


Abstract
Several different models of the linker histone (LH)–nucleosome complex have been proposed, but none of them has unambiguously revealed the position and binding sites of the LH on the nucleosome. Using Brownian dynamics-based docking together with normal mode analysis of the nucleosome to account for the flexibility of two flanking 10 bp long linker DNAs (L-DNA), we identified binding modes of the H5-LH globular domain (GH5) to the nucleosome. For a wide range of nucleosomal conformations with the L-DNA ends less than 65 Å apart, one dominant binding mode was identified for GH5 and found to be consistent with fluorescence recovery after photobleaching (FRAP) experiments. GH5 binds asymmetrically with respect to the nucleosomal dyad axis, fitting between the nucleosomal DNA and one of the L-DNAs. For greater distances between L-DNA ends, docking of GH5 to the L-DNA that is more restrained and less open becomes favored. These results suggest a selection mechanism by which GH5 preferentially binds one of the L-DNAs and thereby affects DNA dynamics and accessibility and contributes to formation of a particular chromatin fiber structure. The two binding modes identified would, respectively, favor a tight zigzag chromatin structure or a loose solenoid chromatin fiber.
INTRODUCTION
In the cell nucleus, the DNA molecules compact to highly ordered chromatin structures, assembling a biological network (1). Within this network, the DNA interacts with histone core proteins and they together form complexes called nucleosomes that in turn interact with each other. The conformation and compaction of the chromatin depend on the interactions between the nucleosomes as well as on the presence of other factors influencing chromatin structure and dynamics. One of these factors is the so-called linker histone (LH) H5 protein, whose key role in chromatin fiber formation is well established (2). The H1/H5 family LH proteins contribute not only to the compaction of chromatin into a 30 nm fiber, but also participate in the regulation of processes such as replication and transcription (2, 3). The existence of two proposed structures of chromatin, the one-start (solenoid) (4) and the two-start (zigzag) (5) helices, implies that the linker DNA (L-DNA) connecting successive nucleosomes varies not only in length (6) but also in conformation. It is known that the LH binds to the nucleosome, forming a chromatosome, but exactly how the two interact, and how this interaction is affected by, and itself affects, the conformation and dynamics of the L-DNA, is not yet understood.
From in vivo FRAP experiments, Brown et al. (7) identified two binding sites and one nonbinding site on the globular domain of the H1 LH, GH1 and modeled a complex of GH1 with the nucleosome to fit this data. Two binding sites were also suggested on the basis of in vitro photocrosslinking data for GH5 (8), and molecular modeling of GH1d (9), but, in these studies, different binding modes to the nucleosome were deduced. Syed et al. (10) proposed a stem structure of the GH1-nucleosome complex based on a combination of experimental techniques, while George et al. (11) indicated that the two structurally similar LHs, H1 and H1c, have different binding orientations. On the other hand, two computational docking studies (12,13) of GH5 (which is 97% homologous to GH1) showed three binding sites on GH5 and different docking positions with respect to the nucleosome. However, in all these studies the nucleosome was modeled either without any linker DNAs or with different lengths of linker DNAs. Due to the highly dynamic nature of DNA in chromatin (14), the binding of the LH can be influenced by the different nucleosome conformations and this can contribute to the different binding modes proposed in the literature. In light of these inconsistencies and in order to obtain a unified model of the linker histone–nucleosome interactions, the aim of this study was to determine the position and orientation of GH5 with respect to the nucleosome, and how binding of GH5 is influenced by and influences the DNA conformation and dynamics. This was achieved by systematic computational modeling and simulation, taking into account the nucleosome flexibility and the available experimental data.
To provide a detailed description of the LH–nucleosome interactions, including the conformational variability of the nucleosome, Normal Mode Analysis (NMA) was applied to the nucleosome with 167 bp of DNA (see ‘Materials and Methods’ section). The physically relevant conformations obtained by the NMA were selected and used separately in a series of Brownian Dynamics (BD) simulations with the globular domain of the LH H5, GH5. These docking simulations revealed a dominant binding position for a wide range of nucleosomal conformations and a second binding location for more open nucleosome conformations. The simulation results were compared to experimental FRAP data indicating a very good agreement for the residue-dependent binding strengths of the LH. A subsequent NMA of the docked dominant complex uncovered the important role of the GH5 in tuning the DNA accessibility by suppression of the motion of the adjacent L-DNA. The results suggest models for the LH-nucleosome interactions that depend on the DNA conformation and flexibility, and which are consistent with the two existing models of the structure of chromatin.
MATERIALS AND METHODS
Structure preparation
The crystal structure of the nucleosome core particle (15) (NCP, Protein Data Bank—PDB code 1kx5, 1.9 Å resolution) was used as a reference structure. The histone tails were removed from the structure because they are much more mobile than other parts of the nucleosome and a recent experimental study (16) showed that histone tail removal does not affect binding of the H5 linker histone significantly. To include the linker DNAs, 20 bp of DNA from the tetranucleosome structure (5) (PDB code 1zbb, 9 Å resolution), 10 bp at each entry/exit, were added to the reference structure and the nucleosome structure obtained (which we will refer to as ‘nucleosome’) was used as an equilibrium conformation in the NMA. The globular domain of the H5 linker histone (GH5) was obtained from its crystal structure (17) (GH5, PDB code 1hst, 2.5 Å resolution). Chain B of GH5 was used for the simulations.
NMA
The crystal structure of the nucleosome was assumed to be in a global energy minimum. We used the Nomad-Ref web server (18) (http://lorentz.immstr.pasteur.fr/nomad-ref.php) to calculate the first 20 normal modes of the nucleosome. The calculation was done for all non-hydrogen atoms. The default parameters of a cutoff of 10 Å, distance weight parameter of 5 Å and average output root mean square deviation (RMSD) of 3 Å were used (18). For calculating the frequencies, a force constant of 100 kcal/mol/Å2 was applied (18). The same NMA procedure was applied to the nucleosome with the GH5 docked to it: nucleosome+GH5. The complex used was the best ranked one obtained from the BD simulation of GH5 and equilibrium nucleosome conformation (70, see Figure 1a). To analyze the motions of the linker DNAs, the atoms from chains I (1–10) and J (347–338), corresponding to L-DNA1, and I (158–167) and J (181–190), corresponding to L-DNA2 (in the tetranucleosome structure 1zbb), were selected for comparison of the modes obtained for the nucleosome and the nucleosome+GH5 complex. The angle between the n-dimensional (n atoms selected) eigenvectors was calculated for each mode. The motions were considered similar if the angles between the eigenvectors of the same order lie in the range [0–45; 135–180] deg. The frequencies of the modes in the free and complexed nucleosome are compared in Supplementary Figure S4.
Figure 1.

Nucleosome and LH crystal structures and the dominant diffusional encounter complex. (a) Nucleosome conformations generated by NMA. The L-DNA conformations chosen from the 7th (left) and the 8th modes (right) are shown by cylinders with the blue points marking the cylinder axes. The equilibrium structure, 70 (80) (corresponding to the crystal structure), is colored red. (b) Geometric parameters for specifying each nucleosome conformation. The green (dyad) point lies on the dyad axis and the red point is located at the center of mass of the nucleosome. α is the angle between L-DNA1 and L-DNA2 in the xy plane; γ1 (γ2) is the angle between L-DNA1 (L-DNA2) and the y-axis in the yz plane; A is the area of the triangle formed by the ends of L-DNA1 and L-DNA2 and the dyad point with angle β and sides d1, d2, d3. (c) The docked position of GH5 (blue) on the nucleosome (red) shown with 13 superimposed conformations generated by NMA. (d) The charged sites on GH5 (shown in the same orientation as in (c)): K69site (orange), R42site (green) and K59site (gray), and Val87 (yellow).
Brownian dynamics docking
Polar hydrogen atoms were added to the structures and partial charges and atomic radii were assigned to all atoms using the PDB2PQR program (19). The electrostatic potential was computed for each nucleosome conformation (70 : 76; 8−3 : 83) (see Figure 1a) and for GH5 by solving the nonlinear Poisson–Boltzmann equation on grids with a grid spacing of 1 Å and with 2573 and 2003 points, respectively. The programs used were APBS (20) and UHBD (21). The temperature was set to 300 K, the solvent dielectric constant to 78, the solute dielectric constant to 2 and the ionic strength to 100 mM. The dielectric boundary was defined by the van der Waals surface. The net formal charges were −237e for nucleosome and +11e for GH5. Before running BD simulations, the ECM program (22) was applied to derive effective charges by fitting them to reproduce the electrostatic potential in a uniform dielectric medium. On the protein, the effective charges were assigned to selected atoms on the charged residues. On the DNA, the effective charges were assigned to the P atoms only. The BD docking simulations were carried out with the SDA package (23) modified so that the docked complexes were only recorded if they satisfied predefined geometric requirements (Supplementary Figure S1). Here, we used two criteria: (i) the center-to-center distance between the nucleosome and GH5 should be <73 Å and (ii) the nucleosome dyad point-GH5 center distance should be <40 Å. In the simulations, the molecules are modeled as rigid bodies with the short-range attractive interactions neglected. An exclusion volume grid with 0.5 Å spacing was assigned to the nucleosome conformations to avoid overlaps. The simulation method has been described in detail in references (23–25). The trajectories were started at a center-to-center distance b = 300 Å and terminated at a distance c = 640 Å. The time step was set to 0.25 ps for center-to-center distances up to 130 Å and it increased linearly for larger distances being 20 ps at 180 Å. If GH5 spent more time than thit(rhit)=0.2 ms within 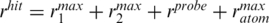 for all sampled trajectories, the BD run was truncated (a run typically contained 10–100 trajectories). This criterion prevents very long sampling of bound configurations. The probe radius rprobe for GH5 was assigned to 1.6 Å, the maximum radius of an atom
for all sampled trajectories, the BD run was truncated (a run typically contained 10–100 trajectories). This criterion prevents very long sampling of bound configurations. The probe radius rprobe for GH5 was assigned to 1.6 Å, the maximum radius of an atom 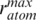 was 1.9 Å, the maximum distance from the center of mass to the furthest atom
was 1.9 Å, the maximum distance from the center of mass to the furthest atom 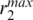 of the GH5 was 19.9 Å while
of the GH5 was 19.9 Å while 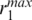 had values depending on the nucleosome conformations (
had values depending on the nucleosome conformations (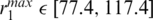 Å). The interaction energies as well as the coordinates of the complexes satisfying the constraints were recorded. A complex was recorded only if it had an RMSD >1 Å from any previously recorded complex. GH5 was docked to 13 nucleosome conformations generated by NMA separately (see ‘Results and Discussion’ section) and, for each system, five different runs with different random generators, i.e. different starting positions and orientations, were performed. For each nucleosome structure, 25 000 complexes were recorded and the 2500 lowest energy docked complexes were clustered according to the backbone RMSD values between them using the PDPIPE software criterion (26). After clustering, the representative of the top ranking cluster (with the greatest number of docked complexes) was designated as the ‘docked position’. The RMSD between the docked position for the 70 conformation and the docked position for the other nucleosome conformations was computed for all atoms and plotted in Figure 2a.
Å). The interaction energies as well as the coordinates of the complexes satisfying the constraints were recorded. A complex was recorded only if it had an RMSD >1 Å from any previously recorded complex. GH5 was docked to 13 nucleosome conformations generated by NMA separately (see ‘Results and Discussion’ section) and, for each system, five different runs with different random generators, i.e. different starting positions and orientations, were performed. For each nucleosome structure, 25 000 complexes were recorded and the 2500 lowest energy docked complexes were clustered according to the backbone RMSD values between them using the PDPIPE software criterion (26). After clustering, the representative of the top ranking cluster (with the greatest number of docked complexes) was designated as the ‘docked position’. The RMSD between the docked position for the 70 conformation and the docked position for the other nucleosome conformations was computed for all atoms and plotted in Figure 2a.
Figure 2.

Analysis of the BD and NMA results. (a) Comparison of the docking modes to the 13 nucleosome conformations showing the RMSD of the docked positions of GH5 from that to the 70 conformation of the nucleosome (circles) and the percentage of docked configurations in which the K69site (squares) or the R42site (diamonds) is within 6 Å of the N-DNA and L-DNA on the nucleosome, respectively. (b) The distance d1 between the dyad point and L-DNA1 with (squares) and without (circles) GH5 docked for NMA modes 7 and 8. (c) Comparison of the eigenvectors of modes 7-20 of L-DNA1 (upper) and L-DNA2 (lower) computed for the nucleosome with (abscissa) and without (ordinate) GH5 docked.
Data analysis
GH5 has 12 positive residues (R or K) on its surface (Figure 1d). In addition, there are 10 hydrophobic residues (A, V, L) on the surface (25 hydrophobic residues in total). The distance of each residue to either the nucleosomal DNA (N-DNA) or the L-DNA in the configurations generated in the BD simulations was monitored. The atoms Nζ and Cζ on Lys and Arg, respectively, were chosen for the distance calculation, whereas Cβ (Ala, Val) and Cγ (Leu) were used for the hydrophobic residues. For each residue, its binding strength to N-DNA and/or L-DNA was described by weight factors defined by
| (1) |
where NN−DNA and NL−DNA give the number of complexes (out of 2500) for which a certain residue is close (<15 Å) to the N-DNA or L-DNA, respectively, while 〈d 〉 is the average distance of the residue to either N-DNA or L-DNA. The maximum value of ω was 1093.5 (1/Å) for d = 2.29 Å(2500/2.29). The higher the weight factor, the closer the residue is to the nucleosome, i.e. the more favorable it is for it to interact with the DNA.
RESULTS AND DISCUSSION
Normal modes of the nucleosome
NMA is a technique for studying the lower frequency motions in dynamic systems. The normal modes (eigenvectors) and their associated amplitudes and frequencies (eigenvalues) around an energy minimum are obtained from the second derivative matrix (Hessian) of the potential energy of a given structure. The elastic network model (ENM) represents the energy landscape by a harmonic potential such that the atoms in the structure within a specified interatomic distance cutoff are connected by springs (27). To determine possible conformations of the nucleosome, we performed a NMA on the crystal structure of the nucleosome modeled with 167 bp of DNA. The assumption that the nucleosome crystal structure is at a global energy minimum was justified by the extremely stable nucleosome core structure on the nanosecond time scale (28). The largest motions were exhibited by the two 10 bp long L-DNAs while the main core of the nucleosome remained stable. To represent the principal structural variations of the nucleosome, 13 different conformations were chosen from the two lowest frequency modes: modes 7 and 8 (modes 1–6 describe rigid body translation and rotation). These modes together with mode 9 are characterized by distinctly lower relative frequencies in comparison with the other modes (Supplementary Figure S4). Although the frequencies themselves are physically meaningless, their relative contribution is a measure of collective motion. Such motion could be relevant for the LH–nucleosome interactions. Since modes 7 and 8 sample extensively the conformational space around the binding region of the GH5, mode 9 was neglected due to its similar dynamics. For the 7th mode, only open conformations with respect to the equilibrium structure (70) were chosen whereas the 8th mode was represented by three conformations on each side of the equilibrium structure (80 = 70). The choice of only 13 conformations is based on the possible deformability of the DNA as well as on computational demands. One of the L-DNAs (L-DNA1) showed larger fluctuations than the other (L-DNA2) (Figure 1a and Supplementary Table S1) for both modes.
Docked complexes of linker histone GH5 and the nucleosome
Brownian dynamics docking of GH5 to each of these 13 nucleosome conformations was then carried out. During the simulations, diffusional encounter complexes were recorded when the center of GH5 was within 40 Å of the nucleosome dyad point because experimental studies indicate that GH5 binds between the L-DNAs and protects 20 bp of the L-DNAs from nuclease digestion (29).
For 8 of the 13 nucleosome conformations, the simulations revealed a single binding mode in which GH5 binds approximately one helical turn away from the dyad point, close to L-DNA1 (Figure 1c). For these conformations, with geometry defined by α ∈ (66.4, 75.1) deg, β ∈ (68.5, 92.3) deg and A ∈ (468, 689)Å2 (Figure 1b and Supplementary Table S1), the RMSD of GH5 from its docked position on the 70 conformation is within 6 Å (Figure 2a).
GH5 approaches the N-DNA with helix 3, containing R47, K69, R73 and R74 (the K69site), and L-DNA1 with R42, R94 and K97 (the R42site). The third charged site, containing K52, K55 and K59 (the K59site), does not contact the nucleosome (Figure 1d). The hydrophobic loop containing V87 lies between the N-DNA and L-DNA1. It lies adjacent to a loop that was considered as a separate binding site interacting with AT nucleotides in the major groove of L-DNA by Cui and Zhurkin (13). These authors claimed first that an AT-rich L-DNA facilitates LH binding, and second that the hydrophobic interactions between the V87 loop and the AT nucleotides lead to bending of the L-DNA. Our results are in agreement with this statement. The GH5 preferably contacts the L-DNA1 (10 bp), which has 6 AT base pairs in contrast to only 2 on the L-DNA2, and NMA showed that the L-DNA1 is more flexible than L-DNA2 and the reason could be the presence of more A and T nucleotides. The generality of such asymmetry is supported by analysis of nucleosome positioning data of drosophila H2A.Z (30) for asymmetry in the flanking linker DNA sequences. We compared AT content percentages in the first 5 bp either side of each nucleosome core particle position in the chromosome sequences. It was found that the nucleosome neighboring sequences are systematically different from each other, with AT content 61.4% versus 54.5% (see Supplementary Table S3). This means that the adjacent 5 bp extensions of 147 bp-long nucleosome-related sequences tend to be asymmetric in AT content. If one considers 30 bp from the nucleosome ends, the asymmetry disappears and AT content is 54.5% on both sides.
Our BD docking procedure identified the encounter complex based only on electrostatic and steric interactions between molecules and this primary, initial interaction involved the charged binding sites, K69site and R42site, on the GH5. Due to short-range hydrophobic interactions, a subsequent second interaction of GH5 might involve the V87 loop that could turn into the major groove of L-DNA1 as suggested by Cui and Zhurkin (13) (Supplementary Figures S2 and S3). Such an interaction with its accompanying conformational relaxation is beyond the present study and requires higher resolution modeling with treatment of conformational flexibility, e.g. by atomic detail molecular dynamics simulation.
For the extreme conformation in the 7th mode (76), the same two sites on GH5 contact the DNA, but the contacts are different, i.e. R42site contacts the N-DNA and K69site contacts L-DNA2 (Figure 2a and Supplementary Figure S5a), similarly to the docking mode proposed by Zhou et al. and Bharath et al. (8,9). For conformations 82 and 83, GH5 also binds closer to L-DNA2 than to L-DNA1, but contacts L-DNA2 with the R42site (Figure 2a and Supplementary Figure S5b) and the hydrophobic loop. The positively charged K69site lies between L-DNA2 and N-DNA contributing to the overall stability of the complex. Since 8−3 and 8−2 are closed nucleosome conformations in which the dyad point is not freely accessible to GH5, the docked positions are outside rather than in-between the L-DNAs for these two conformations.
To identify the residues most important for binding and compare with experimental data, scaled weight factors (averaged over the configurations with RMSD <6 Å, see Figure 2a, ‘Materials and Methods’ section were plotted against the inverse FRAP half-time for recovery measured for H1 and a set of H1 mutants with single point mutations in the GH1 domain (7) (Figure 3). Both, the weight factor and the FRAP half-time are indicators of binding strength, but there is no direct relation between them. One can, however, observe a qualitative agreement. A value of 1 for the scaled weight factor would mean that the corresponding residue ‘touches’ the DNA for all recorded complexes. As a consequence, this would suggest that this residue spends more time interacting with the nucleic acid, i.e. they strongly attract each other. A FRAP experiment for an unfavorable mutation of this residue would decrease its half-time for recovery in comparison with the wild type (WT) (Figure 3). Not only do the two predicted binding sites (K69site and R42site) agree with experimental data, but both the simulation and the experiment show that K69 contributes most and K59 least to binding. The dominant docking mode is thus consistent with the models proposed by Brown et al. (7) and Cui et al. (13) using FRAP and sequence analysis, respectively, but differs from other proposed models (8,9,12).
Figure 3.

Analysis of the FRAP results. Computed weight factor for the complexes with the dominant GH5 docking position plotted against the measured FRAP inverse half-time for recovery. The positions of the 12 positive and 10 hydrophobic residues on the surface of GH5 with respect to the nucleosome were quantified by weight factors ωn and ωl, which are indicators of binding strength to the N-DNA and L-DNAs, respectively (see ‘Materials and Methods’ section).
Normal modes of the chromatosome
We next addressed the question of how H5 tunes DNA accessibility by performing a NMA of the nucleosome with GH5 docked to it in the dominant binding position obtained from the Brownian dynamics simulations (Supplementary Table S2) and comparing the motions of the L-DNAs in the NMA with and without GH5 (Figure 2b). GH5 influences the motion of both L-DNAs, but the motion of L-DNA1 is more suppressed by GH5 than that of L-DNA2. For L-DNA2, the diagonal pattern indicates similar motion in corresponding or neighboring modes, whereas the modes of L-DNA1 have different directions in the presence and absence of GH5 (Figure 2c). Zlatanova et al. (31) proposed that GH5 binds close to one of the L-DNAs while the C-terminal domain acts as a bridge between the L-DNAs and thus locks the nucleosomal gate and shuts down DNA transcription and replication. Our models suggest that, due to the spontaneous accessibility of the DNA (32), H5 could first bind strongly to the nucleosome in the dominant position identified here (see Figure 1c) and then its globular and C-terminal domains could bring the L-DNAs together and H5 would remain bound for a long time because the L-DNA motion is suppressed. It should be noted that in our models the N- and C-termini of GH5 are positioned so that attachment of the N- and C-terminal unstructured domains would be possible without clashing with the nucleosome structure. Furthermore, binding of the flexible C-terminal domain of the LH, or indeed of a second LH (see Supplementary Figure S6) can be easily accommodated without the need to push the GH5 into an energetically unfavorable position (33). In summary, in our models, the binding of H5 depends not only on the interactions with the nucleosome, but also on the geometry and sequence of the L-DNAs. The nucleosome is asymmetric with respect to the L-DNAs and so is the binding of GH5, even though LH binding has been modeled as symmetric in several studies (34,35).
The chromatosome structures obtained can contribute differently to the formation of higher-order chromatin fiber. Recently, an experimental electron microscopy (EM) and theoretical Monte Carlo study (36) reported that the chromatin fiber can exist in a heteromorphic state, i.e. simultaneously having zigzag and solenoidally connected nucleosomes. It was argued that the transition between zigzag and solenoid chromatin states is tuned by the presence of linker histones and divalent ions; the LHs contributing to a tight zigzag structure (36). In addition, the Monte Carlo simulations showed that the linker DNA positional distribution around the nucleosome influences the formation of structurally different chromatin fibers and that the linker DNAs conformations vary significantly in the presence and absence of linker histone. This is in accord with our NMA data showing large linker DNA fluctuations. For example, changing the angle α (Figure 1b) can lead to different intersection points between L-DNA1 and L-DNA2 and, hence if extended, to different linker lengths. A recent EM study (37) showed that the chromatin compaction for 167 bp and 197 bp (the most common in nature) nucleosome repeat lengths has low and high dependence on H5 (0.5 and 1 H5 per nucleosome), respectively. The authors claimed that the ‘30 nm’ fiber is an ordered interdigitated solenoid structure when nucleosome repeat length >177 bp (4), whereas the 167 bp nucleosome repeat length fiber is a zigzag structure in the presence of H5 (37). The compaction of chromatin fiber by the presence of LH has also been confirmed by fluorescence resonance energy transfer (FRET) data (38) and by theoretical models (34,35,39,40). The results provided in this study also clearly show that the LH acts as ‘glue’ between the linker DNA and the nucleosomal DNA. In the dominant binding mode, the linker DNAs, if extended, would result in steric crossing and therefore it is more likely that the dominant LH–nucleosome complex contributes to a zigzag 167 nucleosome repeat length chromatin fiber. On the other hand, enough room is available for the most open linker DNA conformations (76, 82, 83) and they could be extended without steric clashes occurring. In such a way, longer nucleosome repeat length could be formed and, respectively more H5 could be bound per nucleosome. These features are characteristic for a one-start (solenoid) chromatin fibers (35,37) and, thus, the most open LH–nucleosome complexes could be related to such chromatin structures. In addition, the dynamic nature of chromatin (14) implies that DNA takes different conformations and interacts in a dynamic fashion with DNA binding proteins (41). The wrapping and unwrapping of DNA (42–44) from the nucleosome provides a means by which other proteins can gain access to its genetic information. In regard to all these studies, our modeling of different nucleosome conformations with a LH bound can shed light on the compaction of ‘30 nm’ chromatin fiber. In the dominant binding mode, GH5 binds to L-DNA1 but, in the open 76, 82 and 83 conformations, GH5 binds to L-DNA2, even though L-DNA1 is accessible (Figure 4). This suggests that GH5 binding to L-DNA1 favors the more compact form of chromatin whereas binding to L-DNA2 tends to prevent chromatin fiber compaction.
Figure 4.

Schematic figure of the most populated GH5 binding modes on the nucleosome. The dominant binding position (left) on the nucleosome and LH orientation (orange, K69site; green, R42site; grey, K59site) found for 8 nucleosome conformations with a distance between L-DNA ends less than 65 Å. The three most open nucleosome conformations with a L-DNA distance more than 65 Å attract LH to L-DNA2 (right). However, two different LH orientations contribute to the most populated docked solutions.
CONCLUSION
The results presented here give nucleosome conformation-dependent models of the chromatosome particle. Using a three-step sequence of NMA–Brownian dynamics simulation–NMA we were able to reveal not only the interaction sites of the globular domain of the LH H5 (GH5) on the nucleosome, but also the dynamic nature of the nucleosome and its influence on GH5 binding. We show that for a triangle area, A, comprising the ends of the nucleosomal linker DNAs and the dyad point, in the range A ∈ (468, 689) Å2, the GH5 has the dominant binding mode to the nucleosome. It binds asymmetrically with respect to the nucleosome, and contacts N-DNA and L-DNA1 with its helix 3 (K69site) and R42site, respectively. Additionally, our results reveal the most and least important charged residues for binding the nucleosome, K69 and K59, respectively, which are in agreement with FRAP experimental data (7). We expect the binding modes found here to be relevant not just for H5 but for other LHs. GH5 is very similar in sequence to GH1 and the globular domain is better conserved than the flanking N- and C-terminal domains of the LHs that vary in sequence and in post-translational modifications [for review, see e.g. (45)]. Some of the residues important for nucleosome binding vary among the different histone variants, however, these residues are more conserved in human H1 variants than those at non-binding sites (45). The effects of sequence variations and post-translational modifications could be investigated in docking studies of mutants and other variants.
In contrast, on the most open nucleosome conformations, GH5 binds to the more rigid linker DNA, L-DNA2, even though L-DNA1 is accessible. This implies a selectivity mechanism of the H5, dependent on the structural orientation of the L-DNAs, by which the LH preferably binds to the highly flexible L-DNA1 only if the mutual distance between the L-DNAs, d3, is < 65 Å (Figure 4). Otherwise, the LH contacts the more rigid L-DNA, L-DNA2 that is closer to the nucleosome. This shift of GH5 between the two asymmetric L-DNAs provides a model by which the dominant binding mode might contribute to a more compact chromatin fiber, while the modeled complexes with the most open nucleosome conformations might be relevant for a loose chromatin fiber.
The NMA of the chromatosome structure showed suppression of L-DNA1 motion due to the presence of the GH5, which suggests a mechanism by which H5 can tune the DNA accessibility and, thus, play an important role in DNA replication and transcription. The long living chromatosome particle implies that H5 together with its C-terminal domain can bridge both linker DNAs due to the favorable electrostatic interactions. The level of ‘bridging’ can induce different amounts of compaction of chromatin fiber and can permit a simultaneous formation of different chromatin structures (36).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Research Foundation (grant. no. WA 1381/1-1); Klaus Tschira Foundation and BIOMS (Center for Modelling and Simulation in the Biosciences, Heidelberg). Funding for open access charge: Klaus Tschira Foundation.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.van Holde KE. Chromatin. Berlin: Springer; 1989. [Google Scholar]
- 2.Robinson PJJ, Rhodes D. Structure of the ‘30 nm’ chromatin fibre: a key role for the linker histone. Curr. Opin. Struct. Biol. 2006;16:336–343. doi: 10.1016/j.sbi.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Catez F, Ueda T, Bustin M. Determinants of histone H1 mobility and chromatin binding in living cells. Nat. Struct. Mol. Biol. 2006;13:305–310. doi: 10.1038/nsmb1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robinson PJJ, Fairall L, Huynh VAT, Rhodes D. EM measurements define the dimensions of the “30-nm” chromatin fiber: evidence for a compact, interdigitated structure. Proc. Natl Acad. Sci. USA. 2006;103:6506–6511. doi: 10.1073/pnas.0601212103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schalch T, Duda S, Sargent DF, Richmond TJ. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 2005;436:138–141. doi: 10.1038/nature03686. [DOI] [PubMed] [Google Scholar]
- 6.Perišić O, Collepardo-Guevara R, Schlick T. Modeling Studies of Chromatin Fiber Structure as a Function of DNA Linker Length. J. Mol. Biol. 2010;403:777–802. doi: 10.1016/j.jmb.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown DT, Izard T, Misteli T. Mapping the interaction surface of linker histone H1(0) with the nucleosome of native chromatin in vivo. Nat. Struct. Mol. Biol. 2006;13:250–255. doi: 10.1038/nsmb1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou YB, Gerchman SE, Ramakrishnan V, Travers A, Muyldermans S. Position and orientation of the globular domain of linker histone H5 on the nucleosome. Nature. 1998;395:402–405. doi: 10.1038/26521. [DOI] [PubMed] [Google Scholar]
- 9.Bharath MMS, Chandra NR, Rao MRS. Molecular modeling of the chromatosome particle. Nucleic Acids Res. 2003;31:4264–4174. doi: 10.1093/nar/gkg481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Syed SH, Goutte-Gattat D, Becker N, Meyer S, Shukla MS, Hayes JJ, Everaers R, Angelova D, Bednar J, Dimitrov S. Single-base resolution mapping of H1-nucleosome interactions and 3D organization of the nucleosome. Proc. Natl Acad. Sci. USA. 2010;107:9620–9625. doi: 10.1073/pnas.1000309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.George EM, Izard T, Anderson SD, Brown DT. Nucleosome interaction surface of linker histone H1c is distinct from that of H10. J. Biol. Chem. 2010;285:20891–20896. doi: 10.1074/jbc.M110.108639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan L, Roberts VA. Complex of linker histone H5 with the nucleosome and its implications for chromatin packing. Proc. Natl Acad. Sci. USA. 2006;103:8384–8389. doi: 10.1073/pnas.0508951103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui F, Zhurkin VB. Distinctive sequence patterns in metazoan and yeast nucleosomes: implications for linker histone binding to AT-rich and methylated DNA. Nucleic Acids Res. 2009;37:2818–2829. doi: 10.1093/nar/gkp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.von Hippel PH. From “simple” DNA-protein interactions to the macromolecular machines of gene expression. Annu. Rev. Biophys. Biomol. Struct. 2007;36:79–105. doi: 10.1146/annurev.biophys.34.040204.144521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002;319:1097–1113. doi: 10.1016/S0022-2836(02)00386-8. [DOI] [PubMed] [Google Scholar]
- 16.Robinson PJJ, Ann W, Routh A, Martino F, Chapman L, Roeder RG, Rhodes D. 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J. Mol. Biol. 2008;381:816–825. doi: 10.1016/j.jmb.2008.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramakrishnan V, Finch JT, Graziano V, Lee PL, Sweet RM. Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature. 1993;362:219–223. doi: 10.1038/362219a0. [DOI] [PubMed] [Google Scholar]
- 18.Lindahl E, Azuara C, Koehl P, Delarue M. NOMAD-Ref: visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. Nucleic Acids Res. 2006;34:W52–W56. doi: 10.1093/nar/gkl082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis ME, Madura JD, Luty BA, McCammon JA. Electrostatic and diffusion of molecules in solution: simulations with the University-of-Houston-Brownian Dynamics program. Comput. Phys. Commun. 1991;62:187–197. [Google Scholar]
- 22.Gabdoulline RR, Wade RC. Effective charges for macromolecules in solvent. J. Phys. Chem. 1996;100:3868–3878. [Google Scholar]
- 23.Gabdoulline RR, Wade RC. Brownian dynamics simulation of protein-protein diffusional encounter. Methods. 1998;14:329–341. doi: 10.1006/meth.1998.0588. [DOI] [PubMed] [Google Scholar]
- 24.Ermak D, McCammon JA. Brownian Dynamics with Hydrodynamic Interactions. J. Chem. Phys. 1978;69:1352–1360. [Google Scholar]
- 25.Gabdoulline RR, Wade RC. Protein-protein association: investigation of factors influencing association rates by brownian dynamics simulations. J. Mol. Biol. 2001;306:1139–1155. doi: 10.1006/jmbi.2000.4404. [DOI] [PubMed] [Google Scholar]
- 26.Motiejunas D, Gabdoulline RR, Wang T, Feldman-Salit A, Johann T, Winn PJ, Wade RC. Protein-protein docking by simulating the process of association subject to biochemical constraints. Proteins. 2008;71:1955–1969. doi: 10.1002/prot.21867. [DOI] [PubMed] [Google Scholar]
- 27.Tirion M. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 1996;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- 28.Roccatano D, Barthel A, Zacharias M. Structural flexibility of the nucleosome core particle at atomic resolution studied by molecular dynamics simulation. Biopolymers. 2007;85:407–421. doi: 10.1002/bip.20690. [DOI] [PubMed] [Google Scholar]
- 29.Duggan MM, Thomas JOJ. Two DNA-binding sites on the globular domain of histone H5 are required for binding to both bulk and 5 S reconstituted nucleosomes. J. Mol. Biol. 2000;304:21–33. doi: 10.1006/jmbi.2000.4205. [DOI] [PubMed] [Google Scholar]
- 30.Mavrich TN, Jiang C, Ioshikhes IP, Li X, Venters BJ, Zanton SJ, Tomsho LP, Qi J, Glaser RL, Schuster SC, et al. Nucleosome organization in the Drosophila genome. Nature. 2008;453:358–362. doi: 10.1038/nature06929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zlatanova J, Seebart C, Tomschik M. The linker-protein network: control of nucleosomal DNA accessibility. M. Trends Biochem. Sci. 2008;33:247–253. doi: 10.1016/j.tibs.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Li G, Levitus M, Bustamante C, Widom J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2005;12:46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- 33.Stasevich TJ, Mueller F, Brown DT, McNally JG. Dissecting the binding mechanism of the linker histone in live cells: an integrated FRAP analysis. EMBO J. 2005;29:1225–1234. doi: 10.1038/emboj.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langowski J, Heermann DW. Computational modeling of the chromatin fiber. Semin. Cell Dev. Biol. 2007;18:659–667. doi: 10.1016/j.semcdb.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 35.Kepper N, Foethke D, Stehr R, Wedemann G, Rippe K. Nucleosome geometry and internucleosomal interactions control the chromatin fiber conformation. Biophys. J. 2008;95:3692–3705. doi: 10.1529/biophysj.107.121079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grigoryev SA, Arya G, Correll S, Woodcock CL, Schlick T. Evidence for heteromorphic chromatin fibers from analysis of nucleosome interactions. Proc. Natl Acad. Sci. USA. 2009;106:13317–13322. doi: 10.1073/pnas.0903280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Routh A, Sandin S, Rhodes D. Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure. Proc. Natl Acad. Sci. USA. 2008;105:8872–8877. doi: 10.1073/pnas.0802336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bussiek M, Tòth K, Schwarz N, Langowski J. Trinucleosome compaction studied by fluorescence energy transfer and scanning force microscopy. Biochemistry. 2006;45:10838–10846. doi: 10.1021/bi060807p. [DOI] [PubMed] [Google Scholar]
- 39.Aumann F, Lankas F, Caudron M, Langowski J. Monte Carlo simulation of chromatin stretching. Phys. Rev. E Stat. Nonlin. Soft. Matter. Phys. 2006;73:041927. doi: 10.1103/PhysRevE.73.041927. [DOI] [PubMed] [Google Scholar]
- 40.Wong H, Victor J, Mozziconacci J. An all-atom model of the chromatin fiber containing linker histones reveals a versatile structure tuned by the nucleosomal repeat length. PLoS ONE. 2007;2:e877. doi: 10.1371/journal.pone.0000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Catez F, Ueda T, Bustin M. Determinants of histone H1 mobility and chromatin binding in living cells. Nat. Struct. Mol. Biol. 2006;13:305–310. doi: 10.1038/nsmb1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poirier MG, Bussiek M, Langowski J, Widom J. Spontaneous access to DNA target sites in folded chromatin fibers. J. Mol. Biol. 2008;379:772–786. doi: 10.1016/j.jmb.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kruithof M, Chien F, Routh A, Logie C, Rhodes D, van Noort J. Single-molecule force spectroscopy reveals a highly compliant helical folding for the 30-nm chromatin fiber. Nat. Struct. Mol. Biol. 2009;16:534–540. doi: 10.1038/nsmb.1590. [DOI] [PubMed] [Google Scholar]
- 44.Engeholm M, de Jager M, Flaus A, Brenk R, van Noort J, Owen-Hughes T. Nucleosomes can invade DNA territories occupied by their neighbors. Nat. Struct. Mol. Biol. 2009;16:151–158. doi: 10.1038/nsmb.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Happel N, Doenecke D. Histone H1 and its isoforms: Contribution to chromatin structure and function. Gene. 2009;431:1–12. doi: 10.1016/j.gene.2008.11.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.