

Summary
Commonly used as a treatment for Type II diabetes, sulfonylureas (SUs) stimulate insulin secretion from pancreatic β cells by binding to sulfonylurea receptors. Recently, SUs have been shown to also activate exchange protein directly activated by cAMP 2 (Epac2), however little is known about this molecular action. Using biosensor imaging and biochemical analysis, we show that SUs activate Epac2 and the downstream signaling via direct binding to Epac2. We further identify R447 of Epac2 to be critically involved in SU binding. This distinct binding site from cAMP points to a new mode of allosteric activation of Epac2. We also show that SUs selectively activate Epac2 isoform, but not the closely related Epac1, further establishing SUs as a new class of isoform-selective enzyme activators.
Introduction
Type II diabetes is a rising epidemic worldwide, and is characterized by insulin resistance, insulin deficiency, or both (Zimmet, Alberti, and Shaw, 2001). In the case of insulin resistance, cells fail to respond properly to insulin, the hormone primarily responsible for stimulating the uptake of glucose from the blood stream into systemic tissues, whereas insulin deficiency is a condition in which insufficient insulin is produced by the pancreas (Lin, and Sun, 2010). Accordingly, one of the strategies for the treatment of Type II diabetes is to stimulate insulin secretion from pancreatic β cells thereby alleviating the symptoms of diabetes (DeFronzo, 1999). This was first accomplished orally with the class of drugs known as sulfonylureas (SUs) (Duhault, and Lavielle, 1991) which remain a frequently prescribed class of drugs to treat Type II diabetes (Hanefeld, 2007).
SUs exert their anti-diabetic effects by indirectly increasing the cytosolic concentration of Ca2+, the molecular prerequisite for insulin secretory vesicle fusion and release (Holz, 2004). In a normal pancreatic β cell, glucose uptake induces the closure of ATP-sensitive potassium channels (KATP channels), which depolarizes the membrane, resulting in the opening of plasma membrane Ca2+ channels and Ca2+ influx. SUs exploit the same signaling cascade to stimulate insulin secretion as they trigger closure of KATP channels via direct binding to the sulfonylurea receptor (SUR), a subunit of KATP channels (Eliasson, Ma, et al, 2003). In both normal and diseased β cells, insulin secretion is potentiated by increases in intracellular cAMP which activates two primary effectors, cAMP-dependent protein kinase (PKA) and Exchange protein directly activated by cAMP (Epac) (Holz, 2004). Interestingly, recent data suggests that SUs can also activate Epac2, leading to activation of Rap1 (Zhang, Katoh, et al, 2009), a well characterized downstream target of Epac proteins (Holz, 2004). It was further demonstrated that Epac2−/− mice treated with glucose plus SU or SU alone show diminished insulin secretion and elevated blood glucose levels when compared to WT mice receiving identical treatments. These data led the authors to suggest that maximal SU-induced insulin secretion requires binding to both SUR1 and Epac2 (Zhang, Katoh, et al, 2009). While SU action via SUR1 is well characterized, little is known about the interaction between SU and Epac or the mechanisms by which SUs activate Epac2 and the downstream signaling.
Epac proteins are cellular sensors of cAMP and function as guanine nucleotide exchange factors for the small GTPases Rap1 and Rap2. There exist two Epac isoforms, Epac1 and Epac2, which have different tissue expression patterns (Holz, Kang, et al, 2006) – Epac1 is ubiquitously expressed while Epac2 is found at high levels in the brain, adrenal glands and in endocrine tissues, including the pancreas (Kawasaki, Springett, et al, 1998; Roscioni, Elzinga, and Schmidt, 2008). Structurally, while both contain a regulatory domain consisting of a disheveled, Egl-10, and pleckstrin (DEP) domain for membrane targeting and a high affinity cAMP binding domain (CNB-B, Kd = 2.8 μM and 1.2 μM, for Epac1 and Epac2, respectively) as well as a catalytic domain to promote Rap1 binding and nucleotide exchange, Epac2 contains an additional N-terminal low affinity cAMP binding domain (CNB-A, Kd = 87 μM) that Epac1 lacks (Holz, Kang, et al, 2006). Despite these differences, Epac1 and Epac2 share a great deal of sequence similarity and have many of the same interaction partners, including SUR1 in insulin secreting cells (Holz, 2004). The mechanism of activation by the endogenous agonist cAMP is also shared between Epac1 and Epac2. In the auto-inhibited state of both isoforms, the regulatory domain sterically blocks the Rap1 binding interface in the catalytic domain, and cAMP binding induces a conformational change in the “hinge helix” of the regulatory domain that relieves this auto-inhibition (Rehmann, Das, et al, 2006; Tsalkova, Blumenthal, et al, 2009). SUs represent a class of novel activators of Epac2 (Zhang, Katoh, et al, 2009), however several key questions remain as to whether SUs directly bind to Epac2 (Gloerich, and Bos, 2010), how the activation is accomplished, or whether the same activation occurs to Epac1.
In this study, we investigated SU-induced activation of Epac by employing FRET based biosensors to monitor a series of molecular events such as the activation of Epac and Rap1. These FRET-based biosensors can serve as powerful tools both for analyzing complex cellular processes in the native context of the living cell and for assaying direct molecular interactions in vitro. Combining this approach with biochemical analysis, we show that SU directly binds to and activates Epac2, leading to activation of Rap1 and Extracellular signal Regulated Kinase-1/2 (pERK1/2), a downstream target of Rap1. We further identified a putative SU binding site on Epac2 and demonstrated isoform selectivity for SU induced activation of Epac2 over Epac1. These findings establish SUs as a new example of isoform-selective activating drugs that bind to a site distinct from that of the endogenous agonist but exploit a similar conformational change to activate the enzyme.
Results
SUs activate Epac2 through direct binding
We first examined the effect of four clinically approved SUs, namely glibenclamide (GLB), acetohexamide (ACT), tolbutamide (TOL), or glipizide (GLP), on the activity of Epac2 by using a FRET-based reporter of Epac2 activation in HEK293T cells. In this reporter, full length Epac2 is sandwiched between Cerulean, a cyan fluorescent protein variant (Rizzo, Springer, et al, 2004), and Venus, a yellow fluorescent protein (Nagai, Ibata, et al, 2002) (Fig. S1A). The cAMP-induced conformational change of Epac2 that accompanies its activation can be detected as a decrease in FRET, which was depicted as an increase in the emission ratio of cyan over yellow in Figure S1B. In agreement with the recent finding that SUs activate Epac2 in cells (Zhang, Katoh, et al, 2009), we observed an increase in cyan over yellow emission ratio of 7.6 ± 1.3% (N = 10) [mean ± s.d. (N = number of cells)] in cells treated with GLB (Fig. 1A,B, Fig. S1C) and 9.3 ± 1.3% (N = 15), 5.6 ± 0.4% (N = 15), and 11.8 ± 1.6% (N = 10) in response to ACT, TOL, and GLP, respectively (Fig. S1D). After each of the SU responses plateaued, we then added a cAMP-elevating cocktail of the adenylyl cyclase activator forskolin (Fsk) and general phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX) to the cells to induce the maximal response from the biosensor. In the case of GLB, ACT, and TOL, this treatment generated a further emission ratio increase of 5.2 ± 1.8%, 5.8 ± 2.0%, and 10.5 ± 1.8%, respectively (Fig. 1A,B, Fig. S1D), indicating that these compounds do not maximally activate Epac2 at the concentrations used. Conversely, there was no further cAMP-induced change in FRET after GLP addition since GLP treatment alone generated a maximal response from the Epac2 biosensor (Fig. S1D). Importantly, the SU-mediated effect on the Epac2 biosensor was not due to SU-induced cAMP accumulation since neither GLB nor ACT induced a response from the A-Kinase Activity Reporter (AKAR), a FRET-based Protein Kinase A activity biosensor that is known to detect cAMP increases with a high sensitivity (Allen, DiPilato, et al, 2006) (Fig S1E–G). Since all four of the SUs tested activate Epac2, we chose to focus our further studies primarily on one SU, and we decided to proceed with GLB since this is the most clinically relevant SU.
Figure 1. SUs activate Epac2 and downstream signaling by binding directly to Epac2.
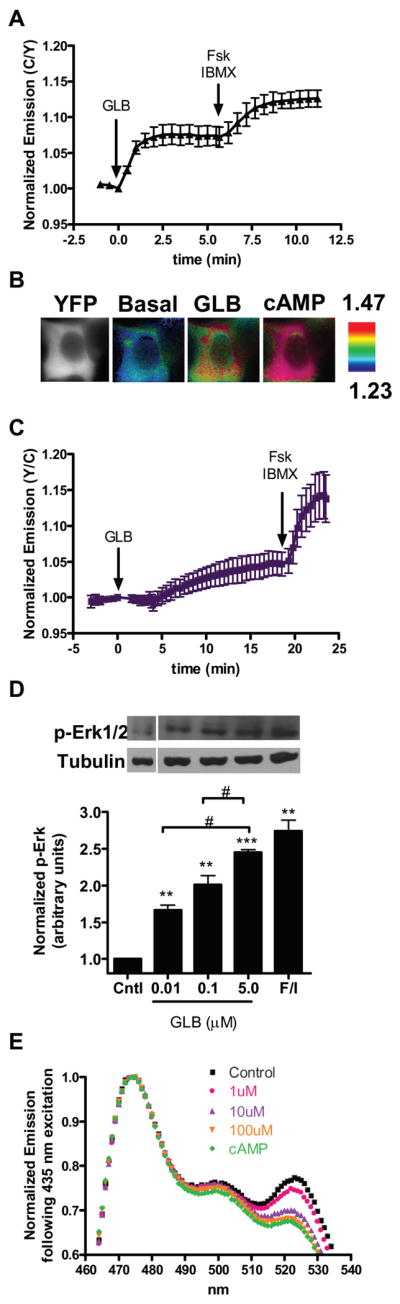
(A) HEK293T cells expressing the Epac2 biosensor were first treated with GLB (5 μM) followed by Fsk (50 μM) and IBMX (100 μM) (N = 10). Data depicted as mean ± s.d. (B) In HEK293T cells, CFP direct (far left panels) and ratiometric images of the Epac2 biosensor before GLB addition (middle left panels), after GLB addition (middle right panels), and after Fsk/IBMX addition (far right panels). Cooler colors correspond to higher FRET and warmer colors correspond to lower FRET. (C) HEK293T cells expressing Raichu-Rap1 and Epac2-mCherry show Rap1 activation after treatment with GLB (5 μM) and Fsk (50 μM) plus IBMX (100 μM) (N = 15), data is mean ± s.d. (D) HEK293T cells expressing Epac2-mCherry were treated as indicated and cell lysates were separated by SDS-PAGE. Immunoblots (top) are representative of a single experiment, and quantification of pERK1/2 levels over tubulin for each treatment (bottom) were plotted as mean ± s.e.m., N = 3–5 individual experiments for each treatment. **P ≤ 0.01, ***P < 1E-4 compared to control, #P < 0.05 between treatments. (E) Emission spectrum of the Epac2 biosensor excited at 435 nm in the basal state (■), after GLB [1 μM (
 ); 10 μM (
); 10 μM (
 ); 100 μM (
); 100 μM (
 )] addition, and after cAMP (500 μM) addition (
)] addition, and after cAMP (500 μM) addition (
 ).
).
Next, we examined the signaling events downstream of Epac2 activation in response to SU treatment, beginning with the activation of Rap1, a signaling event that is involved in insulin secretion (Shibasaki, Takahashi, et al, 2007). To do this we turned to a FRET-based biosensor for Rap1 activation, Raichu-Rap1 (Mochizuki, Yamashita, et al, 2001) (Fig. S1H), which detects cAMP-induced Rap1 activation in HEK293T cells only when Epac is overexpressed, presumably because these cells do not express enough endogenous Epac to activate Rap1 (data not shown). To this end, we co-transfected HEK293T cells with Raichu-Rap1, along with Epac2 tagged C-terminally with mCherry as a control for expression. In these cells, GLB treatment caused a 4.6 ± 1.7% (N = 15) increase in yellow over cyan emission ratio of Raichu-Rap1 (Fig. 1C), which is consistent with the notion that SUs act through Epac2 to activate Rap1. Subsequent treatment of these cells with Fsk/IBMX showed a further increase in emission ratio of 9.1 ± 3.7%, suggesting that SU treatment does not lead to full activation of Rap1. Since similar trends were observed in cells treated with ACT or GLP, the SU capable of maximally activating our Epac2 biosensor, it seems that while SUs can activate Epac2 to stimulate Rap1, this stimulation is not capable of inducing maximal Rap1 activation (Fig. SI-J). This finding is in contrast to the study by Zhang, et. al. in which all three SUs activated Rap1, as detected by immunoblotting assay, to levels comparable to that induced by cAMP at the doses tested. This discrepancy could be due to the different sensitivity of the two assays used. While live cell imaging is a technique to study signal transduction in real time on a single cell level, it lacks the amplification of signal that is often coupled to the detection used in biochemical assays. Thus, to further examine the signaling downstream of Epac2 activation, we analyzed the activation of ERK1/2 in response to GLB treatment at doses of 0.01, 0.1 or 5 μM by examining the level of phosphorylated ERK1/2 (pERK1/2) in HEK293T cells overexpressing Epac2-mCherry. An increase in pERK1/2 was observed as a result of SU treatment, with 5 μM GLB and 750 μM ACT inducing phosphorylation of ERK1/2 to a level comparable to that induced by cAMP (Fig. 1D and Fig. S1K). Together, this data shows that GLB can activate Epac2 and initiate a signaling cascade leading to activation of Rap1 and ERK1/2.
While there is evidence that SUs act through Epac2 to induce insulin secretion and glucose uptake (Zhang, Katoh, et al, 2009), it remains unclear if SUs directly bind to and activate Epac2 (Gloerich, and Bos, 2010). To elucidate this, we tested the effect of SU on Epac2 in vitro. The Epac2 biosensor was purified from HEK293T cell lysates and subjected to SU treatment. With an excitation wavelength of 435 nm for Cerulean, addition of increasing doses of GLB led to a dose-dependent decrease in yellow over cyan emission ratio of the Epac2 biosensor (Fig. 1E), followed by a further decrease upon addition of 500 μM cAMP, consistent with what was observed in cells. This SU-induced FRET decrease in the purified Epac2 biosensor was also observed with ACT treatment (Fig. S1L) and indicates that SU directly binds to and activates Epac2.
R447 is specifically involved in SU binding and activation of Epac2
With the identification of Epac2 as a direct target for SU, we set out to identify the key residues of Epac2 involved in SU binding and/or activation. To begin, we investigated how known SU binding proteins bind to SUs. At the time of our study, the best characterized SU binding protein was the plant protein acetohydroxyacid synthase (AHAS) which is inhibited by SU binding (McCourt, Pang, et al, 2006; Wang, Lee, et al, 2009). Analysis of the crystal structure of AHAS in complex with various SUs identifies two residues, an arginine and a serine, in the SU binding site that make hydrogen bonds to the core structure of the SU, and an aspartic acid which both stabilizes the arginine and forms π stacking interaction with the SU core structure (McCourt, Pang, et al, 2006); (McCourt, Pang, et al, 2005). We thus examined the crystal structure of Epac2 in the auto-inhibited state for properly oriented Arg/Ser/Asp triads that we reasoned could serve as potential SU binding sites. As an additional criterion, we only chose those triads that were at least partially solvent exposed for two reasons: first, the SU binding site on AHAS is relatively exposed, and second, by excluding buried triads, we reduce the chance of causing misfolding of Epac2 when point mutations are introduced. We then set out to mutate the arginine, the residue that makes the most contacts with the SU core, of each triad to alanine.
Among the putative SU-binding triads that we identified in Epac2, we selected four from different locations on the protein and mutated these arginine residues to alanine – R53 in the CNB-A domain, R447 and R466, both in the CNB-B domain but on either side of the hinge region, and R819 in the catalytic region (Fig. S2A). These mutations were introduced into our Epac2 biosensor and the effect of each of these point mutations on the SU-induced conformational change was analyzed. Specifically, we expressed each mutated biosensor in HEK293T cells and first treated the cells with GLB to test the effect of SU binding, followed by treatment with a cocktail of Fsk and IBMX to ensure that introducing the point mutation into Epac2 did not abolish its inherent ability to bind cAMP and adopt the active conformation. If a mutation affected SU binding but did not affect Epac2 folding or cAMP-induced conformational change, we would expect it to cause a reduction in SU response compared to WT (7.6 ± 1.3%, N = 10), while maintaining cAMP sensitivity comparable to that of WT (12.6 ± 1.2%) (Fig. 2A).
Figure 2. R447 is specifically involved in SU binding to and activation of Epac2.

(A) The GLB (5 μM) and cAMP (50 μM Fsk plus 100 μM IBMX) responses of WT (N = 10), D32A/R53A (N = 7), R447A (N = 12), R466A (N = 8), and R819A (N = 8) Epac2 biosensors in HEK293T cells. Values are depicted as mean ± s.d. ***P <1E-08. (B) Time course depicting the diminished GLB-induced response of the R447A mutant (□) Epac2 biosensor compared to the WT (■) Epac2 biosensor. (C) Emission spectrum of the R447A biosensor excited at 435 nm in the basal state (■), after GLB [1 μM (
); 10 μM (
); 100 μM (
)] addition, and after cAMP (500 μM) addition (
). (D) Molecular docking studies identify a putative SU binding site where ACT hydrogen bonds with R447 of Epac2 and makes Van der Waals contacts with F374.
Of the four mutants that we generated, only one, R819A, showed reduced sensitivity to cAMP with a response of 1.4 ± 0.9% (N = 8) (Fig. 2A and Fig. S2B), indicating that this mutation renders Epac2 non-functional. Of all the triads that we studied, this was the least solvent-exposed, and we thus suspect that the R819A mutation may have affected Epac2 folding. Another mutant, R53A, is present in the CNB-A domain and initially showed variable responses to SU treatment. We thus decided to clarify these findings by also mutating the aspartic acid in this particular Arg/Ser/Asp triad (D32) to generate the D32A/R53A double mutant. When compared to WT, the D32A/R53A mutant displayed no difference in SU binding (7.4 ± 1.3%, N = 7) or cAMP binding (11.4 ± 1.0%) compared to WT (Fig. 2A and Fig. S2B), and we consequently concluded that neither D32 nor R53 contribute to SU binding to Epac2. The third mutant in our study, R466A, is located in the CNB-B domain of Epac2, just on the edge of the hinge helix. While R466A displayed a slightly enhanced response to cAMP (22.1 ± 2.9%, N = 8), it did not show reduced sensitivity to SU (7.0 ± 1.4%) (Fig. 2A and Fig. S2B), indicating that R466 is not important for SU binding to Epac2.
The fourth mutant in our study, R447A is also located in the CNB-B, but on the opposite side of the hinge helix to R466. Unlike R466, when the R447A mutant biosensor was subjected to GLB treatment, it displayed minimal change in emission ratio (0.7 ± 0.8%, N = 12), while maintaining a cAMP-induced change (12.1 ± 4.9%) comparable to that of WT (Fig. 2A,B and Fig. S2B,C). Thus the R447A mutant displays a great reduction in its ability to be activated by SU while maintaining its cAMP-binding capability signifying that R447 is specifically involved in SU-induced activation of Epac2.
The R447A mutant biosensor was also purified from HEK293T cell lysates and subjected to SU treatment. Unlike the WT biosensor, addition of GLB or ACT to the R447A biosensor did not cause any decrease in yellow to cyan emission ratio following CFP excitation (Fig. 2C and Fig. S2D). Thus SUs either do not bind to the R447A mutant or do not effectively induce the conformational change in the R447A mutant, consistent with the idea that R447A is a residue critically involved in SU-induced activation of Epac2. In agreement with our cellular experiments, a FRET decrease was observed after cAMP addition confirming that while the R447A mutant of Epac2 cannot be activated by SU, it maintains its ability to respond to cAMP.
To test the involvement of R447 in SU binding to Epac2, we performed molecular docking studies using ACT and the auto-inhibited conformation of Epac2. By defining a docking cube centered on R447 and running 100 docking simulations, an ACT binding site comprised of R447 and F374 was identified (Fig. 2D) (Morris, Goodsell, et al, 1998; Seeliger, and de Groot, 2010; DeLano W.L, 2002). R447 makes a strong hydrogen bond of less than three angstroms with ACT whereas F374 interacts with ACT through Van der Waals contacts. The Van der Waals interaction between F374 and the R group off the urea group of ACT (R2) could explain why SUs with larger R2 groups such as gliclazide (GLC) do not activate Epac2 (Fig. S2E) (Zhang, Katoh, et al, 2009), as such an interaction would unfavorable. While S393 and D449 were not explicitly identified as residues involved in ACT binding to Epac2, they do appear to play a critical role in positioning R447 into its proper orientation to bind ACT via hydrogen bonding. Taken together, the mutational analysis and molecular docking studies suggest that R447 is necessary for direct SU binding to and activation of Epac2.
A hinge motion is important in both cAMP- and SU-dependent activation of Epac2
To elucidate the molecular mechanisms by which SUs activate Epac2, we investigated whether SUs exploit the same mechanism to activate Epac2 as cAMP. In the aforementioned auto-inhibited state of Epac proteins, the Rap1 binding domain in the catalytic region is sterically blocked by the regulatory domain and this inhibition is relieved by a cAMP-induced hinge motion (Rehmann, Das, et al, 2006; Tsalkova, Blumenthal, et al, 2009). Structural studies of Epac2 identified an invariant leucine (L408 in Epac2 and L273 in Epac1) and phenylalanine (F435 in Epac2 and F300 in Epac1) in the hinge region as key residues in regulating the conformational change (Rehmann, Prakash, et al, 2003; Tsalkova, Blumenthal, et al, 2009). Specifically, in the inactive conformation, the relative positions of L408 and F435 prevent the hinge helix from swinging into the active conformation. cAMP binding, however, induces a structural rearrangement in Epac2 that alters the position of the leucine relative to the phenylalaine, thus permitting the hinge helix to shift into the active conformation. It has been shown that a L273W mutant of Epac1 displays a reduced ability to promote Rap1 nucleotide exchange, although its cAMP binding capability is retained, presumably because the bulky tryptophan residue is sterically blocked by F300 and the reorientation of the hinge region is unfavorable (Rehmann, Prakash, et al, 2003). If SUs exploit a similar hinge motion as cAMP to achieve the active conformation of Epac2, we would expect the L408W mutant of our Epac2 biosensor to show diminished conformational change in response to SU.
To test this hypothesis, we generated a mutant biosensor containing a L426W mutation, where L426 corresponds to L408 in the crystal structure (Table S1). As expected, in response to the β-adrenergic receptor agonist isoproterenol (Iso) or combined Fsk/IBMX treatment, the L426W mutant displayed a significantly reduced response (4.1 ± 0.9% to Fsk/IBMX, N = 12) when compared to WT (13.6 ± 1.8% to Fsk/IBMX, N = 6) (Fig. 3A). This confirms that, as is the case in Epac1 (Rehmann, Prakash, et al, 2003), a tryptophan in the equivalent position in Epac2 is too bulky to permit the hinge motion. Notably, while the previous study utilized Rap1 nucleotide exchange assays to evaluate the effect of L273W on Epac1, this biosensor based approach provides a direct assay for evaluating the effect of various mutations on the conformational change of Epac in living cells.
Figure 3. A leucine involved in cAMP activation of Epac is also involved in SU-induced activation.

In HEK293T cells, (A) The L426W mutation in the hinge region of the Epac2 (□; N = 12) shows reduced activation in response to Iso (1 μM) and Fsk (50 μM) plus IBMX (100 μM) when compared to WT (■; N = 6). (B) The L426W mutant (□; N = 8) displays reduced activation in response to GLB (5 μM) when compared to WT (■; N = 10). Data depicted as mean ± s.d. (C) The response of the L426W biosensor (open bars) 5 minutes after SU addition is diminished compared to the WT (solid bars) for each treatment. ***P<0.0005.
We next tested the ability of SU to activate the L426W biosensor. When stimulated with GLB, the L426W mutant showed a diminished response of 5.0 ± 0.5% (N = 8) when compared to the WT biosensor which generates an emission ratio change of 7.6 ± 1.3% (N = 10) in response to this treatment (Fig. 3B,C). We also tested the ability of ACT and GLP to activate this mutant and found that they both generate similar diminished responses compared to that of WT (Fig. 3C, Fig. S3A,B). Since there is no evidence for direct involvement of L426 in binding to SU from our molecular docking study, we conclude that for both cAMP- and SU- induced activation of Epac2, a tryptophan at residue 426 is too bulky to permit the hinge motion for Epac2 activation. This data thus suggests that both activators pursue the same conformational path that involves the motion of the hinge helix to activate Epac2.
SUs selectively activate Epac2 isoform
Since Epac1 and Epac2 are similar in terms of domain structure and cellular targets, we wanted to test if SU can also activate Epac1. In order to test the effect of SU treatment on Epac1, we used a previously developed FRET-based reporter of cAMP dynamics and Epac1 activation, ICUE1 (DiPilato, Cheng, and Zhang, 2004) (Fig. S4A), which generates a FRET decrease upon Epac1 activation (Fig. S4B). When transfected in HEK293T cells and subjected to GLB treatment, the Epac1 biosensor, unlike its Epac2 counterpart, did not show a FRET decrease, suggesting that GLB is unable to activate Epac1 (Fig. 4A). After addition of Fsk/IBMX, however, the Epac1 biosensor was maximally activated, confirming that the Epac1 biosensor responds to cAMP and is functional. We observed similar trends with ACT, GLP, and TOL as well (Fig. S4C–E) and together these data suggest that SUs are incapable of robustly activating Epac1. Taken with the aforementioned data that SUs can induce robust Epac2 activation (Fig. 1), SUs are identified as selective activators of Epac2 over Epac1.
Figure 4. SUs are selective for Epac2 isoform.

(A) The Epac2 (
 ; N = 10) or Epac1 (○; N = 10) biosensor was expressed in HEK293T cells and treated with GLB (5 μM) followed by addition of Fsk (50 μM) plus IBMX (100 μM). Data is depicted as mean ± s.d. (B) HEK293T cells expressing Epac1-mCherry were treated as indicted and cell lysates were separated by SDS-PAGE. A representative blot is shown (top) and a quantification of pERK1/2 over tubulin was plotted for each treatment. Data is mean ± s.e.m. N = 3–5 individual experiments per treatment. ** P < 0.01 (C) Quantification of GLB-induced pERK1/2 levels via Epac1 (open bars) or Epac2 (closed bars); ** P < 0.01, ***P < 1E-5.
; N = 10) or Epac1 (○; N = 10) biosensor was expressed in HEK293T cells and treated with GLB (5 μM) followed by addition of Fsk (50 μM) plus IBMX (100 μM). Data is depicted as mean ± s.d. (B) HEK293T cells expressing Epac1-mCherry were treated as indicted and cell lysates were separated by SDS-PAGE. A representative blot is shown (top) and a quantification of pERK1/2 over tubulin was plotted for each treatment. Data is mean ± s.e.m. N = 3–5 individual experiments per treatment. ** P < 0.01 (C) Quantification of GLB-induced pERK1/2 levels via Epac1 (open bars) or Epac2 (closed bars); ** P < 0.01, ***P < 1E-5.
We next sought to confirm SU isoform selectivity for Epac2 by monitoring ERK1/2 phosphorylation. In contrast to what was observed with Epac2, cells expressing Epac1-mCherry show no GLB-induced Rap1 activation; they do, however, exhibit a large increase in pERK levels in response to Fsk/IBMX (Fig. 4B). Similarly, ACT treatment did not induce significant increase in pERK1/2 levels (Fig. S4F). This data demonstrates that Epac1 can activate ERK1/2 in response to cAMP, but not SU, and confirms that SUs specifically activate Epac2 over Epac1 (Fig. 4C and Fig. S4G). Since the residues that we identified to make up an SU binding site on Epac2, namely R447 and F374, are conserved in Epac1, it is possible that the other regions of Epac2 form additional interactions to facilitate SU binding, which are not present in Epac1, and it is these interactions that confer the SU selectivity for Epac2 over Epac1. To test this hypothesis we used a biosensor containing only the Epac2 CNB-B domain fused between CFP and YFP (Epac2-camps) (Nikolaev, Bunemann, et al, 2004) (Fig. S4H). This lack of GLB-induced conformational change (Fig. S4I) suggests that while the necessary residues for SU binding are present, they alone are not sufficient to permit SU-induced activation of Epac2, and that other domains of the protein may be necessary to facilitate this activation.
Discussion
The stimulation of insulin secretion from pancreatic β cells by SUs has been suggested to be mediated through two cellular targets: SUR1 and Epac2 (Zhang, Katoh, et al, 2009). While a direct role for Epac2 in SU-induced insulin secretion may be disputed by the observation that SUs are incapable of inducing insulin secretion in SUR1−/− mice (Seghers, Nakazaki, et al, 2000), it is important to note that pancreatic islets isolated from these mice also demonstrate impaired cAMP-mediated, PKA-independent insulin secretion, indicative of disrupted Epac2 signaling in these islets (Eliasson, Ma, et al, 2003). Though several rationalizations for this effect exist, one possible explanation is that in β-cells, SUR1 facilitates proper localization of Epac2 to the molecular machinery necessary for it to exert its effect on insulin secretion (Eliasson, Ma, et al, 2003; Hinke, 2009). Regardless of the reason, the impaired Epac2-mediated insulin secretion in SUR1−/− islets argues that in pancreatic islets, the functions of SUR1 and Epac2 are intricately linked.
While the action of SU through SUR1 has been well characterized, little is known about Epac2 activation by SU. Here we established that SUs bind directly to Epac2 to activate it and its downstream signaling components. This classifies SUs into a group of rare, but emergent, small molecules that serve as enzyme activators (Zorn, and Wells, 2010). Enzymes are inherently powerful signaling molecules because one active enzyme is capable of activating several downstream targets, resulting in amplification of the signal. Consequently, while it is frequently necessary to achieve at least 90% enzyme inhibition in order to effectively diminish a desired biological effect, the biological consequence of activation of an enzyme may be achieved with as little as 10% enhancement of its activity. For this reason, small molecule enzyme activators are important not only for their potential therapeutic applications, but also because they can serve as advantageous tools to establish a better understanding of the causal relationship between different components within a signaling pathway (Zorn, and Wells, 2010). In addition, studies of these unique activators can expand our understanding of allosteric activation of enzymes.
In this study, the identification of the R447-containing SU binding site on Epac2 shows that SUs bind to Epac2 at a site different from cAMP to activate this guanine nucleotide exchange enzyme and downstream signaling. Of the four established mechanisms that a small molecule can pursue to activate a target enzyme, this mechanism is most consistent with the Type A1 mechanism in which binding of a small molecule to an allosteric site on a target enzyme promotes an active enzyme conformation (Zorn, and Wells, 2010). In this study we have shown that the SU-bound Epac2 conformation is capable of activating Rap1 and the downstream signaling. However, it is known that Epac2 can also stimulate insulin secretion via Rap1-independent mechanisms (Holz, 2004), and it is possible that Rap1-dependent and independent events may be differentially regulated by the SU-bound state of Epac2. Future studies will address the molecular discrepancy between the SU-bound and cAMP-bound Epac2 conformations and the ability of the SU-bound form to stimulate Rap1-independent signaling pathways. This information will enhance the basic understanding of Epac2-dependent insulin secretion.
Moreover, we have found that SUs specifically activate Epac2 over Epac1. While this was surprising given the similarities between the Epac isoforms, Type A1 activators can often achieve high specificity for a protein target because the allosteric binding site that they exploit is generally not as conserved as the enzyme active site (Zorn, and Wells, 2010). Notable examples of known Type A1 activators that can distinguish between different enzyme isoforms include the compound SRT1720 which specifically activates SIRT1 isoform (Milne, Lambert, et al, 2007) and xanthenone which potently activates angiotensin-converting enzyme 2 (ACE2) but does not effect ACE activity (Hernandez Prada, Ferreira, et al, 2008). In the case of Epac2, while sequence alignment suggests that R447 and F374 are homologous in Epac1, it is possible that there are additional interactions in Epac2 that are not present in Epac1, and these interactions are necessary for formation of the SU-binding site. The observation that a biosensor containing only the Epac2 CNB-B domain is not effected by SU treatment (Fig. S4I) supports the hypothesis that other domains in Epac2 are necessary for the SU-induced activation. Future structural studies should provide a vital comparison to better understand the isoform selectivity of SUs for Epac2.
Together, the findings here identify Epac2 as a direct target for SU and reveal isoform selectivity of SU for Epac2 over Epac1. To our knowledge, there are no other reports of isoform-selective allosteric activators of Epac or any other GEFs. As Epac1 is widely expressed in a variety of tissues and implicated in diverse functions including cardiac hypertrophy and neuronal disorders (Gloerich, and Bos, 2010; Zarich, 2009), selective activation of Epac2 without affecting Epac1 may be a more viable pharmacological tactic (Hinke, 2009). In order to generate more potent Epac2-specific activators, however, it is important to understand the molecular determinants underlying SU binding to and activation of Epac2. Our findings that SUs bind to Epac2 at a new allosteric site and utilize a similar hinge motion for activation that is used by the endogenous agonist have expanded our understanding of Epac activation as well as laid a foundation for generating new Epac2-selective activators.
Significance
Sulfonylureas (SUs), a frequently prescribed treatment for Type II diabetes, stimulate insulin secretion from pancreatic β cells by signaling through sulfonylurea receptors. More recent evidence, however, also identifies exchange protein directly activated by cAMP 2 (Epac2) as an intracellular target of SUs, but whether this activation is achieved through direct SU binding remains unclear. Additionally, little is known about how molecules other than the endogenous agonist, cAMP, activate Epac2. By using a FRET-based biosensor for Epac2 activation we showed that SUs bind directly to Epac2 to activate it. As a result, the downstream targets Rap1 and ERK1/2 were activated in living cells. To identify the molecular determinants of SU binding to and activation of Epac2, we combined mutational analysis with molecular docking studies and identified R447 in Epac2 to be critically involved in this action. We further show that SU-mediated Epac activation may engage the same hinge motion as cAMP-induced activation. Although both R447 and the hinge region are conserved between Epac2 and the closely related isoform Epac1, SU-induced activation of Epac was found to be preferential for Epac2. Selectivity of SUs for Epac2 over Epac1 may be therapeutically beneficial as Epac1 is ubiquitously expressed and involved in various cellular processes (Hinke, 2009). Collectively, these studies identify SUs as a new class of isoform-selective enzyme activators and have revealed a new mode of allosteric activation of Epac2. As Epac proteins emerge as critical regulators of many cAMP-dependent processes and as new drug targets for treating diseases such as Type II diabetes (Gloerich, and Bos, 2010), these findings should provide a basis for the development of more effective therapeutic agents and for the further enhancement of our understanding of Epac signaling at a molecular and cellular level.
Experimental Procedures
Gene construction
Epac2 and Epac1 were PCR amplified and ligated into the mammalian expression vector pcDNA3.1 already containing properly oriented Cerulean and Venus or mCherry. Epac2 mutants were generated using QuickChange Mutagenesis.
Cellular Culture and Transfection
HEK293T were grown in DMEM cell culture media supplemented with 10% FBS at 37°C with 5% CO2. Cells were plated into 35 mm glass-bottom dishes for imaging and 10 cm culture dishes for protein purification and transfected at 60% confluency via calcium phosphate.
Cellular Imaging and Analysis
Cells were imaged in Hanks’ balanced salt solution on a Zeiss Axiovert 200M microscope with a cooledcharge-coupled device camera (MicroMAX BFT512, Roper Scientific, Trenton, NJ) controlled by METAFLUOR software (UniversalImaging, Downingtown, PA). Using a 420DF20 excitation filter and a 450DRLP dichroic mirror, dual emission ratio imaging was performed using appropriate emission filters, 475DF40 for Cerulean and 535DF25 for Venus. Cells were treated with drug as indicated.
Western Blot
Cells were transfected for 24 hours, washed with PBS and treated with respective drugs for 30 minutes. The treated cells were then lysed in RIPA buffer, incubated on ice for 30 minutes and spun at 13,000 × g at 4°C for 30 minutes. Total protein concentration was detected by BCA assay (Pierce). Proteins were separated on 7.5% SDS-PAGE, and transferred to nitrocellulose membranes, which were blocked in 5% BSA and incubated overnight with primary antibodies. Membranes were washed, incubated with secondary antibody, and ECL western blotting substrate (Pierce) was used for detection. Desitrometric analysis was performed using ImageJ (NIH) and pERK1/2 levels were normalized to tubulin levels within each sample. All experiments were repeated 3–5 times.
In vitro fluorescence assays
After 48 hour transfection, HEK293T cells were spun and lysed in a buffer pH 7.4 containing 50mM Tris-HCl, 100mM NaCl, 1mM EDTA, 1mM PMSF, protease inhibitor cocktail (Roche), and 0.2% Triton X-100 in a Dounce homogenizer. Lysed cells were spun and 50 μL of Ni-NTA beads (Qiagen) was added to the supernatant. The beads were spun, washed once in buffer containing 10 mM imidazole, and eluted in buffer containing 100 mM imidazole. Purified protein was detected by Coomasie stain (Pierce Biotechnology) and western analysis using anti-GFP antibody (eBioscience). Fluorescence spectra of Epac2 biosensors were measured with excitation at 435 nm before and after addition of SU as well as cAMP. Spectra were normalized to the CFP peak at 475 nm.
Molecular Docking
Crystal structures were viewed and images generated in Pymol v1.3 (DeLano W.L, 2002). Molecular docking studies of ACT to Epac2 (PDB ID: 2BYV (Rehmann, Das, et al, 2006)) were performed with AutoDock 4.2 (Seeliger, and de Groot, 2010). ACT was sketched using MOE and minimized with the MMFF94x force field in MOE (Chemical Computing Group). A docking cube of 25 angstroms was centered on ‘CZ’ atom of R447. For the 100 docking runs that were executed, the following Lamarckian genetic algorithm parameters were set: maximum number of energy evaluations was 2,500,000, maximum number of generations was 27,000, mutation rate of 0.02, crossover rate of 0.8, and window size was 10 (Morris, Goodsell, et al, 1998).
Supplementary Material
Highlights.
Sulfonylureas (SUs) activate Epac2 via direct binding to Epac2.
SU-induced activation of Epac2 leads to Rap1 activation and ERK1/2 phosphorylation.
R447 in Epac2 is critical for SU binding to and activation of Epac2.
SUs selectively activate Epac2 isoform.
Acknowledgments
We thank Dr. Michiyuki Matsuda for Raichu-Rap1, Dr. Viacheslav Nikolaev for Epac2-camps, and Dr. Jun Liu for the gift of tolbutamide and helpful discussion. This work was funded by NIH R01 DK073368, DP1 OD006419, and 3M (to J. Z.).
References
- Allen MD, DiPilato LM, Rahdar M, Ren YR, Chong C, Liu JO, Zhang J. Reading dynamic kinase activity in living cells for high-throughput screening. ACS Chem Biol. 2006;1:371–376. doi: 10.1021/cb600202f. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA. Pharmacologic therapy for type 2 diabetes mellitus. Ann Intern Med. 1999;131:281–303. doi: 10.7326/0003-4819-131-4-199908170-00008. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System, Version 1.2r3pre. Vol. 1.3 Schrödinger, LLC; 2002. [Google Scholar]
- DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci U S A. 2004;101:16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhault J, Lavielle R. History and evolution of the concept of oral therapy in diabetes. Diabetes Res Clin Pract. 1991;14(Suppl 2):S9–13. doi: 10.1016/0168-8227(91)90003-v. [DOI] [PubMed] [Google Scholar]
- Eliasson L, Ma X, Renstrom E, Barg S, Berggren PO, Galvanovskis J, Gromada J, Jing X, Lundquist I, Salehi A, Sewing S, Rorsman P. SUR1 regulates PKA-independent cAMP-induced granule priming in mouse pancreatic B-cells. J Gen Physiol. 2003;121:181–197. doi: 10.1085/jgp.20028707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- Hanefeld M. Pioglitazone and sulfonylureas: effectively treating type 2 diabetes. Int J Clin Pract Suppl. 2007;(153):20–27. doi: 10.1111/j.1742-1241.2007.01361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez Prada JA, Ferreira AJ, Katovich MJ, Shenoy V, Qi Y, Santos RA, Castellano RK, Lampkins AJ, Gubala V, Ostrov DA, Raizada MK. Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension. 2008;51:1312–1317. doi: 10.1161/HYPERTENSIONAHA.107.108944. [DOI] [PubMed] [Google Scholar]
- Hinke SA. Epac2: a molecular target for sulfonylurea-induced insulin release. Sci Signal. 2009;2:pe54. doi: 10.1126/scisignal.285pe54. [DOI] [PubMed] [Google Scholar]
- Holz GG. Epac: A new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell. Diabetes. 2004;53:5–13. doi: 10.2337/diabetes.53.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG. Cell physiology of cAMP sensor Epac. J Physiol. 2006;577:5–15. doi: 10.1113/jphysiol.2006.119644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- Lin Y, Sun Z. Current views on type 2 diabetes. J Endocrinol. 2010;204:1–11. doi: 10.1677/JOE-09-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCourt JA, Pang SS, Guddat LW, Duggleby RG. Elucidating the specificity of binding of sulfonylurea herbicides to acetohydroxyacid synthase. Biochemistry. 2005;44:2330–2338. doi: 10.1021/bi047980a. [DOI] [PubMed] [Google Scholar]
- McCourt JA, Pang SS, King-Scott J, Guddat LW, Duggleby RG. Herbicide-binding sites revealed in the structure of plant acetohydroxyacid synthase. Proc Natl Acad Sci U S A. 2006;103:569–573. doi: 10.1073/pnas.0508701103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki N, Yamashita S, Kurokawa K, Ohba Y, Nagai T, Miyawaki A, Matsuda M. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature. 2001;411:1065–1068. doi: 10.1038/35082594. [DOI] [PubMed] [Google Scholar]
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. 1998;19:1639-1639–1662. [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Nikolaev VO, Bunemann M, Hein L, Hannawacker A, Lohse MJ. Novel single chain cAMP sensors for receptor-induced signal propagation. J Biol Chem. 2004;279:37215–37218. doi: 10.1074/jbc.C400302200. [DOI] [PubMed] [Google Scholar]
- Rehmann H, Das J, Knipscheer P, Wittinghofer A, Bos JL. Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature. 2006;439:625–628. doi: 10.1038/nature04468. [DOI] [PubMed] [Google Scholar]
- Rehmann H, Prakash B, Wolf E, Rueppel A, de Rooij J, Bos JL, Wittinghofer A. Structure and regulation of the cAMP-binding domains of Epac2. Nat Struct Biol. 2003;10:26–32. doi: 10.1038/nsb878. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- Roscioni SS, Elzinga CR, Schmidt M. Epac: effectors and biological functions. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:345–357. doi: 10.1007/s00210-007-0246-7. [DOI] [PubMed] [Google Scholar]
- Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24:417–422. doi: 10.1007/s10822-010-9352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J. Sur1 knockout mice. A model for K(ATP) channel-independent regulation of insulin secretion. J Biol Chem. 2000;275:9270–9277. doi: 10.1074/jbc.275.13.9270. [DOI] [PubMed] [Google Scholar]
- Shibasaki T, Takahashi H, Miki T, Sunaga Y, Matsumura K, Yamanaka M, Zhang C, Tamamoto A, Satoh T, Miyazaki J, Seino S. Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci U S A. 2007;104:19333–19338. doi: 10.1073/pnas.0707054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalkova T, Blumenthal DK, Mei FC, White MA, Cheng X. Mechanism of Epac activation: structural and functional analyses of Epac2 hinge mutants with constitutive and reduced activities. J Biol Chem. 2009;284:23644–23651. doi: 10.1074/jbc.M109.024950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JG, Lee PK, Dong YH, Pang SS, Duggleby RG, Li ZM, Guddat LW. Crystal structures of two novel sulfonylurea herbicides in complex with Arabidopsis thaliana acetohydroxyacid synthase. FEBS J. 2009;276:1282–1290. doi: 10.1111/j.1742-4658.2009.06863.x. [DOI] [PubMed] [Google Scholar]
- Zarich SW. Antidiabetic agents and cardiovascular risk in type 2 diabetes. Nat Rev Endocrinol. 2009;5:500–506. doi: 10.1038/nrendo.2009.150. [DOI] [PubMed] [Google Scholar]
- Zhang CL, Katoh M, Shibasaki T, Minami K, Sunaga Y, Takahashi H, Yokoi N, Iwasaki M, Miki T, Seino S. The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science. 2009;325:607–610. doi: 10.1126/science.1172256. [DOI] [PubMed] [Google Scholar]
- Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- Zorn JA, Wells JA. Turning enzymes ON with small molecules. Nat Chem Biol. 2010;6:179–188. doi: 10.1038/nchembio.318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.