510–518. doi: 10.1007/s11892-002-0121-3. [DOI] [PubMed] [Google Scholar]
- 3.Singh N, Armstrong DG, Lipsky BA. Preventing foot ulcers in patients with diabetes. JAMA. 2005;293:217–228. doi: 10.1001/jama.293.2.217. [DOI] [PubMed] [Google Scholar]
- 4.Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736–1743. doi: 10.1016/S0140-6736(05)67700-8. [DOI] [PubMed] [Google Scholar]
- 5.Kane CD, Greenhalgh DG. Expression and localization of p53 and bcl-2 in healing wounds in diabetic and nondiabetic mice. Wound Repair Regen. 2000;8:45–58. doi: 10.1046/j.1524-475x.2000.00045.x. [DOI] [PubMed] [Google Scholar]
- 6.Pierce GF. Inflammation in nonhealing diabetic wounds: the space–time continuum does matter. Am J Pathol. 2001;159:399–403. doi: 10.1016/S0002-9440(10)61709-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lobmann R, Ambrosch A, Schultz G, Waldmann K, Schiweck S, Lehnert H. Expression of matrix-metalloproteinases and their inhibitors in the wounds of diabetic and non-diabetic patients. Diabetologia. 2002;45:1011–1016. doi: 10.1007/s00125-002-0868-8. [DOI] [PubMed] [Google Scholar]
- 8.Trengove NJ, Stacey MC, MacAuley S, et al. Analysis of the acute and chronic wound environments: the role of proteases and their inhibitors. Wound Repair Regen. 1999;7:442–452. doi: 10.1046/j.1524-475x.1999.00442.x. [DOI] [PubMed] [Google Scholar]
- 9.Hehenberger K, Heilborn JD, Brismar K, Hansson A. Inhibited proliferation of fibroblasts derived from chronic diabetic wounds and normal dermal fibroblasts treated with high glucose is associated with increased formation of l-lactate. Wound Repair Regen. 1998;6:135–141. doi: 10.1046/j.1524-475x.1998.60207.x. [DOI] [PubMed] [Google Scholar]
- 10.Wetzler C, Kampfer H, Stallmeyer B, Pfeilschifter J, Frank S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse prolonged persistence of neutrophils and macrophages during the late phase of repair. J Invest Dermatol. 2000;115:245–253. doi: 10.1046/j.1523-1747.2000.00029.x. [DOI] [PubMed] [Google Scholar]
- 11.Loots M, Lamme E, Zeegelaar J, Mekkes J, Bos J, Middelkoop E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers vs acute wounds. J Invest Dermatol. 1998;111:850–857. doi: 10.1046/j.1523-1747.1998.00381.x. [DOI] [PubMed] [Google Scholar]
- 12.Komesu M, Tanga M, Buttros K, Nakao C. Effects of acute diabetes on rat cutaneous wound healing. Pathophysiology. 2004;11:63–67. doi: 10.1016/j.pathophys.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Rai NK, Tripathi K, Sharma D, Shakla V. Apoptosis: a basic physiologic process in wound healing. Int J Low Extrem Wounds. 2005;4:138–144. doi: 10.1177/1534734605280018. [DOI] [PubMed] [Google Scholar]
- 14.Singer A, Clark R. Cutaneous wound healing. N Engl J Med. 1999;341:738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 15.Clark RA. Fibrin and wound healing. Ann N Y Acad Sci. 2001;936:355–367. doi: 10.1111/j.1749-6632.2001.tb03522.x. [DOI] [PubMed] [Google Scholar]
- 16.Darby I, Bisucci T, Hewitson T, MacLellan D. Apoptosis is increased in a model of diabetes-impaired wound healing in genetically diabetic mice. Int J Biochem Cell Biol. 1997;29:191–200. doi: 10.1016/s1357-2725(96)00131-8. [DOI] [PubMed] [Google Scholar]
- 17.Rai NK, Suryabhan, Ansari M, Kumar M, Shukla VK, Tripathi K. Effect of glycaemic control on apoptosis in diabetic wounds. J Wound Care. 2005;14:277–281. doi: 10.12968/jowc.2005.14.6.26792. [DOI] [PubMed] [Google Scholar]
- 18.Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146:56–66. [PMC free article] [PubMed] [Google Scholar]
- 19.Messadi DV, Le A, Berg S, et al. Expression of apoptosis-associated genes by human dermal scar fibroblasts. Wound Repair Regen. 1999;7:511–517. doi: 10.1046/j.1524-475x.1999.00511.x. [DOI] [PubMed] [Google Scholar]
- 20.Ladin DA, Hou Z, Patel D, et al. p53 and apoptosis alterations in keloids and keloid fibroblasts. Wound Repair Regen. 1998;6:28–37. doi: 10.1046/j.1524-475x.1998.60106.x. [DOI] [PubMed] [Google Scholar]
- 21.Taylor PC. Anti-TNF therapy for rheumatoid arthritis and other inflammatory diseases. Mol Biotechnol. 2001;19:153–168. doi: 10.1385/MB:19:2:153. [DOI] [PubMed] [Google Scholar]
- 22.Klimiuk PA, Sierakowski S, Domyslawska I, Chwiecko J. Effect of repeated infliximab therapy on serum matrix metalloproteinases and tissue inhibitors of metalloproteinases in patients with rheumatoid arthritis. J Rheumatol. 2004;31:238–242. [PubMed] [Google Scholar]
- 23.Ardizzone S, Porro G Bianchi. Biologic therapy for inflammatory bowel disease. Drugs. 2005;65:2253–2286. doi: 10.2165/00003495-200565160-00002. [DOI] [PubMed] [Google Scholar]
- 24.Graves DT, Liu R, Alikhani M, Al-Mashat H, Trackman PC. Diabetes-enhanced inflammation and apoptosis—impact on periodontal pathology. J Dent Res. 2006;85:15–21. doi: 10.1177/154405910608500103. [DOI] [PubMed] [Google Scholar]
- 25.Goova MT, Li J, Kislinger T, et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol. 2001;159:513–525. doi: 10.1016/S0002-9440(10)61723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buck M, Houglum K, Chojkier M. Tumor necrosis factor inhibits collagen 1 gene expression and wound healing in a murine model of cachexia. Am J Pathol. 1996;149:195–204. [PMC free article] [PubMed] [Google Scholar]
- 27.Salomon GD, Kasid A, Cromack DT, et al. The local effects of cachectin/tumor necrosis factor on wound healing. Ann Surg. 1991;214:175–180. doi: 10.1097/00000658-199108000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moller DE. Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab. 2000;11:212–217. doi: 10.1016/s1043-2760(00)00272-1. [DOI] [PubMed] [Google Scholar]
- 29.Uno S, Imagawa A, Okita K, et al. Macrophages and dendritic cells infiltrating islets with or without beta cells produce tumour necrosis factor-alpha in patients with recent-onset type 1 diabetes. Diabetologia. 2007;50:596–601. doi: 10.1007/s00125-006-0569-9. [DOI] [PubMed] [Google Scholar]
- 30.Behl Y, Krothapalli P, Desta T, Dipiazza A, Roy S, Graves DT. Diabetes-enhanced tumor necrosis factor-alpha production promotes apoptosis and the loss of retinal microvascular cells in type 1 and type 2 models of diabetic retinopathy. Am J Pathol. 2008;172:1411–1418. doi: 10.2353/ajpath.2008.071070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Navarro-Gonzalez JF, Mora-Fernandez C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol. 2008;19:433–442. doi: 10.1681/ASN.2007091048. [DOI] [PubMed] [Google Scholar]
- 32.Brown D, Kao W, Greenhalgh D. Apoptosis down-regulates inflammation under the advancing epithelial wound edge: delayed patterns in diabetes and improvement with topical growth factors. Surgery. 1997;121:372–380. doi: 10.1016/s0039-6060(97)90306-8. [DOI] [PubMed] [Google Scholar]
- 33.Kayal RA, Tsatsas D, Bauer MA, et al. Diminished bone formation during diabetic fracture healing is related to the premature resorption of cartilage associated with increased osteoclast activity. J Bone Miner Res. 2007;22:560–568. doi: 10.1359/jbmr.070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graves D, Nooh N, Gillen T, et al. IL-1 plays a critical role in oral, but not dermal, wound healing. J Immunol. 2001;167:5316–5320. doi: 10.4049/jimmunol.167.9.5316. [DOI] [PubMed] [Google Scholar]
- 35.Nooh N, Graves D. Healing is delayed in oral compared to dermal excisional wounds. J Periodontol. 2003;74:242–246. doi: 10.1902/jop.2003.74.2.242. [DOI] [PubMed] [Google Scholar]
- 36.Davidson JM. Wound repair. J Hand Ther. 1998;11:80–94. doi: 10.1016/s0894-1130(98)80004-5. [DOI] [PubMed] [Google Scholar]
- 37.Furst DE, Fleischmann R, Kopp E, et al. A phase 2 dose-finding study of PEGylated recombinant methionyl human soluble tumor necrosis factor type I in patients with rheumatoid arthritis. J Rheumatol. 2005;32:2303–2310. [PubMed] [Google Scholar]
- 38.Scallon B, Cai A, Solowski N, et al. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301:418–426. doi: 10.1124/jpet.301.2.418. [DOI] [PubMed] [Google Scholar]
- 39.Kaymakcalan Z, Sakorafas P, Bose S, et al. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol. 2009;131:308–316. doi: 10.1016/j.clim.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Agnholt J, Dahlerup JF, Kaltoft K. The effect of etanercept and infliximab on the production of tumour necrosis factor alpha, interferon-gamma and GM-CSF in in vivo activated intestinal T lymphocyte cultures. Cytokine. 2003;23:76–85. doi: 10.1016/s1043-4666(03)00201-1. [DOI] [PubMed] [Google Scholar]
- 41.Kuroda K, Tajima S. HSP47 is a useful marker for skin fibroblasts in formalin-fixed, paraffin-embedded tissue specimens. J Cutan Pathol. 2004;31:241–246. doi: 10.1111/j.0303-6987.2003.00166.x. [DOI] [PubMed] [Google Scholar]
- 42.Alikhani M, Alikhani Z, Graves DT. FOXO1 functions as a master switch that regulates gene expression necessary for tumor necrosis factor-induced fibroblast apoptosis. J Biol Chem. 2005;280:12096–12102. doi: 10.1074/jbc.M412171200. [DOI] [PubMed] [Google Scholar]
- 43.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goren I, Muller E, Schiefelbein D, et al. Systemic anti-TNFalpha treatment restores diabetes-impaired skin repair in ob/ob mice by inactivation of macrophages. J Invest Dermatol. 2007;127:2259–2267. doi: 10.1038/sj.jid.5700842. [DOI] [PubMed] [Google Scholar]
- 45.Mori R, Kondo T, Ohshima T, Ishida Y, Mukaida N. Accelerated wound healing in tumor necrosis factor receptor p55-deficient mice with reduced leukocyte infiltration. FASEB J. 2002;16:963–974. doi: 10.1096/fj.01-0776com. [DOI] [PubMed] [Google Scholar]
- 46.Rapala K, Laato M, Niinikoski J, et al. Tumor necrosis factor alpha inhibits wound healing in the rat. Eur Surg Res. 1991;23:261–268. doi: 10.1159/000129163. [DOI] [PubMed] [Google Scholar]
- 47.Regan MC, Kirk SJ, Hurson M, Sodeyama M, Wasserkrug HL, Barbul A. Tumor necrosis factor-alpha inhibits in vivo collagen synthesis. Surgery. 1993;113:173–177. [PubMed] [Google Scholar]
- 48.Curtis RK, Oresic M, Vidal-Puig A. Pathways to the analysis of microarray data. Trends Biotechnol. 2005;23:429–435. doi: 10.1016/j.tibtech.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 49.Huang H, Regan K, Lou Z, Chen J, Tindall D. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science. 2006;314:294–297. doi: 10.1126/science.1130512. [DOI] [PubMed] [Google Scholar]
- 50.Al-Mashat HA, Kandru S, Liu R, Behl Y, Desta T, Graves DT. Diabetes enhances mRNA levels of proapoptotic genes and caspase activity, which contribute to impaired healing. Diabetes. 2006;55:487–495. doi: 10.2337/diabetes.55.02.06.db05-1201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.