

Abstract
Diabetes mellitus is a metabolic disorder that leads to the development of a number of complications. The etiology of each diabetic complication is undoubtedly multifactorial. We will focus on one potential component that may be common in many diabetic complications, dysregulation of innate immunity associated with an increased inflammatory response. High glucose levels lead to shunting through the polyol pathway, an increase in diacylglycerol which activates protein kinase C, an increase in the release of electrons that react with oxygen molecules to form superoxides, and the non-enzymatic glycosylation of proteins that result in greater formation of advanced glycation end products. Each of these can lead to aberrant cell signalling that affects innate immunity for example, by activating the MAP kinase pathway or inducing activation of transcription factors such as NF-kappaB. This may be a common feature of several complications including periodontal disease, atherosclerosis, nephropathy, impaired healing and retinopathy. These complications are frequently associated with increased expression of inflammatory cytokines such as TNF-alpha, IL-1beta and IL-6 and enhanced generation of reactive oxygen species. Cause and effect relationship between dysregulation of key components of innate immunity and diabetic complications in many instances have been demonstrated with the use of cytokine blockers and antioxidants.
Keywords: Bacteria, Bone, Fracture, Hyperglycemia, Inflammation, Infection, Microvascular, Periodontitis, Review
2. INTRODUCTION
Diabetes mellitus is a metabolic disease estimated to affect over 19 million people in the United States. Based on an evaluation of NHANES 1999-2002, the prevalence of diagnosed and undiagnosed diabetes was estimated at 9.3% in U.S. adults, with an additional 26% having the prediabetic condition of impaired fasting glucose (1). The two major forms of diabetes mellitus are type 1 and type 2. They share similar diabetic complications but have distinct etiologies. Type 1 diabetes occurs when the beta cells of the pancreas are destroyed and insufficient amounts of insulin are produced. In most cases, type 1 diabetes is due to autoimmune-induced inflammation with destruction and apoptosis of beta cells (2). This is usually associated with infiltration of innate immune cells that produce cytokines such as tumor necrosis factor alpha (TNF-alpha) which promote beta cell apoptosis and increased infiltration of islet-specific T cells which attack and destroy beta cells (3).
Approximately 90% of patients with diabetes have type 2 diabetes (4). The development of type 2 diabetes is directly related to increased amounts of visceral adipose tissue (5, 6). Weight gain in early adulthood is linked to a higher risk and earlier onset of type 2 diabetes and is particularly troublesome considering that the incidence of obesity has increased progressively over the last 5 decades (7, 8). Adipose tissue is viewed as an active hormone-regulating organ that releases metabolically active molecules that can inhibit the body’s ability to respond to insulin contributing to insulin resistance (9). Early in the development of type 2 diabetes, insulin resistance requires the production of extra insulin to maintain normal blood glucose levels. In the majority of obese individuals, hyperinsulinemia can occur with an increase in beta-cell mass that facilitates increased production of insulin (10). As a result these individuals do not develop diabetes. However, approximately one third of obese individuals exhibit a decrease in beta-cell mass caused by beta-cell apoptosis rendering these individuals incapable of compensating for the insulin resistant state. It is at this point that hyperglycemia becomes evident, leading to a diagnosis of type 2 diabetes. Thus, type 2 diabetes is caused by a combination of insulin resistance coupled with insufficient production of insulin to overcome the insulin resistance. (4). It is striking that both type 1 and type 2 diabetes are negatively affected by the death of beta cells in the pancreas resulting in inadequate insulin production. For this reason the term, non-insulin dependent diabetes (NIDDM) has been replaced by the term, type 2 diabetes.
3. EFFECTS OF HYPERGLYCEMIA ON METABOLIC AND SIGNALLING PATHWAYS
Diabetes is associated with a number of factors that can cause complications including: 1) shunting through the polyol pathway, 2) enhanced oxidative stress, 3) high levels of protein kinase C activation (PKC), and 4) enhanced formation of advanced glycation endproducts (AGEs). Abnormalities in each of these pathways may affect a number of important cellular and molecular events, which combined lead to pathologic changes. One of these disturbances may be dysregulation in innate immunity as exemplified by the excessive production of TNF-alpha in diabetic compared to normals (11-16). Excessive production of TNF-alpha is associated with many diabetic complications. It is expressed at higher levels in type 1 and type 2 diabetic mice in response to bacterial inoculation, in response to lipopolysaccharide (LPS) and in complications such as diabetic retinopathy, nephropathy, neuropathy and impaired wound healing (17-22). Enhanced levels of TNF-alpha and high glucose conditions have additive effects in stimulating NF-kappaB activation and may be important in diabetes-enhanced inflammation (23), (24). Thus, when TNF–alpha is inhibited high levels of chemokine expression observed in diabetic mice is reversed (25) diabetes impaired wound healing is improved (26, 27) and sodium retention and renal hypertrophy associated with diabetic nephropathy is reduced (28).
3.1. Polyol Pathway
During hyperglycemia excess glucose levels lead to the activation of the polyol pathway and enhanced formation of sorbitol and fructose through the activation of aldose reductase and sorbitol dehydrogenase (14, 29). An informative diagram can be found in ref (30). Shunting through the polyol pathway enhances the formation of advanced glycation endproducts and leads to greater production of reactive oxygen species (ROS) and nitric oxide (NO) (12, 31, 32). Each of these factors can contribute to inflammation. Inhibition of aldose reductase in hyperglycemic conditions reduces high-glucose induced PKC activation, nuclear translocation of NF-kappaB, phosphorylation of IKK, and reduces high glucose stimulated intracellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 expression (33-35). The diversion of glucose to the polyol pathway affects innate immunity by reducing neutrophil opsonophagocytosis, which is improved by blocking aldose reductase (36). Inhibiting aldose reductase also decreases the neutrophil respiratory burst and production of oxygen free radicals which are elevated by high glucose levels (37, 38). Lastly, neutrophils from diabetic patients exhibit defects in bacterial killing which are reversed by treatment with an aldose reductase inhibitor (39). Thus, the use of aldose reductase inhibitors demonstrates that increased shunting through the polyol pathway contributes to altered innate immunity in diabetics.
3.2. Oxidative Stress
Diabetes results in an increase in oxidative stress through enhanced formation of superoxides as well as a decrease in anti-oxidant capacity (40, 41). One of the effects of hyperglycemia is that electron transport chains in mitochondria become overloaded, which leads to the release of electrons that react with oxygen producing superoxides, increasing oxidative stress inside the cell (42). Oxidative stress can damage cell membranes, proteins and DNA (43, 44). Reactive oxygen species (ROS) can also lead to stimulation of an innate immune response through the induced production of proinflammatory cytokines. It has been shown that ROS can stimulate cytokine production through activation of MAP kinase signaling (45, 46). ROS can also enhance inflammation by increasing the phosphorylation of I-kappaB, which leads to activation of NF-kappaB (47). Activation of NF-kappaB is necessary for the expression of key elements of the innate immune system. The functional role of diabetes enhanced ROS in contributing to dysregulation of innate immunity and diabetic complications have been demonstrated with antioxidant therapy. Treatment of STZ induced diabetic rats with the anti-oxidant butylated hydroxyanisole decreased production of TNF-alpha and IL-1beta by peritoneal macrophages. It also inhibited the release of superoxides and nitric oxide from macrophages (48). Treatment of diabetic rats with the antioxidant astragalus saponin reduced oxidative stress, renal hypertrophy, and microalbuminuria that occur in diabetic nephropathy (49).
3.3. Protein kinase C
Diabetes elevated PKC has been linked to diabetic complications. One of the products of glycolysis, which is elevated when glucose levels are high, is dihydroxyacetone phosphate, which in some tissues is reduced to glycerol phosphate and converted to diacylglycerol. Diacylglycerol in turn activates PKC (15, 16, 50). Diabetes associated PKC activity is also enhanced by increasing the NADH/NAD+ ratio (15, 51). Various PKC isoforms are involved in NF-kappaB activation and subsequent production inducible nitric oxide synthase and TNF-alpha (52-54). Increased PKC activity in neutrophils from diabetic patients stimulates the release of superoxide ions (55). A cause and effect relationship between diabetes, increased PKC activity and activation of innate immunity has been shown with inhibitors. Hyperglycemia leads to greater MAP kinase signalling, NF-kappaB activity and production of cytokines such as IL-6, which is reversed with PKC-alpha/PKC-beta inhibitors (56). Inhibition PKC-beta in diabetic rats decreases macrophage infiltration to the kidney compared with diabetic animals which were not treated with the inhibitor (57).
3.4. Advanced glycation endproducts
Hyperglycemia as well as aging is associated with the enhanced formation of advanced glycation endproducts (AGEs) (13, 15). The non-enzymatic glycosylation of proteins that occurs as a result of hyperglycemia can cause cross-linking of molecules such as basement membrane proteins that alter tissue function (13, 15, 58). Glycosylation also causes proteins to acquire functions that they normally do not possess by binding to receptors for AGEs on cell surfaces. For example, glycosylation of collagen and albumin confers signalling properties to these molecules so that they then possess pro-inflammatory and pro-apoptotic activity (50, 59-62) AGEs also have the capacity to stimulate migration of mononuclear phagocytes (63, 64). Blockage of RAGE in diabetic mice reduces TNF-alpha and IL-6 production and decreases activity of matrix metalloproteinases which degrade extracellular matrix proteins (22, 65).
4. DIABETIC COMPLICATIONS
Dysregulation of metabolic or signalling pathways may contribute to diabetic complications by affecting a number of critical cellular events. For the purpose of this review we will focus on one aspect, the potential impact on innate immunity. There is evidence that periodontal disease, cardiovascular disease, nephropathy, impaired fracture healing and retinopathy are negatively affected by diabetes and that alterations in innate immune responses may participate in the pathologic process. Periodontal disease
4.1. Periodontal disease
Periodontal disease is one of the known complications of diabetes (66). The innate immune system is thought to play a major role in the progression of periodontal disease in part through the production of proinflammatory cytokines (67, 68). There is considerable evidence that diabetes enhances the innate immune response in the periodontium. IL-1beta, TNF-alpha and the chemokine MCP-1 are elevated in the gingival crevicular fluid or serum of diabetic patients and are correlated with increased periodontal tissue destruction (69-72). Monocytes from patients with type 1 diabetes produce significantly greater amounts of TNF–alpha, IL-1beta and PGE2 in response to LPS compared to matched normoglycemic individuals (72, 73). Diabetics have enhanced gingival bleeding, increased loss of attachment, greater pocket depth formation and increased loss of alveolar bone (74, 75). Periodontal ligament and gingival fibroblast had enhanced collagenolytic activity when exposed to IL-1beta produced by LPS stimulated phagocytes (76). There is evidence that there is a bidirectional relationship between periodontal disease and hyperglycemia whereby periodontitis enhances the level of hyperglycemia in diabetics (77). However, this concept is controversial since there are several studies which dispute it (77). After A. actinomycetemcomitans inoculation, diabetic NOD mice showed significantly higher alveolar bone loss compared with pre-diabetic and nondiabetic NOD mice. They also showed significantly higher receptor activator of nuclear factor kappaB ligand (RANKL) expression and when treated with osteoprotegerin (OPG), diabetic mice showed a significant reversal of alveolar bone loss, as well as reduced RANKL expression. (78). Mechanisms by which diabetes may impact aspects of the innate immune response associated with periodontal tissue loss such as increased cytokine and superoxide production are discussed below.
In vivo studies have been carried out to examine the effect of diabetes on the response of leukocytes to inflammatory stimuli such as LPS. Prolonged expression of the chemokines MCP-1 and MIP-2 as well as the cytokines TNF–alpha and IL-6 in diabetic compared to normal animals has been shown to occur during wound healing (79). More persistent high levels of TNF–alpha have also been observed with an experimental buccal Streptococcal infection (80). We examined the innate immune response to P. gingivalis in type 2 diabetic and normal control mice (25). At a high dose, a severe inflammatory infiltrate of PMNs formed one day after inoculation that was largely resolved after three days in the control group. In the diabetic, the initial lesion formation was similar but resolution was significantly less three days after inoculation (Figure 1A). A smaller bacterial inoculum gave a similar pattern of more prolonged inflammation in the diabetic mice in response to P. gingivalis. Cytokine expression was measured at the RNA level from tissue obtained at the site of bacterial inoculation. P. gingivalis stimulated TNF expression to a similar extent in both diabetic and control animals one day after inoculation. On day 3, TNF expression was still high in the diabetic but had decreased considerably in the control mice. A similar pattern was observed for the mRNA levels of the chemokines MIP-2 and MCP-1. The finding that early expression of MIP-2, which is functionally equivalent to human IL-8, was similar in normal and diabetic mice is consistent with histologic results quantifying PMN infiltration. To determine whether TNF dysregulation played a significant role in the prolonged expression of chemokines, the TNF specific inhibitor, etanercept was used (Figure 1B). Diabetic mice treated with TNF inhibitor had significantly reduced levels of both MCP-1 and MIP-2 expression compared to control diabetic mice treated with PBS (p<0.05).
Figure 1.
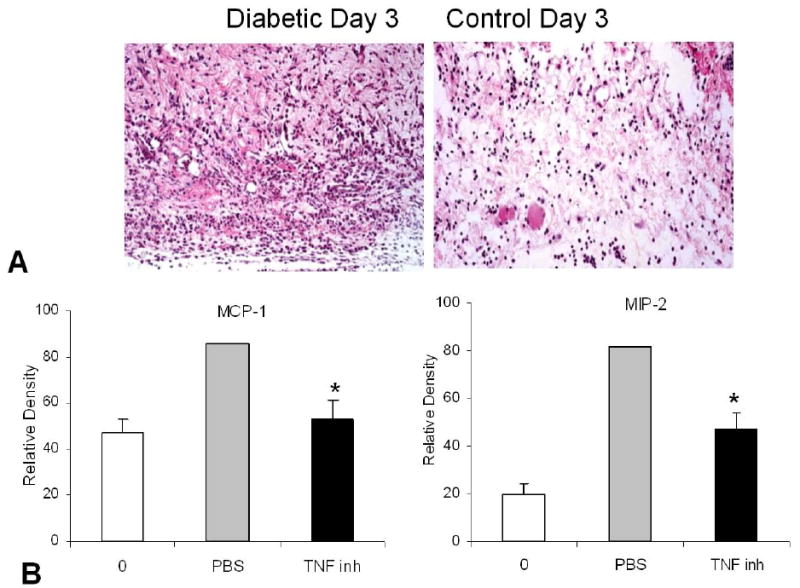
Diabetes leads to a more persistent inflammatory infiltrate P. gingivalis was inoculated subcutaneously into the scalp. One day after inoculation there was a pronounced and equivalent inflammatory infiltrate in type 2 diabetic (db/db) mice and normoglycemic (db/+) mice (data not shown). A. Three days later the infiltrate was more pronounced in the diabetic mice than control group. B. Inhibition of TNF substantially reverses diabetes-enhanced chemokine dysregulation. Diabetic db/db mice were inoculated with P. gingivalis and treated with etanercept (TNF-inh) or vehicle alone (PBS). RNA was extracted 3 days following inoculation and compared to the zero time point (0). MIP-2 and MCP-1 mRNA levels were substantially improved in the etanercept treated diabetic mice indicating that TNF plays an important role in diabetes-elevated chemokine expression. Adapted with permission from 25).
We also investigated whether diabetes affects inflammation in a type 2 diabetic rat in which periodontal bone loss is induced by the placement of ligatures around the teeth. In this model the ligature was kept in place for 7 days to promote plaque accumulation, gingival inflammation and periodontal bone loss. Animals were also observed 4 and 9 days after the ligature removal to assess the resolution of the inflammatory events and the capacity to repair resorbed bone. As shown in Figure 2A, diabetes increased the intensity and duration of the inflammatory infiltrate (P<0.05). This was also associated with greater osteoclastogenesis (81) Once bone is resorbed it is repaired by new bone formation by a process referred to as coupling (82). If the amount of new bone formed is equal to the amount loss there is no bone loss, a process which occurs in physiologic bone remodeling in healthy young adults. When the amount of new bone formation following resorption was measured the amount was 2.4 to 2.9 fold higher in the normal versus diabetic rats (P<0.05). Diabetes also increased apoptosis and decreased the number of bone-lining cells, osteoblasts and periodontal ligament fibroblasts. There was a 5 fold increase in osteoblast apoptosis and 2.7 fold increase in PDL fibroblast apoptosis in the diabetic animals (P<0.05) (81). Thus, diabetes caused a more persistent inflammatory infiltrate. Thus, it is our hypothesis that diabetes enhanced inflammation may negatively affect the periodontium by limiting reparative bone formation, i.e. by interfering with coupling. This process may involve inflammation associated death of osteoblasts and their precursors in the diabetic animals (81, 83). However, further investigation is needed to firmly establish these concepts.
Figure 2.
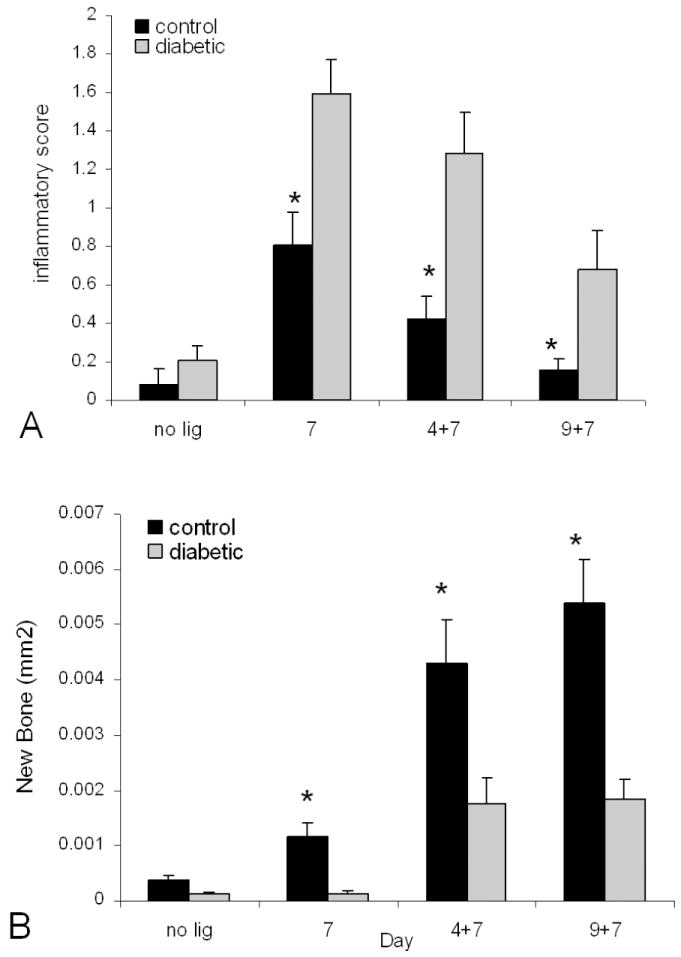
Diabetes prolongs periodontal inflammation and impairs bone formation following resorption. Ligatures were placed around the molar teeth of ZDF type 2 diabetic and normoglycemic control rats and left in place for seven days. Rats were euthanized prior to placement of ligatures (no ligatures), immediately after removal (7 day) or 4 (4+7) or 9 (9+7) days following ligature removal. A. The interproximal gingiva between the 1st and 2nd and 2nd and 3rd molars was examined for the presence of inflammatory cells (PMNs and mononuclear cells) in HandE stained sections using the following scale: 0 (no inflammation), 1 (slight), 2 (moderate), 3 (severe). B. The area of new bone formation was measured in relationship to a reversal line identified in TRAP stained sections. Each value is the mean of five to seven rats + SE. 1 indicates significant difference between diabetics and normoglycemic control rats (P<0.05). Adapted with permission from (81).
Phagocytosis and killing of bacteria are reduced in diabetics and may contribute to increased susceptibility to periodontal pathogens (84-87). Interestingly diabetics are not more susceptible to all infections but seem to be particularly more vulnerable to gram negative infections (88). These may include urinary tract infections, soft tissue infections and periodontal disease (66, 88-90). Soft tissue infections, especially those associated with human or animal bites may involve gram negative oral pathogens, as does periodontitis (91-93). Enhanced susceptibility to periodontal pathogens is one of the significant and characteristic complications of diabetes (66, 94).
Advanced glycation end products have been shown to contribute to diabetes-enhanced bone loss in a murine model of periodontitis (65). Interestingly, when AGEs were blocked there was a reduction in TNF-alpha and IL-6 (65). Thus, a mechanism by which AGEs could potentially enhance periodontitis is through enhanced activation of innate immunity. This possibility is supported by findings that the natural ligand for the AGE receptor, RAGE, has pro-inflammatory activity (95). Moreover, inhibition of AGEs decreases the levels of inflammatory cytokines in diabetic animals during cutaneous wound healing, consistent with results seen in the periodontium (22).
4.2. Cardiovascular disease
Diabetes is known to cause macrovascular complications, which are highly influenced by inflammatory cytokines associated with innate immunity (96) It is noteworthy that inhibiting the activity of pro-inflammatory molecules prevents the early steps in atherogenesis (97). Inflammation can also contribute to morbidity by enhancing the progression from early lesions to culprit lesions by promoting erosion and apoptosis of the plaque making it more likely to rupture (96). Systemic conditions play an import role in exacerbating atherogenesis, such as hypertension and diabetes (98-100). Factors that enhance systemic inflammation may also increase the risk of cardiovascular diseases (101). Anaerobic bacteria, such as Chlamydia pneumoniae and Helicobacter pylori have been linked to higher rates of atherosclerosis (102, 103). Furthermore, Gram-negative pathogens associated with periodontal disease have also been linked to atherosclerotic plaque formation (104, 105). In this regard, it is thought that periodontal disease can lead frequent bacteremia thereby inducing a systemic inflammatory response (106, 107).
Bacteria-induced systemic inflammation may be aggravated by diabetes and represent a mechanism for enhanced atherogenesis. We examined the induction of cytokines of the innate immune response in cardiovascular tissue following subcutaneous inoculation of LPS in db/db mice and matched controls. Subcutaneous inoculation of lipopolysaccharide stimulated a local inflammatory response, as expected. Within 6 hours of subcutaneous LPS administration mRNA levels of pro-inflammatory gene expression had increased several folds in both the diabetic and normoglycemic groups at both the site of inoculation (scalp) and at a distant site, the cardiovascular tissue. However, the values were higher in the diabetic group at both sites (Table 1). Despite similar serum endotoxin levels, diabetic mice had peak TNF–alpha levels that were seven time higher than that of normoglycemic mice (18).
Table 1.
Fold maximum stimulation (Diab/Cont)
| Heart/Aorta | Inoculation Site | |
|---|---|---|
| TNF-alpha | 2.61 | 2.31 |
| MCP-1 | 1.51 | 1.91 |
| ICAM-1 | 1.51 | 2.31 |
| MIP-1 alpha | 2.71 | 1.3 |
The inflammatory response to subcutaneous injection of LPS is greater in diabetic than normoglycemic animals at both the site of inoculation and at a distant site, cardiovascular tissue. LPS was inoculated into the scalp and RNA was extracted from the site of inoculation and the heart/aorta at 1.5, 6 and 24 hours after inoculation. The fold difference between diabetic and normal mice at the time of peak expression is shown.
indicates a significant difference between diabetic and normoglycemic groups (P<0.05)
Interestingly, a link has been established between endotoxemia and atherosclerosis. Higher than normal endotoxin levels are associated with a threefold higher risk of developing atherosclerosis (108). Moreover, endotoxemia enhances formation and progression of early atherosclerotic lesions in a mouse model (109). This suggests that a “chronic” gram negative infection that causes endotoxemia could potentially aggravate the atherosclerotic process. Thus, infections by gram negative organisms that lead to endotoxemia may promote an inflammatory response that affects cardiovascular tissue. If this occurs in diabetics the response may be even greater than that expected in normal individuals and may in part, contribute to enhanced atherogenesis in diabetics.
4.3. Nephropathy
Diabetic nephropathy is another diabetic complication associated with dysregulation of the innate immune response. It is characterized by a thickening of the basement membrane, expansion of the mesangium, reduced filtration, albuminuria and ultimately renal failure. The innate immune system contributes to development of diabetic nephropathy from damage associated with increased infiltration of leukocytes and production of proinflammatory cytokines (110-113). Studies showed increased infiltration of macrophages and neutrophils throughout the renal parenchyma of both db/db mice and STZ induced diabetic mice (110-112). The inflammatory cytokines TNF-alpha, IL-1beta and INF-gamma as well as iNOS are overexpressed in diabetic kidneys (112-115). STZ induced diabetic rats showed that renal cortical mRNA levels of TNF-alpha, IL-1 and IL-6 were 2.4-, 1.2- and 3.4- fold higher respectively than normal control. Kidney weight and urinary albumin excretion were significantly associated with renal mRNA expression of TNF-alpha and IL-6. (116). Treatment of diabetic mice with an antagonist to the chemokine receptor CCR-1 resulted in decreased infiltration of macrophages and decreased expression of proinflammatory mediators (110). Likewise, ICAM-1 deficiency reduces macrophage infiltration and renal injury associated with diabetic nephropathy (117, 118). These findings suggest that enhanced activation of the innate immune response contributes to diabetic nephropathy through increased production of inflammatory cytokines leading to tissue injury. This association is similar to the impact of diabetes on the periodontium and may contain common elements.
4.4. Impaired fracture healing
Diminished bone formation and impaired fracture healing have been observed in animal models and in patients with diabetes. Normal fracture healing is facilitated by a callus which forms a cartilage matrix, which is resorbed and followed by endochondral formation around the fracture site. The callus stabilizes the fracture and allow healing to proceed. To gain insight into how diabetes affects fracture healing experiments were carried out focusing on the impact of diabetes on the resorption of cartilage (119) in a type 1 diabetic model. Counter to expectations there was relatively little difference in the initial formation of the callus. However, diabetes caused an increase in mRNA levels of TNF-alpha, MCSF, and RANKL (Figure 3). The mRNAs encoding ADAMTS 4 and 5, major aggrecanases that degrade cartilage, were also elevated in diabetic animals (119). The increase in factors that participate in the host response and play an important role in stimulating bone resorption resulted in an increase in osteoclastogenesis. The increased osteoclast activity, in turn, led to greater resorption of cartilage and as a result, a smaller callus. This may explain a decrease in callus size and a decrease in the strength of healing bone that is typically found in diabetic fracture healing (120-122). It is also interesting to note that in TNF receptor deficient mice fracture healing is impeded because there is a delay in osteoclast formation and as a result, cartilage is not removed in timely fashion (123). This in turn interferes with endochondral bone formation in the fracture callus. Thus, in diabetes there are high levels of TNF–alpha and increased osteoclastogenesis that leads to excessive removal of cartilage, while in TNF receptor deficient mice there is a delay in osteoclast formation and failure to remove cartilage in timely fashion. In both cases, excessive or delayed removal of cartilage leads to either impaired or delayed fracture healing.
Figure 3.
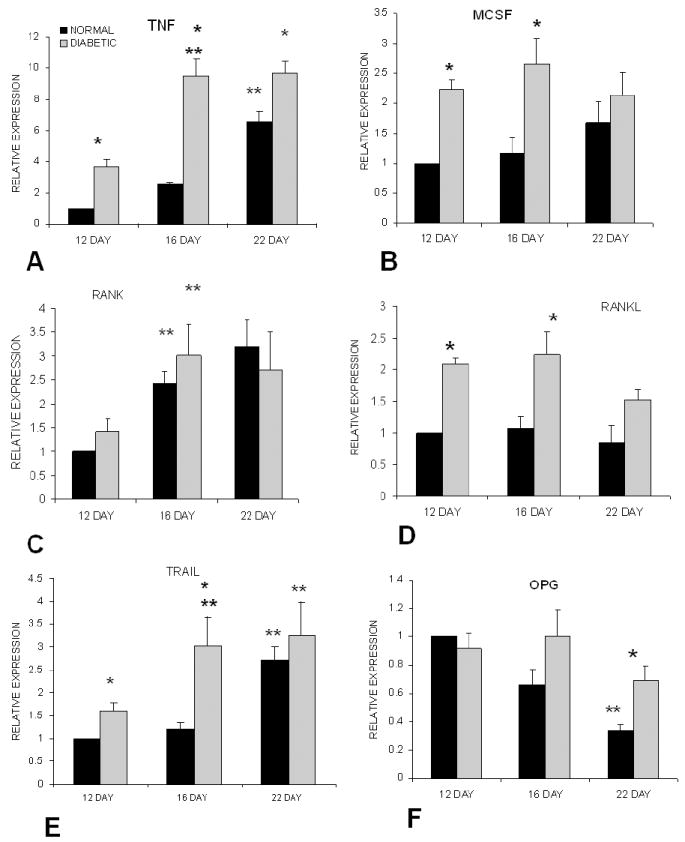
Diabetes results in enhanced expression of cytokines that regulate osteoclast formation during fracture healing. Fractures were induced in the tibia or streptozotocin induced diabetic and normoglycemic mice. Total RNA from diabetic and normoglycemic mice was extracted, purified and evaluated by real-time PCR. Data for each cytokine were normalized to the value obtained from normoglycemic mice at Day 12, which was set as relative expression = 1. (1) represents (p<0.05) between normal and diabetic. (2) represents (p<0.05) between one time point and the previous time point. Adapted with permission from (81).
4.5. Retinopathy
One of the principle long-term complications of diabetes is microvascular disease (124). This is manifested in the eye as diabetic retinopathy and in other tissues leading to problems such as neuropathy and nephropathy. Diabetic retinopathy is a disease of the extreme posterior part of the retina. The earliest clinical lesions of diabetic retinopathy, “dot” hemorrhages and microaneurysms are generally asymptomatic and characterized by the loss of microvascular endothelial cells and pericytes (125-127). This stage is termed nonproliferative retinopathy. Since the initial observation of capillary microaneurysms by MacKenzie and Nettleship in 1879 (128), abnormalities of the retinal vasculature have been known to play a central role in the pathogenesis of diabetic retinopathy. The loss of microvascular cells is an early event in diabetic retinopathy that leads to a loss of perfusion and eventual hypoxia (126, 127, 129, 130). Hypoxia in turn induces the expression of angiogenic factors as a compensatory reaction leading to the next phase, proliferative diabetic retinopathy. It is during the later phase that vision is impaired.
It has recently come to light that diabetes increases the level of inflammation in the early stages of diabetic retinopathy and that this inflammation may be critical in the loss of pericytes and endothelial cells (131). One of the inflammatory factors that are elevated is TNF–alpha. TNF levels are higher in diabetic retinas and linked to the early inflammatory events of diabetic retinopathy (20, 132, 133). The best evidence that TNF plays a role in the pathology of the diabetic retina comes from studies using TNF inhibitors. Joussen and colleagues reported that inhibiting TNF caused a significant reduction in the expression of inflammatory molecules such as ICAM-1 that were elevated in diabetic rats (133). TNF inhibition also reduced the effect of diabetes-enhanced leukocyte adhesion, expression of eNOS, activation of NF-kappaB and blood-retinal barrier breakdown. In addition, positive results have been obtained in humans using a TNF inhibitor to treat diabetic macular edema (134). Treatment with the TNF inhibitor etanercept in a related experimental pathology, LPS induced uveitis, significantly reduced inflammation and apoptosis in the retina (135). These studies support the notion that excessive levels of TNF-alpha contribute to the early events of diabetic retinopathy in particular, because it is a potent inducer of apoptosis in microvascular cells (136, 137). Aspirin and other salicylate anti-inflammatory drugs inhibited activation and nuclear translocation of NF-kappaB, expression of inflammatory markers and the loss of microvascular cells, emphasizing the role of inflammation in the early stages of this diabetic complication. (138, 139).
5. CONCLUSIONS
The impact of diabetes on metabolic pathways, their impact on innate immunity and contribution toward diabetic complications including periodontal disease are shown in Figure 4. A number of pathways are affected by diabetes due to hyperglycemia. These include increased oxidative stress, shunting through the polyol pathway, elevated PKC activity and accelerated formation of advanced glycation end products. These disturbances may lead to alterations in innate immunity by enhancing the infiltration by monocytes and PMNS, increased expression of pro-inflammatory cytokines, and deficits in bacterial killing. Each of these parameters may come into play in periodontal diseases aggravated by diabetes. For example, decreased bacterial killing may facilitate the growth of pathogens. An altered innate immune response to periodontal pathogens can lead to greater or prolonged expression of cytokines that stimulate bone resorption and may limit bone repair. Interestingly, similar abnormalities are noted in other diabetic complications such as retinopathy, nephropathy and impaired fracture healing. The advances in specific inhibitors and anti inflammatory drugs provide therapeutic possibilities for these complications and improving the quality of life for diabetic patients.
Figure 4.
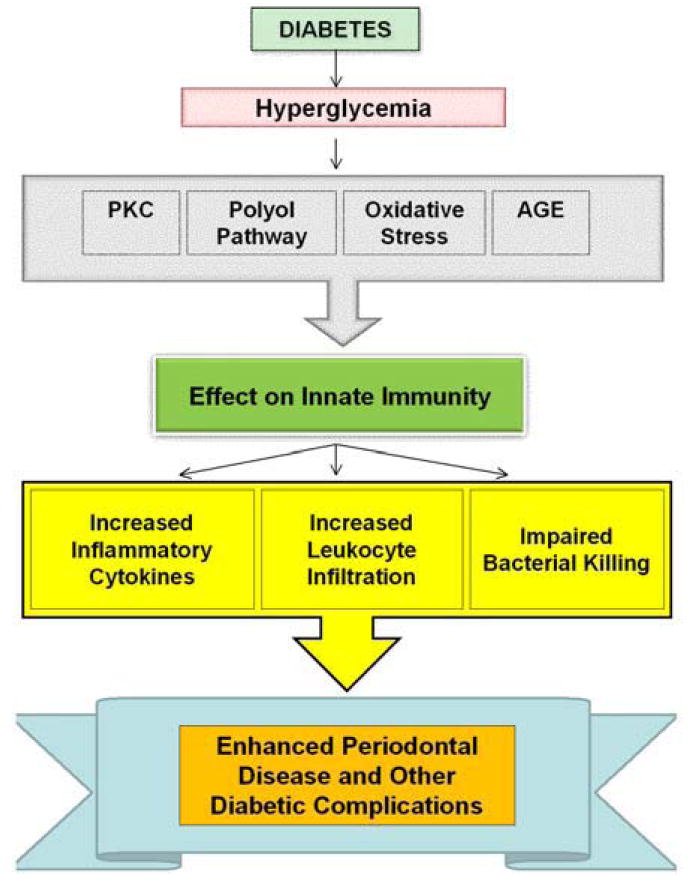
Metabolic dysregulation caused by hyperglycemia alters the innate immune response and contributes to diabetic complications. Due to complexity, the detailed interactions between different pathways are not shown. Hyperglycemia caused by diabetes leads to an increased oxidative stress, activation of the polyol pathway, formation of AGEs and increased PKC activity. These factors affect the innate immune system by increasing proinflammatory cytokine expression such as TNF-alpha, enhancing PMN and monocyte recruitment and impairing bacterial killing. These alterations may contribute to complications seen in diabetes.
Acknowledgments
We would like to thank Alicia Ruff for help in preparing this manuscript and for support from NIH grants DE07559 and P01 AR49920.
References
- 1.Cowie C, Rust K, Byrd-Holt D, Eberhardt M, Flegal K, Engelgau M, Saydah S, Williams D, Geiss L, Gregg E. Prevalence of diabetes and impaired fasting glucose in adults in the U.S. population:National and Nutrition Examination Survey. Diabetes Care. 2006;29:1263–8. doi: 10.2337/dc06-0062. [DOI] [PubMed] [Google Scholar]
- 2.Donathand M, Halban P. Decreased beta-cell mass in diabetes: significance, mechanisms and therapeutic implications. Diabetologia. 2004;47:581–9. doi: 10.1007/s00125-004-1336-4. [DOI] [PubMed] [Google Scholar]
- 3.Green EA, Eynon EE, Flavell RA. Local expression of TNFalpha in neonatal NOD mice promotes diabetes by enhancing presentation of islet antigens. Immunity. 1998;9:733–43. doi: 10.1016/s1074-7613(00)80670-6. [DOI] [PubMed] [Google Scholar]
- 4.Kahnand B, Flier J. Obesity and insulin resistance. J Clin Invest. 2000;106:473–81. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colditz GA, Willett WC, Rotnitzky A, Manson JE. Weight gain as a risk factor for clinical diabetes mellitus in women. Ann Intern Med. 1995;122:481–6. doi: 10.7326/0003-4819-122-7-199504010-00001. [DOI] [PubMed] [Google Scholar]
- 6.Mokdad AH, Ford ES, Bowman BA, Dietz WH, Vinicor F, Bales VS, Marks JS. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. Jama. 2003;289:76–9. doi: 10.1001/jama.289.1.76. [DOI] [PubMed] [Google Scholar]
- 7.Schienkiewitz A, Schulze MB, Hoffmann K, Kroke A, Boeing H. Body mass index history and risk of type 2 diabetes, results from the European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Am J Clin Nutr. 2006;84:427–33. doi: 10.1093/ajcn/84.1.427. [DOI] [PubMed] [Google Scholar]
- 8.Parikh NI, Pencina MJ, Wang TJ, Lanier KJ, Fox CS, D’Agostino RB, Vasan RS. Increasing trends in incidence of overweight and obesity over 5 decades. Am J Med. 2007;120:242–50. doi: 10.1016/j.amjmed.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Greenbergand A, Obin M. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr. 2006;83:461S–465S. doi: 10.1093/ajcn/83.2.461S. [DOI] [PubMed] [Google Scholar]
- 10.Dicksonand L, Rhodes C. Pancreatic B-cell growth and survival in the onset of type 2 diabetes, a role for protein kinase B in the Akt. Am J Physiol Endocrinol Metab. 2004;287:E192–E198. doi: 10.1152/ajpendo.00031.2004. [DOI] [PubMed] [Google Scholar]
- 11.Asnaghi V, Gerhardinger C, Hoehn T, Adeboje A, Lorenzi M. A role for the polyol pathway in the early neuroretinal apoptosis and glial changes induced by diabetes in the rat. Diabetes. 2003;52:506–11. doi: 10.2337/diabetes.52.2.506. [DOI] [PubMed] [Google Scholar]
- 12.Williamson J, Chang K, Frangos M. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42:801–813. doi: 10.2337/diab.42.6.801. [DOI] [PubMed] [Google Scholar]
- 13.Vlassara H. Recent progress in advanced glycation end products and diabetic complications. Diabetes. 1997;46(Suppl. 2):S19–S25. doi: 10.2337/diab.46.2.s19. [DOI] [PubMed] [Google Scholar]
- 14.Greeneand D, Stevens M. The sorbitol-osmotic and sorbitor-redox hypotheses. Diabetes Mellitus Lippincott Raven. 1996:801–809. [Google Scholar]
- 15.Kingand G, Brownlee M. The cellular and molecular mechanisms of diabetic complications. Endocrinol Metab Clin North Am. 1996;25:255–270. doi: 10.1016/s0889-8529(05)70324-8. [DOI] [PubMed] [Google Scholar]
- 16.Koyaand D, King G. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 17.Graves DT, Naguib G, Lu H, Leone C, Hsue H, Krall E. Inflammation is more persistent in Type 1 diabetic mice. J Dent Res. 2005;84:324–328. doi: 10.1177/154405910508400406. [DOI] [PubMed] [Google Scholar]
- 18.Lu H, Raptis M, Black E, Stan M, Amar S, Graves DT. Influence of diabetes on the exacerbation of an inflammatory response in cardiovascular tissue. Endocrinology. 2004;145:4934–9. doi: 10.1210/en.2004-0737. [DOI] [PubMed] [Google Scholar]
- 19.Salvi G, Beck J, Offenbacher S. PGE2, IL-1 and TNF- responses in diabetics as modifiers of periodontal disease expression. Ann Periodontol. 1998;3:40–50. doi: 10.1902/annals.1998.3.1.40. [DOI] [PubMed] [Google Scholar]
- 20.Doganay S, Evereklioglu C, Er H, Turkoz Y, Sevinc A, Mehmet N, Savli H. Comparison of serum NO, TNF-alpha, IL-1beta, sIL-2R, IL-6 and IL-8 levels with grades of retinopathy in patients with diabetes mellitus. Eye. 2002;16:163–70. doi: 10.1038/sj.eye.6700095. [DOI] [PubMed] [Google Scholar]
- 21.Navarro JF, Mora C, Maca M, Garca J. Inflammatory parameters are independently associated with urinary albumin in type 2 diabetes mellitus. Am J Kidney Dis. 2003;42:53–61. doi: 10.1016/s0272-6386(03)00408-6. [DOI] [PubMed] [Google Scholar]
- 22.Goova MT, Li J, Kislinger T, Qu W, Lu Y, Bucciarelli LG, Nowygrod S, Wolf BM, Caliste X, Yan SF, Stern DM, Schmidt AM. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol. 2001;159:513–25. doi: 10.1016/S0002-9440(10)61723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwasaki Y, Kambayashi M, Asai M, Yoshida M, Nigawara T, Hashimoto K. High glucose alone, as well as in combination with proinflammatory cytokines, stimulates nuclear factor kappa-B-mediated transcription in hepatocytes in vitro. J Diabetes Complications. 2007;21:56–62. doi: 10.1016/j.jdiacomp.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 24.Yerneni KK, Bai W, Khan BV, Medford RM, Natarajan R. Hyperglycemia-induced activation of nuclear transcription factor kappaB in vascular smooth muscle cells. Diabetes. 1999;48:855–64. doi: 10.2337/diabetes.48.4.855. [DOI] [PubMed] [Google Scholar]
- 25.Naguib G, Al-Mashat H, Desta T, Graves D. Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation. J Invest Dermatol. 2004;123:87–92. doi: 10.1111/j.0022-202X.2004.22711.x. [DOI] [PubMed] [Google Scholar]
- 26.Liu R, Bal HS, Desta T, Behl Y, Graves DT. Tumor necrosis factor-alpha mediates diabetes-enhanced apoptosis of matrix-producing cells and impairs diabetic healing. Am J Pathol. 2006;168:757–64. doi: 10.2353/ajpath.2006.050907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goren I, Muller E, Pfeilschifter J, Frank S. Severely impaired insulin signaling in chronic wounds of diabetic ob/ob mice: a potential role of tumor necrosis factor-alpha. Am J Pathol. 2006;168:765–77. doi: 10.2353/ajpath.2006.050293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DiPetrillo K, Coutermarsh B, Gesek FA. Urinary tumor necrosis factor contributes to sodium retention and renal hypertrophy during diabetes. Am J Physiol Renal Physiol. 2003;284:F113–21. doi: 10.1152/ajprenal.00026.2002. [DOI] [PubMed] [Google Scholar]
- 29.Gabbay K. The sorbitol pathway and the complications of diabetes. N Engl J Med. 1973;288:831–836. doi: 10.1056/NEJM197304192881609. [DOI] [PubMed] [Google Scholar]
- 30.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 31.Cameronand N, Cotter M. Impaired contraction and relaxation in aorta from streptozotocin-diabetic rats: role of polyol pathway. Diabetologia. 1992;35:1011–1019. doi: 10.1007/BF02221675. [DOI] [PubMed] [Google Scholar]
- 32.Cohen R. Endothelial dysfunction in diabetic vascular disease. Mediographia. 1997;87:31–38. [Google Scholar]
- 33.Ramana KV, Friedrich B, Srivastava S, Bhatnagar A, Srivastava SK. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes. 2004;53:2910–20. doi: 10.2337/diabetes.53.11.2910. [DOI] [PubMed] [Google Scholar]
- 34.Ramana KV, Friedrich B, Bhatnagar A, Srivastava SK. Aldose reductase mediates cytotoxic signals of hyperglycemia and TNF-alpha in human lens epithelial cells. Faseb J. 2003;17:315–7. doi: 10.1096/fj.02-0568fje. [DOI] [PubMed] [Google Scholar]
- 35.Ramana KV, Bhatnagar A, Srivastava SK. Inhibition of aldose reductase attenuates TNF-alpha-induced expression of adhesion molecules in endothelial cells. Faseb J. 2004;18:1209–18. doi: 10.1096/fj.04-1650com. [DOI] [PubMed] [Google Scholar]
- 36.Mazadeand MA, Edwards MS. Impairment of type III group B Streptococcus-stimulated superoxide production and opsonophagocytosis by neutrophils in diabetes. Mol Genet Metab. 2001;73:259–67. doi: 10.1006/mgme.2001.3185. [DOI] [PubMed] [Google Scholar]
- 37.Ihm SH, Yoo HJ, Park SW, Park CJ. Effect of tolrestat, an aldose reductase inhibitor, on neutrophil respiratory burst activity in diabetic patients. Metabolism. 1997;46:634–8. doi: 10.1016/s0026-0495(97)90005-6. [DOI] [PubMed] [Google Scholar]
- 38.Tebbs SE, Lumbwe CM, Tesfaye S, Gonzalez AM, Wilson RM. The influence of aldose reductase on the oxidative burst in diabetic neutrophils. Diabetes Res Clin Pract. 1992;15:121–9. doi: 10.1016/0168-8227(92)90015-j. [DOI] [PubMed] [Google Scholar]
- 39.Boland OM, Blackwell CC, Clarke BF, Ewing DJ. Effects of ponalrestat, an aldose reductase inhibitor, on neutrophil killing of Escherichia coli and autonomic function in patients with diabetes mellitus. Diabetes. 1993;42:336–40. doi: 10.2337/diab.42.2.336. [DOI] [PubMed] [Google Scholar]
- 40.Wolff S, Crabbe M, Thornalley P. The autoxidation of glyceraldehyde and other simple monosccharides. Experientia. 1984;40:244–248. doi: 10.1016/0304-4165(84)90131-4. [DOI] [PubMed] [Google Scholar]
- 41.Giugliano D, Cerielo A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care. 1996;19:257–267. doi: 10.2337/diacare.19.3.257. [DOI] [PubMed] [Google Scholar]
- 42.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–25. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 43.Baynesand J, Thorpe S. Role of oxidative stress in diabetic complications. Diabetes. 1994;48:1–9. doi: 10.2337/diabetes.48.1.1. [DOI] [PubMed] [Google Scholar]
- 44.Hornalley P, Wolff S, Crabbe M. The oxidation of oxyhaemoglobin by glyceraldehyde and other simple monosaccharides. Biochem J. 1984;217:615–622. doi: 10.1042/bj2170615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Millar TM, Phan V, Tibbles LA. ROS generation in endothelial hypoxia and reoxygenation stimulates MAP kinase signaling and kinase-dependent neutrophil recruitment. Free Radic Biol Med. 2007;42:1165–77. doi: 10.1016/j.freeradbiomed.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 46.Mitraand S, Abraham E. Participation of superoxide in neutrophil activation and cytokine production. Biochim Biophys Acta. 2006;1762:732–41. doi: 10.1016/j.bbadis.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 47.Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 48.Stosic-Grujicic SD, Miljkovic DM, Cvetkovic ID, Maksimovic-Ivanic DD, Trajkovic V. Immunosuppressive and anti-inflammatory action of antioxidants in rat autoimmune diabetes. J Autoimmun. 2004;22:267–76. doi: 10.1016/j.jaut.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 49.Yin X, Zhang Y, Wu H, Zhu X, Zheng X, Jiang S, Zhuo H, Shen J, Li L, Qiu J. Protective effects of Astragalus saponin I on early stage of diabetic nephropathy in rats. J Pharmacol Sci. 2004;95:256–66. doi: 10.1254/jphs.fp0030597. [DOI] [PubMed] [Google Scholar]
- 50.Hiramatsu Y, Sekiguchi N, Hayashi M, Isshiki K, Yokota T, King GL, Loeken MR. Diacylglycerol production and protein kinase C activity are increased in a mouse model of diabetic embryopathy. Diabetes. 2002;51:2804–10. doi: 10.2337/diabetes.51.9.2804. [DOI] [PubMed] [Google Scholar]
- 51.Le Good J, Ziegler W, Parekh D. Protein kinase C isotypes controlled by phosphoinositide 3-kinase throught the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- 52.Zhou X, Yang W, Li J. Ca2+- and protein kinase C-dependent signaling pathway for nuclear factor-kappaB activation, inducible nitric-oxide synthase expression, and tumor necrosis factor-alpha production in lipopolysaccharide-stimulated rat peritoneal macrophages. J Biol Chem. 2006;281:31337–47. doi: 10.1074/jbc.M602739200. [DOI] [PubMed] [Google Scholar]
- 53.Diaz-Guerra MJ, Bodelon OG, Velasco M, Whelan R, Parker PJ, Bosca L. Up-regulation of protein kinase C-epsilon promotes the expression of cytokine-inducible nitric oxide synthase in RAW 264.7 cells. J Biol Chem. 1996;271:32028–33. doi: 10.1074/jbc.271.50.32028. [DOI] [PubMed] [Google Scholar]
- 54.Waki K, Inanami O, Yamamori T, Nagahata H, Kuwabara M. Involvement of protein kinase Cdelta in the activation of NADPH oxidase and the phagocytosis of neutrophils. Free Radic Res. 2006;40:359–67. doi: 10.1080/10715760500539121. [DOI] [PubMed] [Google Scholar]
- 55.Karima M, Kantarci A, Ohira T, Hasturk H, Jones VL, Nam BH, Malabanan A, Trackman PC, Badwey JA, Van Dyke TE. Enhanced superoxide release and elevated protein kinase C activity in neutrophils from diabetic patients: association with periodontitis. J Leukoc Biol. 2005;78:862–70. doi: 10.1189/jlb.1004583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Devaraj S, Venugopal SK, Singh U, Jialal I. Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase c-{alpha} and -{beta} Diabetes. 2005;54:85–91. doi: 10.2337/diabetes.54.1.85. [DOI] [PubMed] [Google Scholar]
- 57.Kelly DJ, Chanty A, Gow RM, Zhang Y, Gilbert RE. Protein kinase Cbeta inhibition attenuates osteopontin expression, macrophage recruitment, and tubulointerstitial injury in advanced experimental diabetic nephropathy. J Am Soc Nephrol. 2005;16:1654–60. doi: 10.1681/ASN.2004070578. [DOI] [PubMed] [Google Scholar]
- 58.Stitt A, He C, Vlassara H. Characterization of the advanced glycation end-product receptor complex in human vascular endothelial cells. Biochem Biophys Res Commun. 1999;256:549–556. doi: 10.1006/bbrc.1999.0291. [DOI] [PubMed] [Google Scholar]
- 59.Cohen MP, Shea E, Chen S, Shearman CW. Glycated albumin increases oxidative stress, activates NF-kappa B and extracellular signal-regulated kinase (ERK), and stimulates ERK-dependent transforming growth factor-beta 1 production in macrophage RAW cells. J Lab Clin Med. 2003;141:242–9. doi: 10.1067/mlc.2003.27. [DOI] [PubMed] [Google Scholar]
- 60.Rashid G, Luzon AA, Korzets Z, Klein O, Zeltzer E, Bernheim J. The effect of advanced glycation end-products and aminoguanidine on TNFalpha production by rat peritoneal macrophages. Perit Dial Int. 2001;21:122–9. [PubMed] [Google Scholar]
- 61.Alikhani Z, Alikhani M, Boyd CM, Nagao K, Trackman PC, Graves DT. Advanced glycation end products enhance expression of pro-apoptotic genes and stimulate fibroblast apoptosis through cytoplasmic and mitochondrial pathways. J Biol Chem. 2005;280:12087–95. doi: 10.1074/jbc.M406313200. [DOI] [PubMed] [Google Scholar]
- 62.Alikhani M, Alikhani Z, Boyd C, MacLellan CM, Raptis M, Liu R, Pischon N, Trackman PC, Gerstenfeld L, Graves DT. Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone. 2007;40:345–53. doi: 10.1016/j.bone.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmidt A, Yan S, Brett J, Mora R, Nowygrod R, Stern D. Regulation of human mononuclear phagocyte migration by cell surface-binding proteins for advanced glycation end products. J Clin Invest. 1993;91:2155–2168. doi: 10.1172/JCI116442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brandtand R, Krantz S. Glycated albumin (Amadori product) induces activation of MAP kinases in monocyte-like MonoMac 6 cells. Biochim Biophys Acta. 2006;1760:1749–53. doi: 10.1016/j.bbagen.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 65.Lalla E, Lamster IB, Feit M, Huang L, Spessot A, Qu W, Kislinger T, Lu Y, Stern DM, Schmidt AM. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J Clin Invest. 2000;105:1117–24. doi: 10.1172/JCI8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Loe H. Periodontal disease. The sixth complication of diabetes mellitus. Diabetes Care. 1993;16:329–334. [PubMed] [Google Scholar]
- 67.Assuma R, Oates T, Cochran D, Amar S, Graves D. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J Immun. 1998;160:403–409. [PubMed] [Google Scholar]
- 68.Graves D, Delima A, Assuma R, Amar S, Oates T, Cochran D. Interleukin-1 and tumor necrosis factor antagonists inhibit the progression of inflammatory cell infiltration toward alveolar bone in experimental periodontitis. J Periodontol. 1998;69:1419–1425. doi: 10.1902/jop.1998.69.12.1419. [DOI] [PubMed] [Google Scholar]
- 69.Engebretson SP, Hey-Hadavi J, Ehrhardt FJ, Hsu D, Celenti RS, Grbic JT, Lamster IB. Gingival crevicular fluid levels of interleukin-1beta and glycemic control in patients with chronic periodontitis and type 2 diabetes. J Periodontol. 2004;75:1203–8. doi: 10.1902/jop.2004.75.9.1203. [DOI] [PubMed] [Google Scholar]
- 70.Engebretson S, Chertog R, Nichols A, Hey-Hadavi J, Celenti R, Grbic J. Plasma levels of tumour necrosis factor-alpha in patients with chronic periodontitis and type 2 diabetes. J Clin Periodontol. 2007;34:18–24. doi: 10.1111/j.1600-051X.2006.01017.x. [DOI] [PubMed] [Google Scholar]
- 71.Kurtis B, Tuter G, Serdar M, Akdemir P, Uygur C, Firatli E, Bal B. Gingival crevicular fluid levels of monocyte chemoattractant protein-1 and tumor necrosis factor-alpha in patients with chronic and aggressive periodontitis. J Periodontol. 2005;76:1849–55. doi: 10.1902/jop.2005.76.11.1849. [DOI] [PubMed] [Google Scholar]
- 72.Salvi G, Yalda B, Collins J, Jones B, Smith F, Arnold R, Offenbacher S. Inflammatory mediator response as a potential risk marker for periodontal diseases in insulin-dependent diabetes mellitus patients. J Periodontol. 1997;68:127–135. doi: 10.1902/jop.1997.68.2.127. [DOI] [PubMed] [Google Scholar]
- 73.Salvi G, Collins J, Yalda B, Arnold R, Lang N, Offenbacher S. Monocytic TNFalpha secretion patterns in IDDM patients with periodontal diseases. J Clin Periodontol. 1997;24:8–16. doi: 10.1111/j.1600-051x.1997.tb01178.x. [DOI] [PubMed] [Google Scholar]
- 74.Bridges RB, Anderson JW, Saxe SR, Gregory K, Bridges SR. Periodontal status of diabetic and nondiabetic men: effects of smoking, glycemic control, and socioeconomic factors. J Periodontol. 1996;67:1185–92. doi: 10.1902/jop.1996.67.11.1185. [DOI] [PubMed] [Google Scholar]
- 75.Salvi GE, Kandylaki M, Troendle A, Persson GR, Lang NP. Experimental gingivitis in type 1 diabetics: a controlled clinical and microbiological study. J Clin Periodontol. 2005;32:310–6. doi: 10.1111/j.1600-051X.2005.00682.x. [DOI] [PubMed] [Google Scholar]
- 76.Richardsand D, Rutherford R. The effects of interleukin 1 on collagenolytic activity and prostaglandin-E secretion by human periodontal-ligament and gingival fibroblasts. Arch Oral Biol. 1988;33:237–43. doi: 10.1016/0003-9969(88)90184-7. [DOI] [PubMed] [Google Scholar]
- 77.Mealeyand BL, Oates TW. Diabetes mellitus and periodontal diseases. J Periodontol. 2006;77:1289–303. doi: 10.1902/jop.2006.050459. [DOI] [PubMed] [Google Scholar]
- 78.Mahamed DA, Marleau A, Alnaeeli M, Singh B, Zhang X, Penninger JM, Teng YT. G(-) anaerobes-reactive CD4+ T-cells trigger RANKL-mediated enhanced alveolar bone loss in diabetic NOD mice. Diabetes. 2005;54:1477–86. doi: 10.2337/diabetes.54.5.1477. [DOI] [PubMed] [Google Scholar]
- 79.Wetzler C, Kampfer H, Stallmeyer B, Pfeilschifter J, Frank S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse prolonged persistence of neutrophils and macrophages during the late phase of repair. J Invest Dermatol. 2000;115:245–53. doi: 10.1046/j.1523-1747.2000.00029.x. [DOI] [PubMed] [Google Scholar]
- 80.Furudoi S, Yoshii T, Komori T. Balance of tumor necrosis factor alpha and interleukin-10 in a buccal infection in a streptozotocin-induced diabetic rat model. Cytokine. 2003;24:143–9. doi: 10.1016/j.cyto.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 81.Liu R, Bal HS, Desta T, Krothapalli N, Alyassi M, Luan Q, Graves D. Diabetes Enhances Periodontal Bone Loss Through Enhanced Resorption and Diminished Bone Formation. Journal of Dental Research. 2006;85:510–4. doi: 10.1177/154405910608500606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Parfitt A. The coupling of bone formation to bone resorption: a critical analysis of the concept and of its relevance to the pathogenesis of osteoporosis. Metab Bone Dis Relat Res. 1982;4:1–6. doi: 10.1016/0221-8747(82)90002-9. [DOI] [PubMed] [Google Scholar]
- 83.Al-Mashat HA, Kandru S, Liu R, Behl Y, Desta T, Graves DT. Diabetes enhances mRNA levels of proapoptotic genes and caspase activity, which contribute to impaired healing. Diabetes. 2006;55:487–95. doi: 10.2337/diabetes.55.02.06.db05-1201. [DOI] [PubMed] [Google Scholar]
- 84.Drachman R, Root R, Wood WJ. Studies on the effect of experimental nonketotic diabetes mellitus on antibacterial defense. I demonstration of a defect in phagocytosis. J Exp Med. 1966;124:227–40. doi: 10.1084/jem.124.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sima A, O’Neill S, Naimark D, Yagihashi S, Klass D. Bacterial phagocytosis and intracellular killing by alveolar macrophages in BB rats. Diabetes. 1988;37:544–9. doi: 10.2337/diab.37.5.544. [DOI] [PubMed] [Google Scholar]
- 86.Cutler CW, Eke P, Arnold RR, Van Dyke TE. Defective neutrophil function in an insulin-dependent diabetes mellitus patients. A case report. J Periodontol. 1991;62:394–401. doi: 10.1902/jop.1991.62.6.394. [DOI] [PubMed] [Google Scholar]
- 87.Gocer P, Gurer US, Erten N, Palanduz S, Rayaman E, Akarsu B, Karan A, Cevikbas A. Comparison of polymorphonuclear leukocyte functions in elderly patients and healthy young volunteers. Med Princ Pract. 2005;14:382–5. doi: 10.1159/000088109. [DOI] [PubMed] [Google Scholar]
- 88.Joshi N, Caputo G, Weitekamp M, Karchmer A. Infections in patients with diabetes mellitus. New England Journal of Medicine. 1999;341:1906–12. doi: 10.1056/NEJM199912163412507. [DOI] [PubMed] [Google Scholar]
- 89.Cutler C, Machen R, Jotwani R, Iacopino A. Heightened gingival inflammation and attachment loss in type 2 diabetics with hyperlipidiemia. J Periodontol. 1999;70:1313–21. doi: 10.1902/jop.1999.70.11.1313. [DOI] [PubMed] [Google Scholar]
- 90.Pattersonand J, Andriole V. Bacterial urinary tract infections in diabetes. Infect Dis Clin North Am. 1997;11:735–50. doi: 10.1016/s0891-5520(05)70383-4. [DOI] [PubMed] [Google Scholar]
- 91.Goldstein E. Bite wounds and infection. Clin Infect Dis. 1992;14:633–8. doi: 10.1093/clinids/14.3.633. [DOI] [PubMed] [Google Scholar]
- 92.Brook I. Microbiology and management of human and animal bite wound infections. Prim Care. 2003;30:25–39. doi: 10.1016/s0095-4543(02)00056-8. [DOI] [PubMed] [Google Scholar]
- 93.Williams R. Periodontal Disease. New Engl J Med. 1990;322:373–382. doi: 10.1056/NEJM199002083220606. [DOI] [PubMed] [Google Scholar]
- 94.Nishimura F, Takahashi K, Kurihara M, Takashiba S, Murayama Y. Periodontal disease as a complication of diabetes mellitus. Ann Periodonto. 1998;3:20–29. doi: 10.1902/annals.1998.3.1.20. [DOI] [PubMed] [Google Scholar]
- 95.Hofmann M, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath M, Slattery T, Beach D, Mcclary J, Nagashima M, Morser J, Stern D, Schmidt A. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypetides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 96.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 97.Takeda R, Suzuki E, Satonaka H, Oba S, Nishimatsu H, Omata M, Fujita T, Nagai R, Hirata Y. Blockade of endogenous cytokines mitigates neointimal formation in obese Zucker rats. Circulation. 2005;111:1398–406. doi: 10.1161/01.CIR.0000158482.83179.DB. [DOI] [PubMed] [Google Scholar]
- 98.Bucciarelli L, Wendt T, Qu W, L Y, Lalla E, Rong L, Goova M, Moser B, Kislinger T, Lee D, Kashyap Y, Stern D, Schmidt A. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 2002;106:246–53. doi: 10.1161/01.cir.0000039325.03698.36. [DOI] [PubMed] [Google Scholar]
- 99.Herrera V, Makrides S, Xie H, Adari H, Krauss R, Ryan U, Opazo N. Spontaneous combined hyperlipidemia, coronary heart disease and decreased survival in Dahl salt-sensitive hypertensive rats transgenic for human cholesteryl ester transer protein. Nature Medicine. 1999;5:1383–1389. doi: 10.1038/70956. [DOI] [PubMed] [Google Scholar]
- 100.Mantovand S, Raev D. Additive effect of diabetes and systemic hypertension on the immune mechanisms of atherosclerosis. Int J Cardiol. 1996;56:145–8. doi: 10.1016/0167-5273(96)02706-4. [DOI] [PubMed] [Google Scholar]
- 101.Epstein S. The multiple mechanisms by which infection may contribute to atherosclerosis development and course. Circ Res. 2002;90:2–4. [PubMed] [Google Scholar]
- 102.Mahonyand J, Coombes B. Chlamydia pneumoniae and atherosclerosis: does the evidence support a casual or contributory role? FEMS Microbiol Lett. 2001;197:1–9. doi: 10.1111/j.1574-6968.2001.tb10574.x. [DOI] [PubMed] [Google Scholar]
- 103.Kustersand J, Kuipers E. Helicobacter and atherosclerosis. Am Heart J. 1999;138:S523–7. doi: 10.1016/s0002-8703(99)70292-9. [DOI] [PubMed] [Google Scholar]
- 104.Beck J, Garcia R, Heiss G, Vokonas P, Offenbacher S. Periodontal disease and cardiovascular disease. J Periodontol. 1996;67(suppl):1123–1137. doi: 10.1902/jop.1996.67.10s.1123. [DOI] [PubMed] [Google Scholar]
- 105.Desvarieux M, Demmer RT, Rundek T, Boden-Albala B, Jacobs DR, Jr, Sacco RL, Papapanou PN. Periodontal microbiota and carotid intima-media thickness: the Oral Infections and Vascular Disease Epidemiology Study (INVEST) Circulation. 2005;111:576–82. doi: 10.1161/01.CIR.0000154582.37101.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sconyers J, Crawford J, Moriarty J. Relationship of bacteremia to toothbrushing in patience with periodontitis. J Am Dent Assoc. 1973;87:616–622. doi: 10.14219/jada.archive.1973.0453. [DOI] [PubMed] [Google Scholar]
- 107.Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V. Endotoxemia, Immune Response to Periodontal Pathogens, and Systemic Inflammation Associate With Incident Cardiovascular Disease Events. Arterioscler Thromb Vasc Biol. 2007 doi: 10.1161/ATVBAHA.106.138743. [DOI] [PubMed] [Google Scholar]
- 108.Wiedermann C, Kiechl S, Dunzendorfer S, Schratzberger P, Egger G, Oberhollenzer F, Willeit J. Association of endoxtoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck Study. J Am Coll Cardiol. 1999;34:1975–81. doi: 10.1016/s0735-1097(99)00448-9. [DOI] [PubMed] [Google Scholar]
- 109.Ostos M, Recalde D, Zakin M, Scott-Algara D. Implication of natural killer T cells in atherosclerosis development during a LPS-induced chronic inflammation. FEBS Lett. 2002;519:23–9. doi: 10.1016/s0014-5793(02)02692-3. [DOI] [PubMed] [Google Scholar]
- 110.Ninichuk V, Khandoga AG, Segerer S, Loetscher P, Schlapbach A, Revesz L, Feifel R, Khandoga A, Krombach F, Nelson PJ, Schlondorff D, Anders HJ. The Role of Interstitial Macrophages in Nephropathy of Type 2 Diabetic db/db Mice. Am J Pathol. 2007;170:1267–76. doi: 10.2353/ajpath.2007.060937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Galkinaand E, Ley K. Leukocyte recruitment and vascular injury in diabetic nephropathy. J Am Soc Nephrol. 2006;17:368–77. doi: 10.1681/ASN.2005080859. [DOI] [PubMed] [Google Scholar]
- 112.Mensah-Brown EP, Obineche EN, Galadari S, Chandranath E, Shahin A, Ahmed I, Patel SM, Adem A. Streptozotocin-induced diabetic nephropathy in rats: the role of inflammatory cytokines. Cytokine. 2005;31:180–90. doi: 10.1016/j.cyto.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 113.Hasegawa G, Nakano K, Sawada M, Uno K, Shibayama Y, Ienaga K, Kondo M. Possible role of tumor necrosis factor and interleukin-1 in the development of diabetic nephropathy. Kidney Int. 1991;40:1007–12. doi: 10.1038/ki.1991.308. [DOI] [PubMed] [Google Scholar]
- 114.Nakamura T, Fukui M, Ebihara I, Osada S, Nagaoka I, Tomino Y, Koide H. mRNA expression of growth factors in glomeruli from diabetic rats. Diabetes. 1993;42:450–6. doi: 10.2337/diab.42.3.450. [DOI] [PubMed] [Google Scholar]
- 115.Sugimoto H, Shikata K, Wada J, Horiuchi S, Makino H. Advanced glycation end products-cytokine-nitric oxide sequence pathway in the development of diabetic nephropathy: aminoguanidine ameliorates the overexpression of tumour necrosis factor-alpha and inducible nitric oxide synthase in diabetic rat glomeruli. Diabetologia. 1999;42:878–86. doi: 10.1007/s001250051241. [DOI] [PubMed] [Google Scholar]
- 116.Navarro JF, Milena FJ, Mora C, Leon C, Garcia J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J Nephrol. 2006;26:562–70. doi: 10.1159/000098004. [DOI] [PubMed] [Google Scholar]
- 117.Okada S, Shikata K, Matsuda M, Ogawa D, Usui H, Kido Y, Nagase R, Wada J, Shikata Y, Makino H. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes. 2003;52:2586–93. doi: 10.2337/diabetes.52.10.2586. [DOI] [PubMed] [Google Scholar]
- 118.Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Tesch GH. Intercellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J Am Soc Nephrol. 2005;16:1711–22. doi: 10.1681/ASN.2004070612. [DOI] [PubMed] [Google Scholar]
- 119.Kayal RA, Tsatsas D, Bauer MA, Allen B, Al-Sebaei MO, Kakar S, Leone CW, Morgan EF, Gerstenfeld LC, Einhorn TA, Graves DT. Diminished bone formation during diabetic fracture healing is related to the premature resorption of cartilage associated with increased osteoclast activity. J Bone Miner Res. 2007;22:560–8. doi: 10.1359/jbmr.070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Beam H, Parsons J, Lin S. The effects of blood glucose control upon fracture healing in the BB Wistar rat with diabetes mellitus. J Orthop Res. 2002;20:1210–6. doi: 10.1016/S0736-0266(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 121.Funk JR, Hale JE, Carmines D, Gooch HL, Hurwitz SR. Biomechanical evaluation of early fracture healing in normal and diabetic rats. J Orthop Res. 2000;18:126–32. doi: 10.1002/jor.1100180118. [DOI] [PubMed] [Google Scholar]
- 122.Kagel E, Majeska R, Einhorn T. Effects of diabetes and steroids on fracture healing. Curr Opin Orthop. 1995;6:7–13. doi: 10.1097/00001433-199510000-00002. [DOI] [PubMed] [Google Scholar]
- 123.Gerstenfeld LC, Cho TJ, Kon T, Aizawa T, Tsay A, Fitch J, Barnes GL, Graves DT, Einhorn TA. Impaired fracture healing in the absence of TNF-alpha signaling: the role of TNF-alpha in endochondral cartilage resorption. J Bone Miner Res. 2003;18:1584–92. doi: 10.1359/jbmr.2003.18.9.1584. [DOI] [PubMed] [Google Scholar]
- 124.Donnelly R, Emslie-Smith AM, Gardner ID, Morris AD. ABC of arterial and venous disease: vascular complications of diabetes. Bmj. 2000;320:1062–6. doi: 10.1136/bmj.320.7241.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cogan DG, Toussaint D, Kuwabara T. Retinal vascular patterns. IV. Diabetic retinopathy. Arch Ophthalmol. 1961;66:366–78. doi: 10.1001/archopht.1961.00960010368014. [DOI] [PubMed] [Google Scholar]
- 126.Stanford M. The pathogenesis of diabetic retinopathy. Br J Opthalmol. 2004;88:444–445. doi: 10.1136/bjo.2003.031443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Frank R. Diabetic Retinopathy. N Engl J Med. 2004;350:48–58. doi: 10.1056/NEJMra021678. [DOI] [PubMed] [Google Scholar]
- 128.MacKenzie S. A case of glycosuric retinitis, with comments. (Microscopical examination of the eyes by Mr. Nettleship) Roy London Ophthal Hosp Rep. 1879;9:134–157. [Google Scholar]
- 129.Neely K, Quillen D, Schachat A. Diabetic retinopathy. Med Clin North Am. 1998;82:847–876. doi: 10.1016/s0025-7125(05)70027-4. [DOI] [PubMed] [Google Scholar]
- 130.Ferris FI, Davis M, Aiello L. Treatment of diabetic retinopathy. New England Journal of Medicine. 1999;341:667–678. doi: 10.1056/NEJM199908263410907. [DOI] [PubMed] [Google Scholar]
- 131.Kowluruand RA, Odenbach S. Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br J Ophthalmol. 2004;88:1343–7. doi: 10.1136/bjo.2003.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Limb G, Chignell A, Green W, LeRoy F, Dumonde D. Distribution of TNF alpha and its reactive vascular adhesion molecules in fibrovascular membranes of proliferative diabetic retinopathy. Br J Ophthalmol. 1996;80:168–73. doi: 10.1136/bjo.80.2.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Joussen A, Poulakl V, Mitsiades N, Kirchhof B, K K, Dohmen S, Adamis A. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-{alpha} suppression. FASEB J. 2002;16:438–440. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 134.Sfikakis P, Markomichelakis N, Theodossiadis G, Grigoropoulos V, Katsilambros N, Theodossiadis P. Regression of sight-threatening macular edema in type 2 diabetes following treatment with the anti-tumor necrosis factor monoclonal antibody infliximab. Diabetes Care. 2005;28:445–447. doi: 10.2337/diacare.28.2.445. [DOI] [PubMed] [Google Scholar]
- 135.Koizumi K, Poulaki V, Doehmen S, Welsandt G, Radetzky S, Lappas A, Kociok N, Kirchhof B, Joussen A. Contribution of TNF-alpha to leukocyte adhesion, vascular leakage, and apoptotic cell death in endotoxin-induced uveitis in vivo. Invest Ophthalmol Vis Sci. 2003;44:2184–91. doi: 10.1167/iovs.02-0589. [DOI] [PubMed] [Google Scholar]
- 136.Kimura H, Gules I, Meguro T, Zhang J. Cytoxicity of cytokines in cerebral microvascular endothelial cell. Brain Res. 2003;990:148–56. doi: 10.1016/s0006-8993(03)03450-4. [DOI] [PubMed] [Google Scholar]
- 137.Romeo G, Liu W-H, Asnaghi V, Kern TS, Lorenzi M. Activation of Nuclear Factor-{kappa}B Induced by Diabetes and High Glucose Regulates a Proapoptotic Program in Retinal Pericytes. Diabetes. 2002;51:2241–2248. doi: 10.2337/diabetes.51.7.2241. [DOI] [PubMed] [Google Scholar]
- 138.Zheng L, Howell SJ, Hatala DA, Huang K, Kern TS. Salicylate-based anti-inflammatory drugs inhibit the early lesion of diabetic retinopathy. Diabetes. 2007;56:337–45. doi: 10.2337/db06-0789. [DOI] [PubMed] [Google Scholar]
- 139.Sun W, Gerhardinger C, Dagher Z, Hoehn T, Lorenzi M. Aspirin at low-intermediate concentrations protects retinal vessels in experimental diabetic retinopathy through non-platelet-mediated effects. Diabetes. 2005;54:3418–26. doi: 10.2337/diabetes.54.12.3418. [DOI] [PubMed] [Google Scholar]