


Abstract
There is an increasing interest in using single nucleotide polymorphism (SNP) genotyping arrays for profiling chromosomal rearrangements in tumors, as they allow simultaneous detection of copy number and loss of heterozygosity with high resolution. Critical issues such as signal baseline shift due to aneuploidy, normal cell contamination, and the presence of GC content bias have been reported to dramatically alter SNP array signals and complicate accurate identification of aberrations in cancer genomes. To address these issues, we propose a novel Global Parameter Hidden Markov Model (GPHMM) to unravel tangled genotyping data generated from tumor samples. In contrast to other HMM methods, a distinct feature of GPHMM is that the issues mentioned above are quantitatively modeled by global parameters and integrated within the statistical framework. We developed an efficient EM algorithm for parameter estimation. We evaluated performance on three data sets and show that GPHMM can correctly identify chromosomal aberrations in tumor samples containing as few as 10% cancer cells. Furthermore, we demonstrated that the estimation of global parameters in GPHMM provides information about the biological characteristics of tumor samples and the quality of genotyping signal from SNP array experiments, which is helpful for data quality control and outlier detection in cohort studies.
INTRODUCTION
One critical feature of cancer genomes is chromosomal aberrations (1). Recurrent genomic aberrations such as copy number gain or loss and loss of heterozygosity (LOH), are often associated with inappropriate expression of oncogenes, tumor suppressor genes, and genes that are involved in cancer development (2). Relationships between clinical outcome and chromosomal aberrations have been established based on the association of either individual genomic abnormalities such as amplification of HER2 (ERBB2) and MYCN (1) or distinct patterns of chromosomal abnormalities from whole genome profiling (3).
Emerging data on the genetic makeup of breast cancers show that particular regions of the genome are commonly amplified and these regions contain genes that drive cancer progression. The best example of an important amplified region is the 17q12 amplicon that harbors the HER2 oncogene. This amplicon leads to a more aggressive type of tumor, which is now the target of a highly successful antibody therapy, trastuzumab (Herceptin®). Several genes have been mapped to the HER2 amplicon based on co-expression and close proximity to the HER2 gene (4–7). It has been observed that RNAi knockdown of coamplified genes within the 17q12 amplicon resulted in decreased cell proliferation and increased apoptosis (8). Therefore, the 17q12 amplicon encodes a concerted genetic program that contributes to tumor phenotype.
Clinically, cytogenetic technologies such as fluorescence in situ hybridization (FISH) have been successfully used to detect chromosomal aberrations in cancer cells (1). Cytogenetic technologies do not allow high resolution genome-wide analysis, and for this reason array comparative genomic hybridization (aCGH) was developed, first using plasmid probes (9) and later using oligonucleotide probes (10). The introduction of single nucleotide polymorphism (SNP) genotyping arrays for copy number analysis is a major advance because they allow for simultaneous detection of copy number and allelic imbalance (including LOH) with high resolution. SNP arrays from two platforms, Affymetrix (11) and Illumina (12), have been widely adopted because of their high quality and number of probes. Despite the technical difference between these two platforms, it is suggested that similar algorithms can be applied if raw data from Affymetrix SNP arrays can be converted into the log R ratio (LRR) and B allele frequency (BAF) signals that are used in the Illumina platform when accompanied by appropriate normalization and transformation (13,14).
Though various computational methods have been proposed for automatic detection of copy number change or LOH from SNP array data (13–25), many of these methods are not specifically designed to study chromosomal aberrations in cancer genomes, and do not address some critical issues that have been encountered in previous studies of tumor samples (13,15,22,24,25). Specifically, recent studies (24,25) have shown that in SNP-array experiments, sample DNA is treated as if it has an approximate normal (near diploid) genome and therefore the normalized intensity signals may fail to correctly indicate the underlying aneuploidy in cancer cells. From LRR signals alone, cancer cells with a complete triploid genome cannot be distinguished from those with a normal diploid genome. To address this issue, new algorithms such as OverUnder (24) and GAP (25) have been proposed to correct erroneously-shifted LRR signal baseline. These algorithms are designed to infer copy number not only from total signal intensity but also allelic imbalance information. OverUnder examines the LRR distribution in regions with discriminative patterns observed from BAF signals; for example, a BAF stretch centering on 0.5 indicates even-numbered copy number (24). For Affymetrix platform, Greenman et al. (26) introduced a preprocessing transformation and hidden Markov model algorithm referring to aneuploid cancer samples. These approaches advanced our understanding of the systematic bias in SNP-array data for complex tumor samples.
Another issue in identification of chromosomal aberrations in cancer cells comes from the fact that biopsies extracted from a tumor usually contain normal, non-tumor cells (such as stroma and lymphocytes), which may lead to a dramatic alteration of both LRR and BAF signals generated from SNP array experiments. It has been reported that a small amount of normal cells admixed with tumor cells can be helpful for identifying somatic deletions (15,22). On the other hand, an increase in the proportion of normal stromal cells in a tumor sample will make both LRR and BAF signals converge to the typical pattern of a diploid genome (23). In other words, normal cell contamination decreases the signal-to-noise ratio in SNP array experiments and chromosomal aberrations can eventually become undetectable when normal cells dominate in a biopsy. Some studies attempted to solve this problem by modeling the dynamic patterns of both LRR and BAF signals in contaminated tumor samples (13,15,22,23,25). For example, SiDCoN provides empirical formulas of LRR/BAF signals in the presence of normal cell contamination (23). The formulas used for BAF signals were later shown to be identical to those adopted in the BAFsegmentation method (15), which was developed to detect LOH and allelic imbalance in cancer cells using only BAF signals. Recently, Sun et al. (13) proposed a different approach to solve this problem. They argued that a HMM with fixed parameters for copy number variation, as used in PennCNV (14), are inappropriate for analyzing tumor samples. Instead, sample-specific HMMs are required due to the varying proportions of normal cell contamination in tumor samples. Therefore they introduced an HMM named genoCNA, which is based on traditional continuous HMMs with a training procedure for parameter estimation.
There is another important issue in the assessment of SNP array data. Diskin et al. (27) identified genomic waves in both Illumina and Affymetrix platforms, which may prevent accurate inference of copy number changes. They further demonstrated that the wavy effects in LRR signals best correlate with GC content and proposed a linear regression model to correct GC content bias. However, an underlying assumption of this model is that SNPs used for regression should have the same copy number since copy number is the most influential factor in determining LRR signals. As a result, this model is suitable for normal genomes with sparse copy number alteration events but may not work well for tumor samples with widespread chromosomal abnormalities.
Critically, all the aforementioned issues strongly affect each other and will dramatically complicate determination of copy number and LOH if they occur in the same sample. For example, the OverUnder method for LRR signal shift in aneuploid tumors may fail because of altered patterns of BAF signals caused by normal cell contamination. Likewise, approaches to determine the normal cell proportion from altered BAF signals or to adjust GC content bias may also fail as copy number cannot be directly inferred from LRR signals if there is an erroneous baseline shift due to aneuploidy. Finally, failure to quantitatively measure the effect of LRR signal shift due to aneuploidy, normal cell contamination, and GC content bias will hamper correct identification of copy number change and LOH in cancer genomes. Therefore it is preferable that all these issues can be integrated together based on their empirical models suggested in (15,17,22,23,25,27) and then addressed simultaneously in a detection method. For example, using the dynamic model of LRR/BAF signals that has been validated in refs. (15,25), Popova et al. (25) pioneered a pattern recognition approach that first models a Genome Alteration Print (GAP) template parameterized with LRR baseline shift and the proportion of normal cells, and then by performing an exhaustive search of all parameter configurations, identify all feasible GAP templates from which the best interpretation is chosen based on pre-defined criteria. By modeling and solving these two issues simultaneously, this method demonstrates better performance than OverUnder on different data sets (25).
We have previously developed a HMM approach for detection of tumor chromosomal aberrations using SNP array data which addresses some of these issues but requires manual annotation of LRR shift and proportion of normal cells (28). In this study, we introduce a novel method, named Global Parameter Hidden Markov Model (GPHMM), which automatically dissects copy number alteration and LOH in SNP array data from tumor sample DNA. Instead of trying to separately address each of the complicating issues discussed above, we propose new observation probability density functions (pdfs) to allow quantitative modeling of all these issues together. Similar to the intent of GAP, GPHMM provides a comprehensive description of the statistical distributions of genotyping signals from tumor samples and a novel approach to address these issues by incorporating them into the HMM statistical framework. GPHMM provides not only improved solutions to these issues but also automatic and accurate identification of copy number and LOH status of each SNP in the assay.
METHODS
Definitions of hidden states
The definitions of the hidden states used in GPHMM are shown in Table 1. Here, we use ‘A’ and ‘B’ to represent the two SNP alleles inherited from parents. Different kinds of chromosomal abnormalities such as copy number gain/loss and LOH are modeled in this study. In addition we use tumor and normal cell genotype pairs to illustrate the intrinsic relationship between tumor genotype and the genotype of normal cells admixed in tumor samples. For example, (AAB, AB) represents the case that tumor genotype is ‘AAB’ while the genotype of the normal cell is ‘AB’. Certain constraints regarding these two genotypes are assumed, which are similar to the assumptions involved in the definition of hidden states used by genoCNA (13). We assume that the tumor genotype is derived from the genotype of normal cells; for example, when the normal genotype is homozygous, the tumor genotype can only be homozygous; when the normal genotype is heterozygous, the tumor genotype can be either heterozygous or homozygous. The maximal copy number state modeled in this study is set to 5, given the fact that due to saturation effects in array hybridization, genotyping signals may not provide adequate discriminative power to detect the difference between hidden states with copy number larger than or equal to 5. Furthermore, a special hidden state denoted as 0, is employed to represent occasional signal fluctuation. The effect of signal fluctuation is uniquely formulated (see next section for details) and independent of other hidden states. whereas it is instead modeled in the observation pdfs of the other states in PennCNV (14) and genoCNA (13). Finally, to avoid a possible overflow issue in practice, the copy number of deletion of two copies is set to a small positive number of 0.01.
Table 1.
Definition of hidden states in GPHMM
| State | Copy number | Allelic information | Copy number alteration status | (Tumor genotype, normal cell genotype) |
|---|---|---|---|---|
| 0 | N/A | N/A | Fluctuation effect | (N/A, AA), (N/A, BB), (N/A, AB) |
| 1 | 0 | Deletion | Deletion of two copies | (N/A, AA), (N/A, BB), (N/A, AB) |
| 2 | 1 | LOH | Deletion of one copy | (A,AA), (B,BB), (A,AB), (B,AB) |
| 3 | 2 | Heterozygous | Normal | (AA,AA), (BB,BB), (AB,AB) |
| 4 | 2 | LOH | Copy neutral with LOH | (AA,AA), (AA,AB), (BB,BB), (BB,AB) |
| 5 | 3 | Heterozygous | Three copies with duplication of one allele | (AAA,AA), (BBB,BB), (AAB,AB), (ABB,AB) |
| 6 | 3 | LOH | Three copies with LOH | (AAA,AA), (AAA,AB), (BBB,BB), (BBB,AB) |
| 7 | 4 | Heterozygous | Four copies with duplication of one allele | (AAAA,AA), (BBBB,BB), (AAAB,AB), (ABBB,AB) |
| 8 | 4 | Heterozygous | Four copies with duplication of both alleles | (AAAA,AA), (BBBB,BB), (AABB,AB) |
| 9 | 4 | LOH | Four copies with LOH | (AAAA,AA), (BBBB,BB), (AAAA,AB), (BBBB,AB) |
| 10 | 5 | Heterozygous | Five copies with duplication of one allele | (AAAAA,AA), (BBBBB,BB), (AAAAB,AB), (ABBBB,AB) |
| 11 | 5 | Heterozygous | Five copies with duplication of both alleles | (AAAAA,AA), (BBBBB,BB), (AAABB,AB), (AABBB,AB) |
| 12 | 5 | LOH | Five copies with LOH | (AAAAA,AA), (BBBBB,BB), (AAAAA,AB), (BBBBB,AB) |
Observation probability density functions
In this study, we propose new observation pdfs in GPHMM that include different quantitative models of the issues in tumor genotyping data analyses. We refer to the parameters used in the observation pdfs as global parameters. They are the key parameters of the proposed statistical framework and essentially control the distributions of both LRR and BAF signals for all hidden states in GPHMM. Five different global parameters are employed in all: proportion of normal cells (denoted as 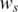 ), LRR baseline shift (denoted as o), coefficient of the GC content of investigated SNPs (denoted as h), and the standard deviation of LRR and BAF signals (denoted as
), LRR baseline shift (denoted as o), coefficient of the GC content of investigated SNPs (denoted as h), and the standard deviation of LRR and BAF signals (denoted as 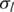 and
and 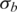 , respectively). These global parameters can be affected by the genetic makeup of a tumor sample, characteristics of chromosomal aberrations, quality of DNA mass, features of SNP array platform, and other technical details during experimentation.
, respectively). These global parameters can be affected by the genetic makeup of a tumor sample, characteristics of chromosomal aberrations, quality of DNA mass, features of SNP array platform, and other technical details during experimentation.
Suppose that the LRR signal (representing the over-all allele intensity) of the ith SNP in the array is 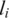 , then its observation pdf for a hidden state c (except state 0, see below) can be formulated as (here we simply assume all hidden states in GPHMM have the same signal variance):
, then its observation pdf for a hidden state c (except state 0, see below) can be formulated as (here we simply assume all hidden states in GPHMM have the same signal variance):
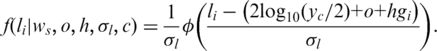 |
(1) |
Here 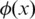 is the pdf of standard normal distribution.
is the pdf of standard normal distribution. 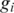 is the GC content associated with the ith SNP (27) and the logarithm part is adopted from the formula introduced in ref. (23), in which a log-linear relationship between the expected mean of LRR signal for each hidden state and the corresponding average copy number. The average copy number
is the GC content associated with the ith SNP (27) and the logarithm part is adopted from the formula introduced in ref. (23), in which a log-linear relationship between the expected mean of LRR signal for each hidden state and the corresponding average copy number. The average copy number 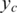 , is defined as:
, is defined as:
| (2) |
Here 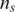 and
and 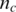 are the copy number of normal cell and the copy number of tumor in state c, respectively. For example, in the case of no normal contamination, the average copy number of state 0 by Equation (2) is 0.01 and the expected theoretical LRR mean (suppose neither GC content bias nor LRR baseline shift occurs) is −4.6, which is consistent with the results reported in refs (23). Using above formulas, the effects of LRR signal shift by aneuploidy, normal cell contamination and GC content bias are simultaneously modeled. As the intensity ratio of two different alleles, BAF signals are not directly affected by aneuploidy or GC content bias. However, they are extremely sensitive to normal cell contamination. Based on existing models described in refs (15) and (23), we derive the pdf of the BAF signal for the ith SNP in state c (c >1) as:
are the copy number of normal cell and the copy number of tumor in state c, respectively. For example, in the case of no normal contamination, the average copy number of state 0 by Equation (2) is 0.01 and the expected theoretical LRR mean (suppose neither GC content bias nor LRR baseline shift occurs) is −4.6, which is consistent with the results reported in refs (23). Using above formulas, the effects of LRR signal shift by aneuploidy, normal cell contamination and GC content bias are simultaneously modeled. As the intensity ratio of two different alleles, BAF signals are not directly affected by aneuploidy or GC content bias. However, they are extremely sensitive to normal cell contamination. Based on existing models described in refs (15) and (23), we derive the pdf of the BAF signal for the ith SNP in state c (c >1) as:
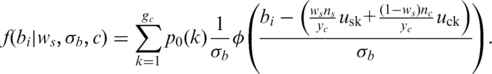 |
(3) |
Here 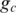 is the number of genotype pairs included in state c.
is the number of genotype pairs included in state c. 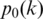 is the prior probability of observing genotype pair k, which is estimated from the BAF in the normal population (14), with respect to the genotype of the normal cell in the genotype pair.
is the prior probability of observing genotype pair k, which is estimated from the BAF in the normal population (14), with respect to the genotype of the normal cell in the genotype pair. 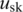 and
and 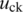 are the theoretical means of BAF signals for normal and pure tumor cells, respectively, in genotype pair k. For example with
are the theoretical means of BAF signals for normal and pure tumor cells, respectively, in genotype pair k. For example with 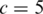 and
and 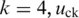 represents the mean value of tumor BAF signals for the forth genotype pair in state 5 [i.e. (ABB, AB), see in Table 1], which is defined as 0.667 in this study. The implementation of GPHMM, Equation (3), is further simplified by using mirrored BAF signals (equal or greater than 0.5), which requires fewer genotype configurations (15). For example, in state 3 only two genotype pairs: (BB, BB) and (AB, AB) are required after this transformation. Finally, it should be pointed out that, due to the concern of model simplicity and computational cost, the effect of BAF signal truncation investigated in ref. (13) is not taken into account in Equation (3).
represents the mean value of tumor BAF signals for the forth genotype pair in state 5 [i.e. (ABB, AB), see in Table 1], which is defined as 0.667 in this study. The implementation of GPHMM, Equation (3), is further simplified by using mirrored BAF signals (equal or greater than 0.5), which requires fewer genotype configurations (15). For example, in state 3 only two genotype pairs: (BB, BB) and (AB, AB) are required after this transformation. Finally, it should be pointed out that, due to the concern of model simplicity and computational cost, the effect of BAF signal truncation investigated in ref. (13) is not taken into account in Equation (3).
For SNPs in state 0, information about tumor genotype is not available from SNP-array experiments. Therefore a uniform distribution is employed to approximate the pdfs of LRR and BAF signals:
| (4) |
| (5) |
In this study, a, b are set to −5, 5 for LRR, and, a′, b′, are set to 0, 1 for BAF.
EM algorithm for GPHMM
For canonical HMMs, algorithms for parameter estimation have been proposed and successfully applied in fields such as speech recognition (29). In this study, we propose an EM algorithm (30) specially designed for GPHMM to update the global parameters in the algorithm. As suggested in ref. (30), for finite mixtures we can estimate global parameters using only partial log-likelihood functions. Specifically, in the nth iteration of the EM algorithm, given the LRR data we can formulate the partial log-likelihood as:
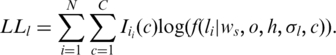 |
(6) |
Here, 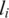 is the observed LRR value of the ith SNP and
is the observed LRR value of the ith SNP and 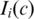 is an indicator function with value 1 if the ith SNP is in state c in tumor cells and value 0 otherwise. C is the total number of hidden states. In the expectation step of the EM algorithm, the expectation of the partial log-likelihood for LRR data is formulated as:
is an indicator function with value 1 if the ith SNP is in state c in tumor cells and value 0 otherwise. C is the total number of hidden states. In the expectation step of the EM algorithm, the expectation of the partial log-likelihood for LRR data is formulated as:
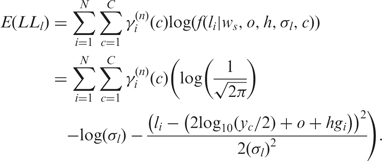 |
(7) |
Here 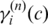 is the posterior probability of the ith SNP to be in state c, which can be calculated by the forward–backward algorithm (29). Similarly, we can derive the expectation of the partial log-likelihood function for BAF data as:
is the posterior probability of the ith SNP to be in state c, which can be calculated by the forward–backward algorithm (29). Similarly, we can derive the expectation of the partial log-likelihood function for BAF data as:
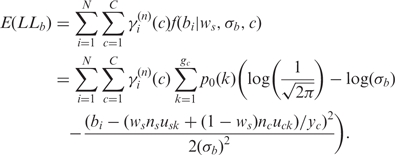 |
(8) |
Here 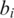 is the observed BAF signal of the ith SNP. In the maximization step of the EM algorithm, we adopted the coordinate ascent algorithm, to increase the expectation of total partial log-likelihood which is the sum of the right-hand sides of Equations (7) and (8), with respect to different global parameters. First, we select
is the observed BAF signal of the ith SNP. In the maximization step of the EM algorithm, we adopted the coordinate ascent algorithm, to increase the expectation of total partial log-likelihood which is the sum of the right-hand sides of Equations (7) and (8), with respect to different global parameters. First, we select 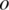 and replace the other global parameters with the corresponding values obtained from the previous iteration. By taking the partial derivative with respect to
and replace the other global parameters with the corresponding values obtained from the previous iteration. By taking the partial derivative with respect to 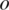 and setting it to 0, we derive the following formula to update global parameter
and setting it to 0, we derive the following formula to update global parameter 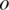 for the next iteration:
for the next iteration:
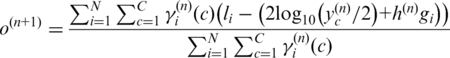 |
(9) |
with
| (10) |
Similarly, we update 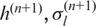 and
and 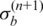 by
by
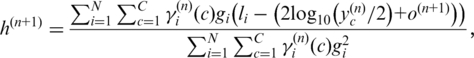 |
(11) |
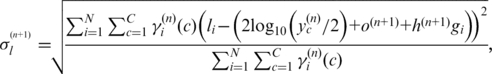 |
(12) |
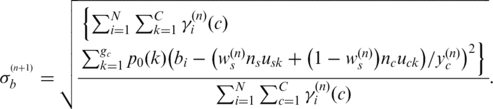 |
(13) |
Finally, we focus on the expected likelihood in Equation (13) for updating global parameter ws, since it has been shown (15,23) that BAF signals are very sensitive to normal cell contamination. They, therefore, can alone provide sufficient information for accurate inference of normal cell proportion if information about the copy number states is available (in this setting copy number information is obtained from 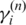 ). Using the strategy described above, we obtain the following formula to update ws by replacing
). Using the strategy described above, we obtain the following formula to update ws by replacing 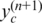 with
with 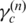 in this procedure:
in this procedure:
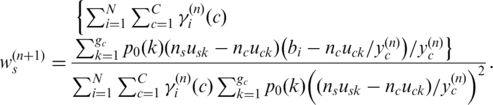 |
(14) |
The algorithm is constrained to identify ws in the interval of [0 0.9], so if 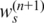 is less than 0 or greater than 0.9, it will be set to 0 or 0.9, respectively. We note that the closed form in Equation (14) only provides an approximate solution even though it works well in practice. Alternatively, some numerical methods, e.g. Newton–Raphson method (31), can also be adopted in GPHMM for estimating ws
is less than 0 or greater than 0.9, it will be set to 0 or 0.9, respectively. We note that the closed form in Equation (14) only provides an approximate solution even though it works well in practice. Alternatively, some numerical methods, e.g. Newton–Raphson method (31), can also be adopted in GPHMM for estimating ws
For estimation of state transition matrix A and initial state distribution 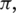 we use the standard approach discussed in ref. (29) since it is unrelated to the global parameters. In practice, the EM algorithm for GPHMM is implemented as follows: (i) start with initial parameters (
we use the standard approach discussed in ref. (29) since it is unrelated to the global parameters. In practice, the EM algorithm for GPHMM is implemented as follows: (i) start with initial parameters (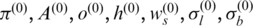 ) and calculate intermediate parameters
) and calculate intermediate parameters 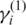 using the standard forward-backward algorithm, (ii) update
using the standard forward-backward algorithm, (ii) update 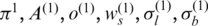 using the aforementioned method, (iii) repeat steps 1 and 2 until the algorithm converges. Once this training procedure is finished, the global parameters in the last iteration will be output as the optimal estimators. At the same time, genotype, copy number and LOH status for each probe in SNP array can be inferred from the hidden state associated with the largest posterior probability.
using the aforementioned method, (iii) repeat steps 1 and 2 until the algorithm converges. Once this training procedure is finished, the global parameters in the last iteration will be output as the optimal estimators. At the same time, genotype, copy number and LOH status for each probe in SNP array can be inferred from the hidden state associated with the largest posterior probability.
Initialization of parameters
In this study, probabilities of initial states in GPHMM are pre-defined using the following formula:
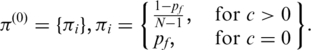 |
(15) |
Here N is the number of hidden states defined in GPHMM. The initial value for probability of fluctuation 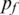 is set to a small value of 10−4. For other states, the initial values are set to be the same, i.e. no prior information. As for the state transition probabilities, the initial values are determined as follows:
is set to a small value of 10−4. For other states, the initial values are set to be the same, i.e. no prior information. As for the state transition probabilities, the initial values are determined as follows:
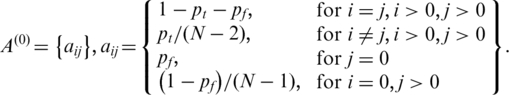 |
(16) |
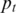 is the initial transition probability between two different non-fluctuation states, which is set to be 10−5 in this study. Initial values of the global parameters h,
is the initial transition probability between two different non-fluctuation states, which is set to be 10−5 in this study. Initial values of the global parameters h, 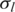 and
and 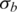 are set to 0, 0.2 and 0.03, which are the expected values of normal SNP array data with good genotyping signal quality, based on our data quality control analysis of various SNP array datasets (data not shown). Moreover, our analyses suggest that the appropriate initial values of the global parameters o and
are set to 0, 0.2 and 0.03, which are the expected values of normal SNP array data with good genotyping signal quality, based on our data quality control analysis of various SNP array datasets (data not shown). Moreover, our analyses suggest that the appropriate initial values of the global parameters o and 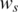 are necessary for modeling training, therefore a simple grid search (31) of these two parameters is adopted in GPHMM in order to find optimal initial parameters.
are necessary for modeling training, therefore a simple grid search (31) of these two parameters is adopted in GPHMM in order to find optimal initial parameters.
Implementation of GPHMM
An efficient implementation of GPHMM using Matlab/C is available at: http://genecube.med.yale.edu:8080/GPHMM. Information about the GC content and BAF for each SNP probe is obtained from the PennCNV package (14). It generally takes less than 10 min to run a Human 610-Quad (∼610 000 SNP probes) sample using a standard desktop PC with 2.33GHz CPU and 2G RAM. This utility provides not only accurate prediction of genotype, copy number and LOH status of each SNP probe, but also estimation of global parameters as well as other information such as the proportion of abnormal chromosomal regions in cancer genome and tumor cell DNA index. It also includes a function that can be used to visualize LRR/BAF signals and copy number/LOH results.
SNP arrays
Fresh tumor core biopsies were taken before and at a 2 week time point after a single dose of trastuzumab (T) (8 mg/m2) from 80 HER2-overexpressing, early breast cancer patients enrolled on a clinical trial of trastuzumab and chemotherapy. Nucleic acids were extracted from 109 core biopsies using a Qiagen AllPrep kit (Qiagen, Valencia, CA, USA). DNA was analyzed with the Human 610-Quad (v1.0) DNA Analysis BeadChip Kits (Illumina Inc., San Diego, CA, USA) with the assistance of the W. M. Keck Foundation Microarray Resource (New Haven, CT, USA). Normalized intensity signals were generated from the Illumina BeadStudio utility and then processed by tQN (32) in order to remove possible asymmetry in BAF signals.
FISH
Tissue preparation and FISH was performed using the manufacturer’s guidelines (Vysis®, Abbott Molecular, Downers Grove, IL, USA). Sections were deparaffinized with Xylenes and pretreated with the Vysis® Pretreatment Kit II. The LSI TOP2A Spectrum Orange/HER2/neu Spectrum Green/ CEP 17 Spectrum Aqua Probe; ProVysion™ Multi-color Probe; LSI Cyclin D1 Spectrum Orange/CEP 11 Spectrum Green Probe was used for hybridizations. Co-denaturation of the probes and tissues was done on a ThermoBrite® (Abbott Molecular) at 75°C for 6 min and hybridization at 39°C for 17–19 h. Rapid wash protocol was used. At least 20 tumor cells (range of 20–80 cells) were enumerated.
RESULTS
Dilution series data
We first tested the performance of GPHMM on a dilution series of breast cancer/normal cell lines admixed with known proportions of cancer cell DNA (15). The genomic DNA from an aneuploid cancer cell line (ATCC: CRL-2324D) was mixed in 0–0.9 proportion with DNA from a normal cell line (ATCC: CRL-2325D) and then hybridized to Illumina Human370K BeadChips. Genotyping data for each sample was examined before testing, and large regions of heterozygous deletion on chromosomes 6 and 16 of the normal cell line were identified (Supplementary Figure S1) and removed from further analysis. All of the mixture samples as well as the cancer cell line were run against GPHMM, and the estimated global parameters are show in Table 2. The standard deviations of LRR/BAF signals (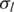 and
and 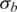 ) for different dilution samples are close to the initial values of 0.2 and 0.03, indicating that all of these samples have good signal quality. Coefficients of GC content (h) for different dilution samples are shown to have small absolute values, suggesting there are no significant GC content bias in this data set. These results are consistent with our finding in manual inspection of the genomic plots of BAF and LRR. On the other hand, we found a strong correlation (correlation coefficient >0.98) between LRR signal shift and the proportion of normal cell DNA (Figure 1). The most significant LRR signal shift occurred in the pure cancer cell line DNA. With the percentage of normal cell DNA increasing, the overall aneuploidy in the mixed sample diminishes and LRR shift tends to decrease correspondingly. When the proportion of normal cell reaches to 0.9, the LRR baseline shift identified by GPHMM becomes barely above zero. These results provide additional support that cancer aneuploidy leads to LRR baseline shift in SNP array experiments (24). Furthermore, we examined the estimated ws by GPHMM and compared them with the actual proportions used on the SNP arrays. As shown in Table 2, the global parameter ws estimated by GPHMM is close to the true normal cell DNA proportion at different dilution levels. For example, in mixed sample ‘CRL2324_10pc_Tum’ which is dominated by normal cell and includes only 10% cancer cell DNA, GPHMM can still accurately determine the proportion of normal cell DNA from the extremely weak signals of chromosomal aberration. Analyses of the global parameters provide useful information about SNP array experiments such as quality of genotyping signal and the genetic makeup of a mixed sample. In comparison, GAP can also correctly estimate low and medium proportions of normal cell DNA in admixed samples, but fails to recognize high normal contamination in samples ‘CRL2324_10pc_Tum’ and ‘CRL2324_14pc_Tum’ by incorrectly treating them as pure diploid samples.
) for different dilution samples are close to the initial values of 0.2 and 0.03, indicating that all of these samples have good signal quality. Coefficients of GC content (h) for different dilution samples are shown to have small absolute values, suggesting there are no significant GC content bias in this data set. These results are consistent with our finding in manual inspection of the genomic plots of BAF and LRR. On the other hand, we found a strong correlation (correlation coefficient >0.98) between LRR signal shift and the proportion of normal cell DNA (Figure 1). The most significant LRR signal shift occurred in the pure cancer cell line DNA. With the percentage of normal cell DNA increasing, the overall aneuploidy in the mixed sample diminishes and LRR shift tends to decrease correspondingly. When the proportion of normal cell reaches to 0.9, the LRR baseline shift identified by GPHMM becomes barely above zero. These results provide additional support that cancer aneuploidy leads to LRR baseline shift in SNP array experiments (24). Furthermore, we examined the estimated ws by GPHMM and compared them with the actual proportions used on the SNP arrays. As shown in Table 2, the global parameter ws estimated by GPHMM is close to the true normal cell DNA proportion at different dilution levels. For example, in mixed sample ‘CRL2324_10pc_Tum’ which is dominated by normal cell and includes only 10% cancer cell DNA, GPHMM can still accurately determine the proportion of normal cell DNA from the extremely weak signals of chromosomal aberration. Analyses of the global parameters provide useful information about SNP array experiments such as quality of genotyping signal and the genetic makeup of a mixed sample. In comparison, GAP can also correctly estimate low and medium proportions of normal cell DNA in admixed samples, but fails to recognize high normal contamination in samples ‘CRL2324_10pc_Tum’ and ‘CRL2324_14pc_Tum’ by incorrectly treating them as pure diploid samples.
Table 2.
Comparison of normal DNA proportions estimated by different methods on dilution series data
| Sample | GPHMM |
GAP p | Normal DNA proportion | ||||
|---|---|---|---|---|---|---|---|
| o | h | 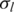 |
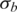 |
ws | |||
| CRL2324_10pc_Tum | 0.011 | 0.027 | 0.20 | 0.02 | 0.90 | 0.01 | 0.90 |
| CRL2324_14pc_Tum | −0.009 | 0.019 | 0.19 | 0.02 | 0.88 | 0.01 | 0.86 |
| CRL2324_21pc_Tum | −0.016 | 0.023 | 0.20 | 0.03 | 0.81 | 0.84 | 0.79 |
| CRL2324_23pc_Tum | −0.067 | 0.023 | 0.23 | 0.03 | 0.69 | 0.73 | 0.77 |
| CRL2324_30pc_Tum | −0.046 | 0.022 | 0.18 | 0.03 | 0.72 | 0.75 | 0.70 |
| CRL2324_34pc_Tum | −0.058 | 0.026 | 0.23 | 0.03 | 0.68 | 0.72 | 0.66 |
| CRL2324_45pc_Tum | −0.069 | 0.016 | 0.22 | 0.03 | 0.63 | 0.66 | 0.55 |
| CRL2324_47pc_Tum | −0.102 | 0.042 | 0.22 | 0.03 | 0.55 | 0.58 | 0.53 |
| CRL2324_50pc_Tum | −0.102 | 0.031 | 0.25 | 0.03 | 0.57 | 0.59 | 0.50 |
| CRL2324_79pc_Tum | −0.189 | 0.032 | 0.24 | 0.03 | 0.19 | 0.20 | 0.21 |
| CRL2324 | −0.283 | 0.024 | 0.24 | 0.02 | 0.02 | 0.00 | 0.00 |
Figure 1.

Strong correlation observed between proportion of normal cell and LRR signal shift in dilution series data. The empirical regression function is also shown in the figure.
Next, we investigated copy number and LOH regions to further evaluate the efficiency of GPHMM. Two state of the art methods, genoCNA (version: 1.0.8 with default parameters, no normal tissue genotypes are used) and GAP, were also employed for comparison. A simple but efficient measurement to evaluate the prediction performance of different methods using dilution series samples is the self-consistency of the results from different dilution samples, with respect to the predicted results of the pure cancer cell line DNA. We calculated the self-consistency percentages based on the predicted results of LOH state, copy number, copy number and LOH state (Figure 2). To make a fair comparison, we grouped results with copy number ≥4 since genoCNA only identifies genotypes with a maximal copy number of 4. GPHMM consistently demonstrates the best performance and a significant advantage over genoCNA and GAP when there is 50% or more normal cell in a mixed sample. For example, even with only 10% cancer cell DNA, >80% of the LOH assignments by GPHMM are consistent with the results from the pure cancer cell line, which is ∼40% higher than the self-consistency obtained by genoCNA and GAP. The self-consistency for GPHMM is 57% for copy number state prediction, which is >15% higher than the scores by genoCNA and GAP. When both copy number and LOH states are considered, significant reduction in self-consistency is observed for both genoCNA and GAP, suggesting that only a small part of the whole cancer genome is perfectly identified, whereas GPHMM retains the same good performance.
Figure 2.

Comparison of the self-consistency percentages for different methods. (a) Self-consistency percentages based on LOH status. (b) Self-consistency percentages based on copy number state. (c) Self-consistency percentages based on both copy number and LOH states.
An example illustrated in Figure 3 further validates the efficiency of GPHMM. It shows the genotyping signals and assignment by GPHMM for two adjacent LOH regions with different copy number on chromosome 17. With the increase of normal cell proportion, BAF signals representing different genotype pairs are dramatically altered. At the same time, the difference of LRR signals between two and three copies diminishes steadily. However, both copy number and LOH assignments are very consistent throughout all four samples as well as other samples in the dilution series data (Supplementary Figure S2). For comparison, we also plotted the results of genoCNA and GAP and showed them in Supplementary Figures S3 and S4. GenoCNA is not specifically designed for aneuploid samples and for this reason failed to correctly identify these chromosomal aberrations in most of the dilution series samples and the results throughout different samples are rather discrepant. This indicates that SNP-array data generated from aneuploid cancer samples significantly differs from diploid cancer samples and the issue introduced by aneuploidy should be carefully addressed in any method for cancer SNP-array data analysis. Also, it should be pointed out that we did not include genotyping information for the matched normal tissue that can be used in genoCNA, in order to compare the three methods and to illustrate the effect of the baseline shift in LRR signals caused by aneuploidy. On the other hand, the results of GAP have very good agreement with those obtained by GPHMM in the pure cancer cell line data. GAP can also detect most of the LOH region when normal cell proportion is no more than 0.66. However, the assignment of copy number state by GAP seems to be sensitive to experimental noise. For example in sample ‘CRL2324_45pc_Tum’ with 45% of cancer cell DNA, GAP provides correct assignment of copy number for the majority of these two regions. However, for sample ‘CRL2324_47pc_Tum’ with approximately the same percentage of cancer cell DNA, the results of GAP become inconsistent. In samples with normal cell proportion >0.66, large blocks of chromosomal regions with inconsistent LOH/copy number assignment are observed in Supplementary Figure S4.
Figure 3.

Plots of LOH regions on chromosome 17 and the results of GPHMM for dilution series data. (a) Plot of sample ‘CRL2324’ (100% cancer cell DNA). Typical LOH patterns are observed in this pure cancer cell line and there is a significant difference in LRR signals for two LOH regions. (b) Plot of sample ‘CRL2324-50pc-Tum’ (50% cancer cell DNA). Due to normal cell contamination, two additional BAF bands and reduction in difference in LRR signals are observed whereas the results of GPHMM remain the same. (c) Plot of sample ‘CRL2324-14pc-Tum’ (14% cancer cell DNA). The results of GPHMM keep unchanged with the increase of normal cell proportion. (d) Plot of sample ‘CRL2324-10pc-Tum’ (10% cancer cell DNA). With 90% of normal cell, the patterns of BAF and LRR signals are barely discernible. However, GPHMM can still accurately identify these two LOH regions.
GAP data
In the recent study of Popova et al. (25), 40 breast cancer samples were profiled using 300 K Illumina SNP-arrays (Human Hap300-Duo). Furthermore, the DNA indices of 18 samples obtained by flow cytometry (FCM) are reported to be very close to the estimated DNA indices by GAP. To test whether other methods can also provide accurate estimation, we downloaded the SNP array data of these 18 samples and performed analyses using GPHMM and genoCNA. First, whole genomic data for each sample was plotted and manually inspected, and most of them are shown to have good data quality. For example the plot of sample ‘BLC_B1_T19’ (Supplementary Figure S5) exhibits very clear LRR/BAF patterns throughout the whole genome, indicating high-quality of genotyping signals. This conclusion is also supported by the LRR/BAF standard deviations estimated by GPHMM (Table 3). However, samples ‘BLC_B1_T14’, ‘BLC_B1_T17’ and ‘BLC_B1_T22’, have substantially increased noise in both LRR and BAF signals (Supplementary Figure S5), and the corresponding 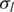 and
and 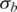 estimated by GPHMM are 0.42, 0.65, 0.46 and 0.06, 0.06, 0.05, respectively (Table 3). These noisy samples can be easily identified from the histograms of these two global parameters (Supplementary Figure S6), suggesting an efficient way for outlier detection and quality control in batch analysis of SNP array data.
estimated by GPHMM are 0.42, 0.65, 0.46 and 0.06, 0.06, 0.05, respectively (Table 3). These noisy samples can be easily identified from the histograms of these two global parameters (Supplementary Figure S6), suggesting an efficient way for outlier detection and quality control in batch analysis of SNP array data.
Table 3.
Comparison of tumor DNA indices estimated by different methods on GAP data
| Sample | GPHMM |
GAP |
FCM | ||||||
|---|---|---|---|---|---|---|---|---|---|
| o | h | 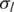 |
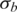 |
ws | DNA index | p | DNA index | DNA index | |
| BLC_B1_T14 | −0.38 | 0.005 | 0.42 | 0.06 | 0.15 | 1.61 | 0.15 | 0.85 | 1.14 |
| BLC_B1_T17 | 0.04 | 0.080 | 0.65 | 0.06 | 0.30 | 0.84 | 0.23 | 0.82 | 0.84 |
| BLC_B1_T19 | −0.18 | −0.013 | 0.18 | 0.03 | 0.55 | 1.56 | 0.60 | 1.63 | 1.60 |
| BLC_B1_T20 | −0.11 | 0.003 | 0.18 | 0.03 | 0.59 | 1.39 | 0.60 | 1.48 | 1.41 |
| BLC_B1_T22 | 0.07 | 0.047 | 0.46 | 0.05 | 0.09 | 0.94 | 0.13 | 0.94 | 1.98 |
| BLC_T07 | −0.15 | 0.012 | 0.18 | 0.03 | 0.56 | 1.45 | 0.56 | 1.49 | 1.68 |
| BLC_T09 | −0.40 | 0.006 | 0.22 | 0.03 | 0.02 | 1.70 | 0.08 | 1.85 | 2.02 |
| BLC_T10 | −0.45 | 0.008 | 0.18 | 0.03 | 0.04 | 1.81 | 0.05 | 1.90 | 1.88 |
| BLC_T12 | −0.20 | −0.003 | 0.19 | 0.03 | 0.35 | 1.48 | 0.35 | 1.54 | 1.51 |
| BLC_T15 | −0.26 | −0.019 | 0.19 | 0.03 | 0.42 | 1.68 | 0.26 | 0.89 | 1.11 |
| BLC_T23 | −0.09 | 0.029 | 0.21 | 0.03 | 0.57 | 1.34 | 0.59 | 1.39 | 1.32 |
| BLC_T31 | −0.38 | −0.011 | 0.23 | 0.04 | 0.07 | 1.72 | 0.16 | 1.84 | 1.91 |
| BLC_T34 | 0.08 | 0.003 | 0.24 | 0.03 | 0.09 | 0.98 | 0.13 | 0.99 | 1.55 |
| BLC_T37 | −0.23 | −0.051 | 0.26 | 0.04 | 0.08 | 1.44 | 0.11 | 1.53 | 1.51 |
| L_B1_T24B | −0.18 | −0.028 | 0.21 | 0.03 | 0.42 | 1.50 | 0.41 | 1.64 | 1.84 |
| L_B1_T25A | 0.00 | −0.032 | 0.17 | 0.03 | 0.58 | 1.00 | 0.61 | 1.04 | 1.00 |
| L_B1_T30 | −0.39 | −0.005 | 0.22 | 0.04 | 0.17 | 1.76 | 0.22 | 1.83 | 1.84 |
| L_B1_T47 | 0.01 | −0.022 | 0.19 | 0.03 | 0.54 | 1.00 | 0.55 | 1.03 | 1.00 |
Next, we estimated the DNA indices from the results of the GPHMM and GAP by following the approach in (25) and compared them with the experimental results from FCM analysis (Table 3). Both methods provide good prediction of DNA index and similar estimations of normal cell proportion for the samples in this data set except for a few discordant cases. For example, similar to the previously discussed results by GAP (25), DNA indices of ‘BLC_B1_T22’ and ‘BLC_34’ predicted by GPHMM are around 1, suggesting approximately diploid genomes. However, the DNA indices determined by FCM indicate cancer chromosomes in these samples are near tetraploid. Another case is tumor sample ‘BLC_T15’, for which GPHMM and GAP have different estimations with diverging tendencies. The DNA index of 1.68 predicted by GPHMM suggests the cancer genome is largely amplified, while the experimental result (1.11 by FCM) indicates it is only moderately duplicated. At the same time, GAP has an estimated DNA index of only 0.89 and reports many deleted chromosomal regions in the results.
The discrepancy between the results of FCM analysis and these two computational methods may actually be caused by tumor sub-clonal losses that are erroneously assigned as three copies in these samples. Therefore further experimental study is required to validate the prediction results as previously suggested (25).
HER2-positive breast cancer data
In addition to the two public data sets discussed above, we applied GPHMM to a SNP array data set from 109 fresh tumor core biopsies that were taken before or after systemic therapy in 80 HER2-overpressing (defined as IHC 3+ by DAKO Herceptest or HER2:CEP 17 ratio of >2.0) early breast cancer patients enrolled on a clinical trial of preoperative therapy. Similar to a previous observation that ∼10% of breast cancers had genomic profiles without discernible abnormalities (3), some of the samples (13 of 109) exhibit no discernable genomic aberrations along all chromosomes and therefore are not included for further analysis in this study. We first examined the distribution of global parameters illustrated in Figure 4. In a few samples of this data set, non-trivial GC coefficients are observed, suggesting GC content bias may be an issue in these samples. For example, the tumor sample with the largest absolute GC coefficient (h = −0.108) exhibits significant GC content bias (shown in Supplementary Figure S7a). After removing the bias of GC content using the linear model described in Equation (1) with the estimated coefficient, the LRR signal becomes much smoother, which further leads to smooth and consistent assignments of both copy number and LOH states (Supplementary Figure S7b). The majority of the samples, however, have good signal quality based on the distributions of the global parameters 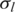 and
and 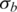 (illustrated in Figure 4). We also identified two outlier samples with notable increase of noise in both LRR and BAF signals. About 91% tumor samples (87 of 96) are mixed with >50% normal cells, of which 60 have normal cell proportions larger than 0.7, and 12 have normal cell proportions greater than 0.85.
(illustrated in Figure 4). We also identified two outlier samples with notable increase of noise in both LRR and BAF signals. About 91% tumor samples (87 of 96) are mixed with >50% normal cells, of which 60 have normal cell proportions larger than 0.7, and 12 have normal cell proportions greater than 0.85.
Figure 4.

Histograms of estimated global parameters for HER2-positive breast cancer data. Top left: (a) histogram of GC coefficient h. Top right: (b) histogram of normal cell proportion 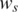 Bottom left: (c) standard deviation of LRR signal
Bottom left: (c) standard deviation of LRR signal 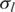 Bottom right: (d) standard deviation of BAF signal
Bottom right: (d) standard deviation of BAF signal 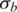 .
.
Since all of the tumor biopsies included in this cohort were taken from HER2-positive breast cancers, it is important to show efficient identification of HER2 amplifications in these samples using SNP array data. Based on the results of GPHMM, the distribution of the maximal copy number in the HER2 region (inferred from the HER2 FISH probe used in this study) is illustrated in Figure 5a. About 95% of the tumor samples (91 of 96) are identified as HER2 amplified with copy number greater than 2. The majority of the identified samples (78%) are assigned with the maximum copy number gain (five or more copies). Interestingly, we found that the genomic patterns of chromosome 17 in most HER2-positive cancers can be classified into three categories based on the copy number assignments provided by GPHMM (demonstrated in Figure 5b). One common genomic pattern is an isolated narrow peak in the HER2 locus with high-level copy number amplification (top of Figure 5b). In other tumors, HER2 amplification spans a much broader chromosomal region (middle of Figure 5b). Finally, in a few cases, amplification covers the whole q arm of the chromosome (bottom of Figure 5b). It is noteworthy that large shifts in the LRR signals illustrated in Figure 5b are observed, indicating that these samples would not be correctly classified as HER2-positive cancer, if correction of LRR signal shift is not performed.
Figure 5.

Identification of HER2 amplification in HER2-positive breast cancer data. (a) Pie chart for the maximal copy numbers of HER2 region estimated by GPHMM. CN <2: maximal copy number <2; CN = 2: maximal copy number equal to 2; CN = 3: maximal copy number equal to 3; CN = 4: maximal copy number equal to 4; CN ≥5: maximal copy number greater than or equal to 5. (b) Different genomic patterns of HER2 amplification identified in HER2-positive breast cancer data, arrows indicate the HER2 locus on chromosome 17.
Additionally, we performed FISH to further evaluate the results from GPHMM. Breast cancer tissue from one patient (YBCID: 184) was prepared and hybridized with three FISH probes specific for six loci: HER2 (17q11.2-q12), TOP2A (17q21-22), CCND1 (11q13), ZNF217 (20q13.2), MYC (8q24) and LPL (8p22). Moreover, the α satellite DNA sequences at the centromeric regions of chromosome 8, 11 and 17 were also identified with chromosome enumeration probes (CEP). Figure 6 shows the results from FISH experiments, for example in Figure 6a the multicolor FISH probes detected high level amplification of the HER2 locus (average copy number 23.1) and normal copy number of both TOP2A locus and the centromeric DNA of chromosome 17, which are consistent with the maximal copy numbers estimated by GPHMM (Figure 6d). Furthermore, Figure 6b indicates that this tumor actually consists of two different clonal subpopulations: one is characterized by three copies of CCND1 locus and two copies of the centromeric DNA of chromosome 11 (indicated by yellow arrow) and the other is shown to have four copies of CCND1 locus and three copies of the centromeric DNA (indicated by green arrow). In this scenario, the genotyping signals are extremely complicated because they are generated from a mixture of three kinds of genotypes (two different tumor subpopulations and normal cells). However, as shown in Figure 6d, GPHMM can still correctly identify the copy numbers of the first tumor subpopulation and the result is close to the copy numbers estimated by FISH, which are approximately the averaged copy numbers of all tumor subpopulations. The FISH analysis on chromosome 8 is quite similar: two different types of tumor cells can be recognized in Figure 6c, and GPHMM provides correct estimations of the copy numbers in one subpopulation and the results are also close to the averaged copy numbers determined by FISH. Based on these results and the fact that the estimated proportion of normal cells in this tumor sample is nearly 0.8, we conclude that GPHMM is effective for analyses of tumor SNP array data.
Figure 6.

Validation of GPHMM results in HER2-positive breast cancer sample. (a) FISH image of HER2 (green), TOP2A (red) and CEP 17 (aqua) probe signals in tumor sample nuclei. HER2 locus is highly amplified (average copy number 23.1). (b) FISH image of CCND1 (red) and CEP 11 (green) probe signals in tumor nuclei. Yellow and green arrows show two different tumor subpopulations. (c) FISH image of MYC (green), LPL (red) and CEP 8 (aqua) probe signals in tumor sample nuclei. Yellow and green arrows show two different tumor subpopulations. (d) Comparison of the copy numbers estimated from FISH probes and the results of GPHMM using SNP array data.
DISCUSSION
In this study, we introduced a novel hidden Markov model for detection of chromosomal aberrations in tumor samples using whole-genome SNP genotyping data. Our proposed method demonstrates several advantages compared with other methods. GPHMM is a novel method elaborated to decode the extremely complicated SNP-array signals generated from tumor samples, in which analysis has been shown to be very sensitive to normal cell contamination of a tumor biopsy (13,15,22,23), different types of chromosomal aberration (24,25), as well as other factors such as DNA quantity in experimentation (27). A significant difference between GPHMM and previous HMM methods is that by taking all these effects into account, new quantitative models were employed as the observation density functions in GPHMM, which provide more accurate and comprehensive description of the statistical behavior of genotyping signals generated from tumor samples. Second, these models are automatically optimized in GPHMM during the execution of the EM training algorithm. The global parameters are estimated by fitting these quantitative models, and the state transition matrix and the initial state distribution in the Markov chain of the GPHMM model are updated simultaneously. These two parameter estimation procedures cooperate together to maximize the likelihood of the observed SNP-array data. Based on the well-established theory of HMM and the EM algorithm, GPHMM achieves superior performance in identification of chromosomal aberrations in the cancer genome. Its advantages are most pronounced when SNP-array data is extremely contaminated by normal cells or considerably shifted due to aneuploidy. Third, instead of updating individual mean and standard deviation for each hidden state as in traditional continuous HMMs, the global parameters shared by all hidden states are directly estimated specifically for each sample, and therefore provide useful information regarding the tumor sample and SNP array experiment. Global parameters 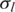 and
and 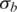 estimate the standard deviations of LRR and BAF signals that are associated with the quality of genotyping data. Another global parameter ws provides accurate estimation of the proportion of admixed normal cells and allows a better understanding of the genetic makeup of a tumor biopsy. Moreover, LRR baseline shift o is correlated with the overall aneuploidy in the tumor sample and GC coefficient h is an indicator of possible GC content bias in LRR signals. Information obtained from these global parameters can be used in the pre-processing procedure of a cohort study and is especially helpful in data quality control and outlier detection. Finally, as a HMM approach our proposed method does not require preliminary segmentation of genotyping data that is used in ref. (25) as a part of overall fitting procedure, and therefore is advantageous in fitting extremely contaminated and noisy samples. Taken together, these unique modeling and optimizing strategies endow GPHMM with superior performance.
estimate the standard deviations of LRR and BAF signals that are associated with the quality of genotyping data. Another global parameter ws provides accurate estimation of the proportion of admixed normal cells and allows a better understanding of the genetic makeup of a tumor biopsy. Moreover, LRR baseline shift o is correlated with the overall aneuploidy in the tumor sample and GC coefficient h is an indicator of possible GC content bias in LRR signals. Information obtained from these global parameters can be used in the pre-processing procedure of a cohort study and is especially helpful in data quality control and outlier detection. Finally, as a HMM approach our proposed method does not require preliminary segmentation of genotyping data that is used in ref. (25) as a part of overall fitting procedure, and therefore is advantageous in fitting extremely contaminated and noisy samples. Taken together, these unique modeling and optimizing strategies endow GPHMM with superior performance.
In this study, we also made following assumptions and simplifications in statistical modeling of GPHMM: (i) there is a log-linear relationship between average copy number and expected mean of LRR signals; (ii) all hidden states defined in GPHMM have the same signal variance; (iii) possible truncations of BAF signals (13) are not taken into account. These approaches can greatly improve the robustness and computational efficiency of our proposed model, and are advantageous for challenging cancer samples with severe normal cell contamination and samples with noisy genotyping signals caused by aneuploidy and GC content bias. These assumptions and simplifications may also reduce the sensitivity of the detection algorithm and even become a disadvantage for less challenging cancer samples with only slight normal cell contamination and good genotyping signals. However, with the cancer samples we analyzed, even in the case that aforementioned assumptions are considerably violated, e.g. data that has different signal variance in some states or is influenced by the effect of signal truncation, GPHMM can still provide accurate prediction results. For example in Supplementary Figure S8, we show the genotyping data for chromosome 1 of sample ‘BLC_T10′ in the GAP data, which is complicated by both effects. The BAF signals of two LOH regions on the left have smaller variances than those of the heterozygous regions on the right, and are very close to the boundaries, suggesting some of them are probably truncated to 0 or 1, respectively. Although the estimated global parameter 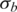 may become inaccurate in this case, the whole statistical framework including estimation of other global parameters is barely affected. The estimated mean values of genotyping signals, calculated by the empirical formulas in Equations (1) and (3), are illustrated by the red lines in the two panels at the bottom, which clearly demonstrate that GPHMM can still precisely grasp the statistical characteristics of the genotyping data and provides accurate information about copy number and LOH status for every chromosomal region. This conclusion is also supported by the summarized results from GAP and FCM analysis on this sample (see in Table 3).
may become inaccurate in this case, the whole statistical framework including estimation of other global parameters is barely affected. The estimated mean values of genotyping signals, calculated by the empirical formulas in Equations (1) and (3), are illustrated by the red lines in the two panels at the bottom, which clearly demonstrate that GPHMM can still precisely grasp the statistical characteristics of the genotyping data and provides accurate information about copy number and LOH status for every chromosomal region. This conclusion is also supported by the summarized results from GAP and FCM analysis on this sample (see in Table 3).
Quantitative SNP arrays generate high-resolution genotyping data with total signal intensity as well as information about individual alleles. They therefore allow accurate identification of copy number change and LOH in cancer genome by using both LRR and BAF signals. Despite the success reported in this article and previous studies, there are still some concerns about interpreting SNP array genotyping data from tumor samples. One concern is that chromosomal aberrations will not be correctly identified if global parameters and copy number/LOH states cannot be uniquely determined. As we previously mentioned, there are >10% tumor samples with no discernable chromosomal aberrations in the HER2-positive breast cancer data. As pointed out by Attiyeh et al. (24), due to the technical limitation of SNP arrays, we cannot determine the proportion of normal cells in these tumors. Therefore additional investigation by histopathological examination and other biological techniques such as FISH are necessary.
Another obstacle to the application of SNP arrays in cancer research is tumor heterogeneity. Recently Navin et al. (33) investigated genomic heterogeneity in breast tumors and showed more than a half of the tumors studied (11 of 20) were polygenomic tumors with multiple clonal subpopulations. This discovery suggests that tumor heterogeneity should not be ignored in interpreting tumor SNP array data. However, so far there are few efficient approaches for identification of polygenomic tumors using SNP arrays, since the genotyping signals will then be representative of the variation of both subpopulation genotypes and their proportions in the tumor. As we showed in the analyses of HER2-positive breast cancer data set, GPHMM provides reasonable estimations of the tumor subpopulation with the greatest degree of copy number aberration, even though the model is not specifically designed for polygenomic tumors. This conclusion will still hold if there is another tumor subpopulation that closely resembles normal cells but has sparse focal abnormalities, except that in this case estimation of normal cell proportion may be inaccurate since there is little genomic information that can help to distinguish these ‘normal-like’ tumor cells. Comprehensive evaluation of the performance of GPHMM under the effect of tumor heterogeneity is beyond the scope of the current work. In fact, it is possible that tumor subpopulations have distinct aberrations in the same region, for example, that one tumor clone has amplification in a chromosomal region and another has deletion in the same region. In this case, it is almost unsolvable to elucidate the genotypes of all tumor subpopulations using SNP array alone. Therefore additional experiments such as FISH are required, especially to estimate small proportion clonal populations (33). However, the results suggest that GPHMM can provide reasonable estimates of copy number for tumors with a low proportion of polygenotypes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Department of Defense (grant W81XWH-04-1-0549 to L.H.); Yale Center of Excellence in Molecular Hematology P30 DK072442-03 NIDDK (to D.T. and V.S.); Susan G. Komen Foundation (grant number FAS0703853 to D.L.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Drs Min Chen and Yunxiao He for critical reading of the article.
REFERENCES
- 1.Albertson DG, Collins C, McCormick F, Gray JW. Chromosome aberrations in solid tumors. Nat. Genet. 2003;34:369–376. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- 2.Bentires-Alj M, Gil SG, Chan R, Wang ZC, Wang Y, Imanaka N, Harris LN, Richardson A, Neel BG, Gu H. A role for the scaffolding adapter GAB2 in breast cancer. Nat. Med. 2006;12:114–121. doi: 10.1038/nm1341. [DOI] [PubMed] [Google Scholar]
- 3.Hicks J, Krasnitz A, Lakshmi B, Navin NE, Riggs M, Leibu E, Esposito D, Alexander J, Troge J, Grubor V, et al. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 2006;16:1465–1479. doi: 10.1101/gr.5460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jarvinen TA, Liu ET. HER-2/neu and topoisomerase IIalpha in breast cancer. Breast Cancer Res. Treat. 2003;78:299–311. doi: 10.1023/a:1023077507295. [DOI] [PubMed] [Google Scholar]
- 5.Keith WN, Douglas F, Wishart GC, McCallum HM, George WD, Kaye SB, Brown R. Co-amplification of erbB2, topoisomerase II alpha and retinoic acid receptor alpha genes in breast cancer and allelic loss at topoisomerase I on chromosome 20. Eur. J. Cancer. 1993;29A:1469–1475. doi: 10.1016/0959-8049(93)90022-8. [DOI] [PubMed] [Google Scholar]
- 6.Smith K, Houlbrook S, Greenall M, Carmichael J, Harris AL. Topoisomerase II alpha co-amplification with erbB2 in human primary breast cancer and breast cancer cell lines: relationship to m-AMSA and mitoxantrone sensitivity. Oncogene. 1993;8:933–938. [PubMed] [Google Scholar]
- 7.Tanner M, Isola J, Wiklund T, Erikstein B, Kellokumpu-Lehtinen P, Malmstrom P, Wilking N, Nilsson J, Bergh J. Topoisomerase IIalpha gene amplification predicts favorable treatment response to tailored and dose-escalated anthracycline-based adjuvant chemotherapy in HER-2/neu-amplified breast cancer: Scandinavian Breast Group Trial 9401. J. Clin. Oncol. 2006;24:2428–2436. doi: 10.1200/JCO.2005.02.9264. [DOI] [PubMed] [Google Scholar]
- 8.Kao J, Pollack JR. RNA interference-based functional dissection of the 17q12 amplicon in breast cancer reveals contribution of coamplified genes. Genes Chromosomes Cancer. 2006;45:761–769. doi: 10.1002/gcc.20339. [DOI] [PubMed] [Google Scholar]
- 9.Solinas-Toldo S, Lampel S, Stilgenbauer S, Nickolenko J, Benner A, Dohner H, Cremer T, Lichter P. Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer. 1997;20:399–407. [PubMed] [Google Scholar]
- 10.Park PJ. Experimental design and data analysis for array comparative genomic hybridization. Cancer Invest. 2008;26:923–928. doi: 10.1080/07357900801993432. [DOI] [PubMed] [Google Scholar]
- 11.McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, Shapero MH, de Bakker PI, Maller JB, Kirby A, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat. Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 12.Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F, Haden K, Li J, Shaw CA, Belmont J, et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–1148. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun W, Wright FA, Tang Z, Nordgard SH, Van Loo P, Yu T, Kristensen VN, Perou CM. Integrated study of copy number states and genotype calls using high-density SNP arrays. Nucleic Acids Res. 2009;37:5365–5377. doi: 10.1093/nar/gkp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Staaf J, Lindgren D, Vallon-Christersson J, Isaksson A, Goransson H, Juliusson G, Rosenquist R, Hoglund M, Borg A, Ringner M. Segmentation-based detection of allelic imbalance and loss-of-heterozygosity in cancer cells using whole genome SNP arrays. Genome Biol. 2008;9:R136. doi: 10.1186/gb-2008-9-9-r136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J, Wei W, Chen J, Zhang J, Liu G, Di X, Mei R, Ishikawa S, Aburatani H, Jones KW, et al. CARAT: a novel method for allelic detection of DNA copy number changes using high density oligonucleotide arrays. BMC Bioinformatics. 2006;7:83. doi: 10.1186/1471-2105-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laframboise T, Harrington D, Weir BA. PLASQ: a generalized linear model-based procedure to determine allelic dosage in cancer cells from SNP array data. Biostatistics. 2007;8:323–336. doi: 10.1093/biostatistics/kxl012. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto G, Nannya Y, Kato M, Sanada M, Levine RL, Kawamata N, Hangaishi A, Kurokawa M, Chiba S, Gilliland DG, et al. Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of affymetrix single-nucleotide-polymorphism genotyping microarrays. Am. J. Hum. Genet. 2007;81:114–126. doi: 10.1086/518809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scharpf RB, Parmigiani G, Pevsner J, Ruczinski I. Hidden Markov models for the assessment of chromosomal alterations using high-throughput SNP arrays. Ann. Appl. Stat. 2008;2:687–713. doi: 10.1214/07-AOAS155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, Bassett AS, Seller A, Holmes CC, Ragoussis J. QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35:2013–2025. doi: 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Assie G, LaFramboise T, Platzer P, Bertherat J, Stratakis CA, Eng C. SNP arrays in heterogeneous tissue: highly accurate collection of both germline and somatic genetic information from unpaired single tumor samples. Am. J. Hum. Genet. 2008;82:903–915. doi: 10.1016/j.ajhg.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nancarrow DJ, Handoko HY, Stark MS, Whiteman DC, Hayward NK. SiDCoN: a tool to aid scoring of DNA copy number changes in SNP chip data. PLoS One. 2007;2:e1093. doi: 10.1371/journal.pone.0001093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Attiyeh EF, Diskin SJ, Attiyeh MA, Mosse YP, Hou C, Jackson EM, Kim C, Glessner J, Hakonarson H, Biegel JA, et al. Genomic copy number determination in cancer cells from single nucleotide polymorphism microarrays based on quantitative genotyping corrected for aneuploidy. Genome Res. 2009;19:276–283. doi: 10.1101/gr.075671.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Popova T, Manie E, Stoppa-Lyonnet D, Rigaill G, Barillot E, Stern MH. Genome Alteration Print (GAP): a tool to visualize and mine complex cancer genomic profiles obtained by SNP arrays. Genome Biol. 2009;10:R128. doi: 10.1186/gb-2009-10-11-r128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenman CD, Bignell G, Butler A, Edkins S, Hinton J, Beare D, Swamy S, Santarius T, Chen L, Widaa S, et al. PICNIC: an algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostatistics. 2010;11:164–175. doi: 10.1093/biostatistics/kxp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diskin SJ, Li M, Hou C, Yang S, Glessner J, Hakonarson H, Bucan M, Maris JM, Wang K. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. Nucleic Acids Res. 2008;36:e126. doi: 10.1093/nar/gkn556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Z, Li A, Schulz V, Chen M, Tuck D. MixHMM: inferring copy number variation and allelic imbalance using SNP arrays and tumor samples mixed with stromal cells. PLoS ONE. 2010;5:e10909. doi: 10.1371/journal.pone.0010909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rabiner LR. A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE. 1989;77:257–286. [Google Scholar]
- 30.Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via em algorithm. J. Roy. Stat. Soc. B-Methodol. 1977;39:1–38. [Google Scholar]
- 31.RAO SS. Engineering Optimization: Theory and Practice. New York, NY: Wiley-Interscience; 2009. [Google Scholar]
- 32.Staaf J, Vallon-Christersson J, Lindgren D, Juliusson G, Rosenquist R, Hoglund M, Borg A, Ringner M. Normalization of Illumina Infinium whole-genome SNP data improves copy number estimates and allelic intensity ratios. BMC Bioinformatics. 2008;9:409. doi: 10.1186/1471-2105-9-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Navin N, Krasnitz A, Rodgers L, Cook K, Meth J, Kendall J, Riggs M, Eberling Y, Troge J, Grubor V, et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20:68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.