

Abstract
Enolase is a dimeric enzyme that catalyzes the interconversion of 2-phospho-D-glycerate and phosphoenolpyruvate. This reversible dehydration is effected by general acid-base catalysis that involves, principally, Lys345 and Glu211 (numbering system of enolase 1 from yeast). The crystal structure of the inactive E211Q enolase shows that the protein is properly folded. However, K345 variants have, thus far, failed to crystallize. This problem was solved by crystallization of an engineered heterodimer of enolase. The heterodimer was composed of an inactive subunit that has a K345A mutation and an active subunit that has N80D and N126D surface mutations to facilitate ion-exchange chromatographic separation of the three dimeric species. The structure of this heterodimeric variant, in complex with substrate/product, was obtained at 1.85 Å resolution. The structure was compared to a new structure of wild-type enolase obtained from crystals belonging to the same space group. Asymmetric dimers having one subunit exhibiting two of the three active site loops in an open conformation and the other in a conformation having all three loops closed appear in both structures. The K345A subunit of the heterodimer is in the loop-closed conformation; its Cα carbon atoms closely match those of the corresponding subunit of wild-type enolase (root-mean-squared deviation of 0.23 Å). The kcat and kcat/Km values of the heterodimer are approximately half those of the N80D/N126D homodimer, which suggests that the subunits in solution are kinetically independent. A comparison of enolase structures obtained from crystals belonging to different space groups suggests that asymmetric dimers can be a consequence of the asymmetric positioning of the subunits within the crystal lattice.
Keywords: enolase, homodimer, heterodimer, asymmetry, independent subunits
Introduction
Enolase (EC 4.2.1.11; 2-phospho-D-glycerate hydrolase) catalyzes the interconversion of 2-phospho-D-glycerate (2-PGA) and phosphoenolpyruvate (PEP) in glycolysis and in gluconeogenesis. The enzyme uses general acid-base catalysis to effect this interconversion, the mechanism of which is depicted in Scheme 1. Several lines of evidence support this mechanism.1–4 In particular, the roles of Lys345 and Glu211 in general acid-base catalysis are supported by site-specific mutagenesis experiments.3 Near neutral pH, catalysis in the forward direction is supported by a small fraction of the enzyme that exists in a state of reverse protonation, namely that in which the amino side-chain of Lys345 is unprotonated and the carboxyl side-chain of Glu211 is protonated.5
Scheme 1.

Previous experiments showed that K345A and K345M variants and the E211Q variant are inactive in the overall reaction.3,6 A recent structure of E211Q showed proper folding,5 but all previous attempts to obtain crystals of K345A (or other variants of this critical residue) have proven unsuccessful. Since enolase exists as a homodimer in most organisms, a new strategy involving engineered heterodimeric enolases was adopted.
The engineered heterodimers contained an inactive subunit that had the K345A mutation and an active subunit that had N80D and N126D surface mutations. The surface changes in the active subunit of the heterodimer altered the charge of the protein and facilitated separation of the following dimeric species: homodimers of K345A, heterodimers of (K345A)/(N80D/N126D), and homodimers of N80D/N126D. (The N80D/N126D variant is subsequently referred to as wt*.) The creation and purification of the heterodimeric enzyme provided the opportunity for crystallization screens and subsequent structural and kinetic analyses, which are reported herein. In addition, new crystals of wild-type enolase, belonging to the same space group as crystals of the heterodimer, were obtained, and the structure solved at high resolution.
Results
Formation and purification of the engineered heterodimer
The strategy employed in engineering heterodimeric enolases was adopted from previous studies in which “charge-tagged” subunits were used to create mixed, separable dimers (or other oligomers).7,8 Asparagine residues that were changed in the wt* protein were chosen because these residues are not part of the dimer interface. Furthermore, a comparison of the structure of wild-type enolase that is substrate-bound4 versus that which is substrate-free9 showed that these surface residues do not move appreciably upon substrate binding. The incubation conditions (2 mM Tris-HCl (pH 8), 1 mM EDTA-Tris, and 1 M KCl) were sufficient to disrupt interactions at the dimer interface and allow, following dialysis to remove excess KCl, formation of the heterodimer. The heterodimer was successfully purified with ion-exchange chromatography (Figure 1).
Figure 1.
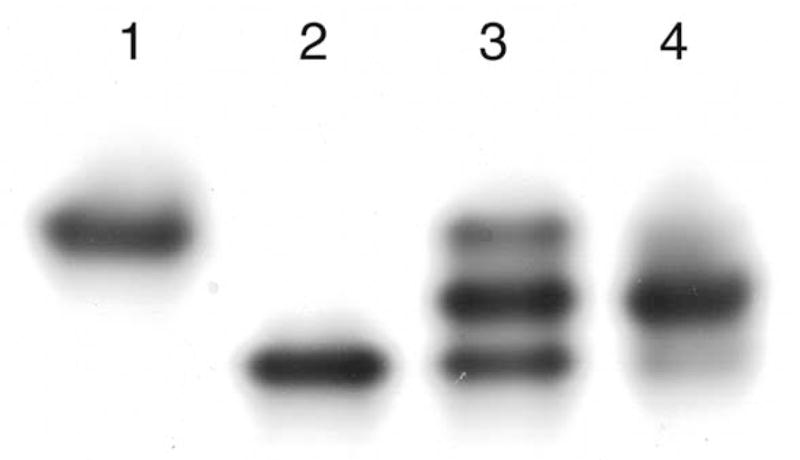
Native gel from the isolation of the K345A/wt* heterodimer. Lane 1 is a sample of the K345A homodimer; lane 2 is a sample of the wt* homodimer; lane 3 is a sample from the hybridization reaction; and lane 4 is a sample (overloaded) of the isolated heterodimer.
A different variant at position 345 (K345E) also was tested. The corresponding variant of Escherichia coli enolase was reported to have significant activity.10 When the yeast K345E variant was overexpressed and purified, it was found to have <1 part in 105 activity of wild-type enolase, consistent with the critical importance of this lysine in general acid-base catalysis. Because the change in charge of K345E parallels that of the wt* variant, visualization of K345E/wt* heterodimers in native gels was impeded, so efforts to isolate these heterodimers were not pursued.
Crystal structures
The crystallization conditions, which included MgCl2 and PEP (3.5 mM each), were sufficient to stabilize the K345A/wt* heterodimers and prevent reshuffling of the subunits. In the structure, the electron density explicitly shows an alanine at position 345 in one of the subunits (Figure 2(a)) and a lysine at position 345 of the other subunit (Figure 2(b)).
Figure 2.

(a) Stereoview of residues 344–346, 2-PGA, and magnesium ions (green spheres) in the K345A subunit of the heterodimer. (b) Stereoview of residues 344–346, PEP, and magnesium ions in the wt* subunit of the heterodimer. An Fo−Fc electron density map, contoured at 3.5 σ, is shown for residue 345 in both subunits.
The structure of the heterodimer shows an intact dimer interface. An overlay (not shown) of the Cα carbon atoms of the heterodimer and the Cα carbon atoms of wild-type enolase (determined here) indicates that the two structures share the same overall fold and dimer interface. The rms deviation between the Cα carbon atoms of the K345A subunit of the heterodimer and those of the wild-type subunit that has three closed loops was 0.23 Å, with a maximum deviation of 1.5 Å (Asp280). The rms deviation between the Cα carbon atoms of the active subunit of the heterodimer and those of the wild-type subunit that has two out of three loops opened up was 0.18 Å, with a maximum deviation of 1.1 Å (Gly157). The three main loops include residues 37–43 (loop 1), 153–166 (loop 2), and 251–275 (loop 3). A comparison of the loop-open versus loop-closed subunits indicates that loops 2 and 3 move in a hinge-like manner such that large portions of each loop move in unison. Figure 3 highlights the loop regions of the K345A subunit versus those of the wt* subunit. Aside from these differences in the loop regions, the two subunits are superimposable.
Figure 3.

Stereoview of a ribbon representation of the K345A subunit (teal). An overlay of the K345A subunit and the wt* subunit shows that the loop regions are the only regions of significant difference. Loop 1 (residues 37-43) is labeled L1. Loop 2 (residues 153-166) is labeled L2. Loop 3 (residues 251–275) is labeled L3. The violet loops represent the K345A subunit and the red loops (parts of which are disordered) represent the wt* subunit. The 2-PGA, shown in ball and stick representation, and the magnesium ions, shown as green spheres, are from the K345A subunit. The C terminus is labeled C and the N terminus is labeled N.
A stereoview of the active site of the K345A subunit of the heterodimer overlaid with that of the subunit of wild-type enolase having closed loops (Figure 4(a)) shows that, except for position 345, all key residues are essentially in the same position, as are the two magnesium ions of each active site. A stereoview of the active site of the wt* subunit of the heterodimer overlaid with that of the subunit of wild-type enolase having open loops (Figure 4(b)) shows that Lys345, Glu168, and Glu211 are in the same position, but the region around Ser39, which is part of a movable loop, is disordered in the wt* subunit of the heterodimer but not in the analogous subunit of wild-type enolase. In addition, residues 158–162 in loop 2 are disordered in both structures. The two magnesium ions are, however, essentially in the same positions in the two active sites. For the 345A subunit of the heterodimer and for the subunit of wild-type having closed loops, 2-PGA fits into the electron density corresponding to the position of the substrate. When SHELX11 was used to refine the occupancy, however, the ratio of 2-PGA:PEP was ~2:1 (total occupancy of 1) for the K345A subunit of the heterodimer and ~1:1 (total occupancy of 1) for the subunit of the wild-type enzyme having closed loops. The occupancy of the wild-type subunit is consistent with the internal thermodynamics of enolase in solution.12 Figure 5 shows the Fo−Fc density map for the substrate in the K345A subunit. The presence of the hydroxyl group of 2-PGA is clearly defined. Figure 6 shows the Fo−Fc density map for the substrate of the closed subunit of wild-type enolase. In Figure 6(a) the density, contoured at 4 σ, clearly shows that the hydroxyl group of 2-PGA is represented. However, as shown in Figure 6(b), the density for the hydroxyl group weakens selectively when contoured at 6 σ, while the density for phosphate and carboxylate groups remain strong. Similarly, SHELX refinement of the substrate occupancy in the wt* subunit of the heterodimer indicated an ~ 1:1 ratio of 2-PGA:PEP. Except for the missing Lys side-chain at position 345 in the 345A subunit of the heterodimer, the structure is virtually identical with that of wild-type enolase.
Figure 4.

(a) Stereoview of key active-site residues of the subunit of wild-type enolase with closed loops (gray) overlaid with the same residues (except position 345) of the K345A subunit of the heterodimer (dark blue). 2-PGA is shown with gray carbon atoms, red oxygen atoms, and a purple phosphorus atom. Magnesium ions are shown as green spheres. (b) Stereoview of key active-site residues of the subunit of wild-type enolase having open loops (gray) overlaid with the same residues (when well-ordered) of the wt* subunit of the heterodimer (dark blue). Atoms of PEP and magnesium ions are as represented in (a).
Figure 5.

Stereoview of key active-site residues for the K345A subunit. The Fo−Fc electron density map, contoured at 3.7σ, is shown for 2-PGA.
Figure 6.

(a) Stereoview of key active-site residues for subunit 1 wild-type enolase pH 8.0. The Fo−Fc electron density map, contoured at 4 σ, is shown for 2-PGA. (b) The same stereoview as (a), except the Fo−Fc electron density map is contoured at 6 σ.
Kinetic analysis of the engineered heterodimers
Table 1 shows estimates of kcat, Km-PGA, and kcat/Km-PGA for wild-type enolase, and for the wt* homodimers and the K345A/wt* heterodimers. The kcat of the wt* homodimer, isolated in the hybridization experiments, was nearly equal to that of wild-type enolase. The Km-PGA of the wt* homodimer was slightly elevated compared to wild-type enolase, but overall, the kcat/Km-PGA was not significantly different from wild-type enolase. The kcat and the kcat/Km-PGA values of the K345A/wt* heterodimer were 43% and 49% of the corresponding values of the wt* homodimer, respectively. The absence of perturbations in Km-PGA of the heterodimer suggests that substrate binding to the wt* subunit is not adversely affected by the wt* connection to the “drone” subunit.
Table 1.
Kinetic parameters of homodimeric and heterodimeric enolases
| Form of enolase | kcat (s−1) | KM-PGA (mM) | kcat/KM-PGA (M−1 s−1) |
|---|---|---|---|
| Wild-type enolase | 58±1 | 0.043±0.004 | 1.4(±0.1)×106 |
| wt* homodimera | 56±1 | 0.057±0.005 | 9.9(±0.7)×105 |
| (K345A)/wt* heterodimer | 24±1 | 0.050±0.004 | 4.9(±0.3)×105 |
| K345E homodimer | 2.5(±0.1)×10−4 |
The wt* homodimer was isolated as part of the K345A/wt* hybridization experiment. This protein eluted from the Mono Q column after the heterodimer.
The possibility that the isolated heterodimers reshuffled or completely dissociated during the assays (or when the samples were diluted prior to running the assays) is unlikely because both the storage buffer and dilution buffer contained Mg2+, which previously was shown to stabilize dimers of enolase, even in the presence of high concentrations of KCl.13 As mentioned, the stabilizing effects of Mg2+ (and to some extent 2-PGA/PEP) also were observed in the present work because intact heterodimers were observed in the crystal structures despite the presence of 0.25 M KCl in the crystallization solution. In addition, native gel electrophoresis of samples of heterodimer showed that the heterodimers remained intact for several months after first being isolated, which suggests that reshuffling, to the extent that it occurs, is a slow process. Finally, in separate experiments with a different heterodimer (composed of an E211Q subunit and the same wt* used above), a tenfold excess of the E211Q homodimer was included as a control in the assay cocktails such that, if transient dissociation occurred upon dilution, an active subunit would be far more likely to encounter and re-associate with an inactive subunit. These heterodimers also were ~50% as active as the wt* homodimers, and no differences in activity in these control experiments were detected.
Discussion
Structural and kinetic properties of the engineered heterodimer
The crystal structure clearly shows that the K345A subunit of the heterodimer is folded properly. Proper folding of the K345A subunit is consistent with the previous finding that homodimeric K345A can catalyze a reaction analogous to the first step of the reverse reaction (addition of OH− to 3-Cl-PEP).3 Thus, the inactivity of the K345A variant is not due to misfolding but rather to loss of a critical acid/base catalyst, Lys345. Solution studies of the catalytic properties of other engineered oligomeric enzymes have been reported.7,8 The present work shows that this strategy of engineering and isolating hetero-oligomers can also be useful in obtaining structural information on difficult-to-crystallize variants.
Although there may be lingering suspicions that subunits of enolase work in a negatively cooperative manner,14 the simplest explanation of the kinetic results of the present study is that the catalytic activity of the heterodimer is essentially that expected for an enzyme that has independent subunits, one of which is active and the other of which is inactive. Extensive kinetic measurements of enolase catalysis have not uncovered hall-marks of cooperative behavior such as non-linear double reciprocal plots (1/v versus 1/[S]).15 Half-of-the-sites activity (the extreme case of negative cooperativity) might escape detection via normal steady-state kinetic criteria.16 In the case of half-of-the-sites behavior, one might expect the specific activity of the heterodimer to match that of wt*, because in the latter only one active site would be functioning at any given time. The present kinetic observations on the heterodimer are consistent with a previous study in which subunits of enolase were suggested to function independently, as indicated by specific activity measurements for monomers.17
The split in occupancy between 2-PGA and PEP that was present in the structure of the heterodimer also was of interest. Although the heterodimer was co-crystallized with PEP, the presence of one active subunit led to an equilibrium mixture of 2-PGA and PEP in solution such that either compound could bind to either active site. The 2:1 ratio of 2-PGA to PEP in the K345A subunit suggests that this inactive subunit has a slight preference for binding of 2-PGA.
The K345A subunit was in a conformation wherein all three of the active site loops were closed. The wt* subunit was in a conformation in which two of the three loops were opened up (Figure 3). The difference in surface charges between the two subunits and/or some other feature(s) of the subunits apparently led to this consistent arrangement of the different subunits within the crystal lattice. Otherwise, the subunits might have alternated such that density corresponding to partial occupancy for lysine at position 345 would have been observed in both subunits.
General features of structures of enolase
Crystal structures of dimeric enolase are intriguing because some structures show symmetric dimers in which both subunits have their flexible loops in a closed conformation,18,19 while other structures show asymmetric dimers in which one subunit has closed loops and the other subunit has one or more of the loops opened up20 (and the present work). One of the “symmetric” structures contains Mg2+ and the inhibitor phosphonoacetohydroxamate (PhAH).18 The other “symmetric” structure contains Mg2+ and an equilibrium mixture of 2-PGA and PEP.4 Both crystals belong to the space group C2. The three asymmetric structures all contain Mg2+, 2-PGA, and/or PEP. All three of these crystals belong to the space group P21.
In structures with symmetric dimers, partner subunits contact surrounding dimers in the crystal lattice in an approximately symmetrical manner. Specifically, loop regions of partner subunits each contact two neighboring dimers. In structures having asymmetric dimers, a similar comparison shows that the loop regions of the closed subunit of a dimer contact three neighboring dimers, whereas the loop regions of the open, partner subunit contact only one neighboring dimer. The positions of flexible loops of proteins in crystals can vary, depending upon the crystal form.21–24 It is possible that the observation of asymmetric dimers in some crystal structures of homodimeric enolase may be due to the packing of the dimers within the crystal lattice. This possibility is supported by the appearance of symmetric dimers in some crystal forms.4,18,19
In conclusion, these experiments have provided a structure for an important positional variant of yeast enolase that had eluded crystallization as a homodimeric protein. The structure removes lingering ambiguities concerning the proper folding of K345A variants. The catalytic properties of the heterodimer are those expected for a dimer having an active and an inactive subunit, the specific activity being about half that of the active homodimer. The program SHELX is useful in assigning occupancies in crystal structures containing equilibrium mixtures of substrate and product.
Materials and Methods
Chemicals and buffers
PEP (monopotassium salt) was purchased from Alfa Aesar (Ward Hill, MA, USA). Streptomycin sulfate, NaN3, PMSF, and 2-PGA were purchased from Sigma-Aldrich (St. Louis, MO, USA). When 2-PGA became unavailable commercially, it was synthesized enzymatically as described.25 Mes and Hepps buffers were purchased from Research Organics (Cleveland, OH, USA). Ammonium sulfate, EDTA, MgCl2 hexahydrate, and the buffers Tris and Hepes were purchased from Fisher (Pittsburgh, PA, USA). PEG 8000 was purchased from Fluka (Milwaukee, WI, USA). High-purity MgCl2 (99.99%) was purchased from Aldrich (Milwaukee, WI, USA).
Chromatography materials
Chelex 100 resin was purchased from Bio-Rad Laboratories (Hercules, CA, USA). DEAE-cellulose, Microgranular Form (Preswollen), was purchased from Sigma. Sephadex™ G-50 Coarse, DEAE Sephadex™ A-50, and CM Sephadex™ C-50 resins were purchased from Pharmacia/Amersham (now part of GE Healthcare, Piscataway, NJ, USA). A Mono Q HR 10/10 anion-exchange column also was from Pharmacia.
Site-directed mutagenesis
The enolase variant N80D/N126D (wt*) was created from the PetEnol plasmid3 as advised by the Quick Change mutagenesis kit from Stratagene (La Jolla, CA, USA). The resulting expression vector was sequenced at the University of Wisconsin–Madison Biotechnology Center.
Enzymes
Lysozyme was purchased from Sigma. DNase I was purchased from Roche (Indianapolis, IN, USA). Wild-type enolase was purified from yeast as described.5,18 Purified E211Q enolase was a gift from Dr Russell Poyner. The K345A and wt* variants of enolase were expressed recombinantly in E. coli as described.3 These variants generally were purified as follows: 60–80 g of cell paste was resuspended in 0.6–0.8 l of buffer A (20 mM Tris–HCl, pH 8), which contained 0.3 g l−1 of lysozyme, 0.2 g l−1 of PMSF, and ~1–2 mg l−1 of DNase I. The resuspended cells were shaken at room temperature for 1.5 h, at which time 10 g l−1 of streptomycin sulfate and EDTA-Tris (pH 8) (0.5 mM final) were added. The mixture was shaken an additional 30 min and then was centrifuged at 4 °C and at 14,000 rcf for 20 min.
The resulting supernatant was brought to 55% saturation with ammonium sulfate and then was centrifuged as above. The 55% supernatant then was brought to 80% saturation with ammonium sulfate and was centrifuged as above. The 80% pellet was redissolved in ~0.05 l of buffer A and was loaded onto a 4 cm × 55 cm column of Sephadex™ G-50, equilibrated in buffer A. (This step and subsequent steps were done at room temperature unless stated otherwise.) The sample was passed through this desalting column, and the fractions that contained protein but no ammonium sulfate were pooled and loaded onto a 5 cm×15 cm column of DEAE Sephadex™ A-50, also equilibrated in buffer A. After the sample was loaded onto the DEAE column, the column was washed with ~0.3 l of buffer A. The sample was eluted from the DEAE column with a 2 l gradient from 0 to 0.3 M KCl in buffer A
Peak fractions that contained the particular variant of enolase were identified by SDS-PAGE in 12% (w/v) polyacrylamide gels. These peak fractions were pooled and diluted fivefold with buffer A. The pH of the pooled, diluted sample was adjusted to 6 by addition of solid Mes. This sample then was loaded onto a 4.5 cm×20 cm column of CM Sephadex™ C-50, which was equilibrated in buffer B (buffer A plus solid Mes, pH 6). Variants of enolase were eluted with a 2 l gradient from 0–0.3 M KCl in buffer B. Fractions that contained the particular variant were identified as above, pooled, and concentrated at ~4 °C in a 0.2 l Amicon pressure cell, which was fitted with a YM 30 membrane. The concentrated sample was diluted to the full volume of the pressure cell with storage buffer (buffer A with 2 mM MgCl2, 0.5 mM Tris–EDTA, and 1 mM NaN3) and re-concentrated. This process was repeated once or twice.
The concentrated samples, which had been exchanged into storage buffer, were loaded onto a 2 cm×10 cm column of DEAE-cellulose (microgranular), which was equilibrated in buffer A and was used as a final, polishing column to remove any remaining contaminants. A 0.5 l gradient from 0–0.25 M KCl in buffer Awas used to elute the variants from the polishing column. Fractions that contained purified variant were identified, pooled, and concentrated/exchanged into storage buffer as described above.
Removal of contaminating metal ions
2-PGA solutions were passed through 1 cm×5 cm columns of Chelex 100 (200–400 mesh) in the K+ form to remove contaminating metal ions. A stock solution of 1 M Hepes–KOH (pH 7.5), which was used in the activity assays, was passed through a 2.5 cm×30 cm column of Chelex 100 (200–400 mesh) K+ form.
Standardization of stock solutions
Concentrations of enolase and the variants of enolase were determined from A280 measurements. For these determinations, an extinction coefficient of 0.89 ml mg−1 cm−126 and a subunit molecular mass of 46,50027,28 were used. The concentration of 2-PGA solutions was determined by enzymatic end-point assays as described.29 PEP concentrations were determined by A240 readings at pH 9, with no added MgCl2. For these determinations, an extinction coefficient of the trianion form of PEP (εPEP=1.7×103 M−1 cm−1) was used as described.5
Heterodimer formation and isolation
The K345A/wt* heterodimer was formed and purified as follows: equal concentrations of the inactive K345A variant and the active, wt* variant (N80D/N126D) were combined (5 mg ml−1 each in 1 ml total volume) with 2 mM Tris–HCl (pH 8), which contained 1 mM Tris–EDTA (pH 8), and 1 M KCl. The samples were incubated for 90 min at room temperature and then were dialyzed against buffer C (2 mM Tris–HCl (pH 9), plus 4 mM MgCl2, 1 mM PEP, and enough solid Tris to raise the pH to 9).
After dialysis, the sample was loaded onto a Mono Q HR 10/10 column that was equilibrated in buffer C. Heterodimers were eluted in 1.5 ml fractions with a ~0.15 l gradient from 0–12 mM ammonium sulfate in buffer C. Fractions that contained protein were identified by A280 readings. The shallow gradient provided baseline separation of the three protein fractions, which eluted in order of increasing charge. The identities of samples in the peak fractions (e. g. homodimers of inactive variant; heterodimers of inactive/active variants; and homodimer of wt*) were confirmed by electrophoresis in native, 6% or 8% polyacrylamide gels. These gels contained 10 mM MgSO4 to stabilize the heterodimers. Gels were electrophoresed at a constant potential difference of 100 V, with the bottom third of the buffer tank immerged in ice. Peak fractions that corresponded to the heterodimer were pooled and concentrated in 20 mM Tris–HCl (pH 8), which contained 4 mM MgCl2. These samples were concentrated such that A280 readings of undiluted samples were within the linear range of the spectrophotometer. Peak fractions that corresponded to the wt* homodimer also were pooled and, when necessary, concentrated as above.
Kinetic measurements of the dehydration reaction
Kinetic assays were run at 25 °C and were done in triplicate as described. The concentration of substrate, 2-PGA, was varied (0.02–1.1 mM), but the concentration of Mg2+ (1 mM MgCl2) was not varied because of its importance in stabilizing dimer contacts. (Initial assays were run such that MgCl2 was varied from 0.5–5 mM; it was found that the measured velocities did not change appreciably in this range of [MgCl2].) Assay mixtures were buffered with 0.05 M Hepes–KOH (pH 7.5). Dilutions of enzyme, when required, were made into buffer that contained 2 mM MgCl2; these dilutions were made fresh before the assay, and the concentration of enzyme used in the assays was 1 nM or 4 nM final. Kinetic data were fitted to the Michaelis–Menten equation or to a modified form of the Michaelis–Menten equation in which kcat/Km is treated as a single variable to obtain a reliable estimate of the error of this parameter. For the heterodimer, the enzyme concentration used to calculate kcat was the subunit concentration (i.e. both subunits were included in the concentration even though the active site in the K345A subunit was disabled).
Crystallization and data collection
Crystals of wild-type enolase were grown by the batch method from solutions that contained 10 mg ml−1 of enzyme, 13% PEG 8000, 0.25 M KCl, 25 mM Hepps–KOH (pH 8.0), 3.5 mM PEP, and 3.5 mM MgCl2. Microseeding from crystals of the E168Q variant of enolase was used to initiate growth of crystals. This procedure led to the growth of rod-like crystals (~0.1 mm×0.3 mm×0.4 mm) within three days
Crystals of the K345A/wt* heterodimer also were grown by the batch method but from solutions that contained ~5 mg ml−1 of enzyme, 15% PEG 8000, 0.25 M KCl, 25 mM Hepps–KOH (pH 8.0), 3.5 mM PEP, and 3.5 mM MgCl2. Growth of crystals was initiated by use of microseeding from crystals of wild-type enolase, which led to the growth of rod-like crystals (~0.05 mm×0.1 mm×0.1 mm) within three days.
Single crystals of wild-type enolase and the K345A/wt* heterodimer were transferred separately into a solution (solution A′) that contained 50 mM Hepps–KOH (pH 8.0), 20% PEG 8000, 0.3 M KCl, 5 mM MgCl2, and 4 mM PEP. Each crystal was allowed to equilibrate in solution A′ for 1 min prior to a five-step serial transfer into solutions that contained increasing concentrations (4% per step) of the cryoprotectant ethylene glycol. A 5 s equilibration time between each of the successive steps was used throughout the serial transfers. After the last serial transfer, each crystal was flash-frozen and stored in liquid nitrogen until acquisition of structural data.
X-ray data for wild-type enolase were collected at the Life Sciences Collaborative Access Team (LS-CAT) ID beam line (Advanced Photon Source, Argonne National Laboratory). X-ray data for the K345A/wt* heterodimer were collected with a Bruker Nonius FR591 MICROSTAR rotating anode generator and a Bruker Nonius Pt-135 CCD detector. X-ray exposure times were 1 min/deg (0.5 deg/image) with a crystal-to-detector distance of 6.0 cm. Data reduction was performed using the programs SAINT and SADABS from the Proteum software suite (Bruker AXS Inc., 2004).
Crystals of the wild-type enolase-Mg2+-substrate complex belonged to the space group P21, contained one dimer per asymmetric unit, and had unit cell dimensions a=72.0 Å, b=65.0 Å, c=85.9 Å, and β=99.5°. The structure of this enolase complex was solved by molecular replacement using the software program MOLREP from the CCP4 suite.30 The search-model coordinates used were from the coordinates from the E168Q variant.5 The initial R factor was 34.9%. The software program SHELX11 was used to further refine the enolase structure, and the graphics program Turbo-Frodo31 was used to observe the structure. Observation using Turbo-Frodo revealed that residues 38–40, 156–164, and 258–274 (the active site loops) in both subunits required additional adjustments. The residues were removed, SHELX refinement was again performed, and the resulting Fo−Fc density maps were used to rebuild the removed residues. Density corresponding to 2-PGA was present in subunit 1, and density corresponding to PEP was present in subunit 2. Further observation of subunit 1 revealed that 2-PGA and PEP could both fit into the density; accordingly, SHELX was used to refine the occupancy of 2-PGA and PEP in subunit 1. After inclusion of the substrates and two magnesium ions in each subunit, ordered water molecules were added using SHELX. Further adjustments were done manually, and additional water molecules were added using Turbo-Frodo. The final refinement statistics are summarized in Table 2.
Table 2.
Crystal and refinement data of the wild-type enolase-Mg2+-PEP/2-PGA complex and the K345A/wt* heterodimer-Mg2+-PEP/2-PGA complex
| Crystallographic data | Wild-type enolase | K345A/wt* |
|---|---|---|
| Space group | P21 | P21 |
| Cell dimensions | ||
| a (Å) | 72.0 | 71.7 |
| b (Å) | 65.0 | 65.6 |
| c (Å) | 85.9 | 85.9 |
| β (deg.) | 99.5 | 99.2 |
| No. of measurements | 470,313 | 715,034 |
| No. of independent reflections | 120,985 | 67,650 |
| Data range (Å) | 30–1.5 | 26–1.85 |
| Completeness (%) | ||
| Overall | 96.9 | 100 |
| Last shell | (1.55–1.50) 95.2 | (1.90–1.85) 100 |
| Rmerge (%) | ||
| Overall | 4.6 | 6.6 |
| Last shell | 24.7 | 29.3 |
| I/σ | ||
| Overall | 23.4 | 24.4 |
| Last shell | 3.7 | 5.0 |
| Refinement statistics | ||
| R cryst (%) | 15.3 | 19.6 |
| R factor (free) 5% data (%) | 21.7 | 26.7 |
| rmsd of bonds (Å) | 0.0055 | 0.0054 |
| rmsd of angles (deg) | 1.2 | 1.2 |
| No. of water molecules | 694 | 538 |
Crystals of the K345A/wt* heterodimer, as a complex with Mg2+ and 2-PGA/PEP, belonged to the space group P21, contained one dimer per asymmetric unit, and had unit cell dimensions a=71.7 Å, b=65.6 Å, c=85.9 Å, and β=99.2°. Initial phases of the heterodimeric complex were calculated from the refined coordinates of the native enolase structure (minus the substrates, Mg2+, and water molecules). The initial R factor was 28.6%. Observation of the structure with the graphics program Turbo-Frodo revealed that the conformations of residues 39–40 and 158–162 in subunit 2, and 259–272 in both subunits required adjustments. These residues were removed and SHELX refinement was performed. The Fo−Fc density maps were then used to rebuild the removed residues. Density corresponding to 2-PGA/PEP was present in subunit 1 and in subunit 2. The same refinement procedure as described for the wild-type enolase complex was done, and the final refinement statistics are given in Table 2.
Protein Data Bank accession numbers
The atomic coordinates of the K345A/wt* heterodimer of enolase and of wild-type enolase have been deposited with the RCSB Protein Data Bank with accession numbers 2AL2 for K345A/wt* and 2AL1 for wild-type enolase.
Acknowledgments
This research was supported, in part, by NIH grant GM35752. S. M. is supported by National Institutes of Health Predoctoral Training Grant T32 GM08293 in Molecular Biophysics. The authors are grateful to Dr George Phillips for helpful comments on the manuscript, to Dr Matthew Benning for assistance, to Dr Russell Poyner for helpful suggestions, and to Professor Dr George Sheldrick for helpful discussions regarding SHELX. Use of the Argonne National Laboratory Life Sciences Collaborative Access Team (LS-CAT) ID beamline at the Advanced Photon Source, was supported by the U. S. Department of Energy, Office of Energy Research, under Contract no. W-31-109-ENG-38. The Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor also are acknowledged for the support of LS-CAT research program (grant 085P1000817).
Abbreviations used
- 2-PGA
2-phospho-D-glycerate
- PEP
phosphoenolpyruvate
- DEAE
diethylaminoethyl
- CM
carboxymethyl
- Mes
2-(N-morpholino)ethanesulfonic acid
- Hepps
N-(2-hydroxyethyl)piperazine-N′-3-propanesulfonic acid
- PEG
polyethylene glycol
- PMSF
phenylmethanesulfonyl fluoride
- PhAH
phosphonoacetohydroxamate
References
- 1.Cohn M, Pearson JE, O’Connell EL, Rose I. Nuclear magnetic resonance assignment of the vinyl hydrogens of phosphoenolpyruvate. Stereochemistry of the enolase reaction. J Am Chem Soc. 1970;92:4095–4098. doi: 10.1021/ja00716a044. [DOI] [PubMed] [Google Scholar]
- 2.Dinovo EC, Boyer PD. Isotopic probes of the enolase reaction mechanism. J Biol Chem. 1971;246:4586–4593. [Google Scholar]
- 3.Poyner RR, Laughlin LT, Sowa GA, Reed GH. Toward identification of acid/base catalysts in the active site of enolase: comparison of the properties of K345A, E168Q, and E211Q variants. Biochemistry. 1996;35:1692–1699. doi: 10.1021/bi952186y. [DOI] [PubMed] [Google Scholar]
- 4.Larsen TM, Wedekind JE, Rayment I, Reed GH. A carboxylate oxygen of the substrate bridges the magnesium ions at the active site of enolase: structure of the yeast enzyme complexed with the equilibrium mixture of 2-phosphoglycerate and phosphoenolpyruvate at 1.8 Å resolution. Biochemistry. 1996;35:4349–4358. doi: 10.1021/bi952859c. [DOI] [PubMed] [Google Scholar]
- 5.Sims PA, Larsen TM, Poyner RR, Cleland WW, Reed GH. Reverse protonation is the key to general acid-base catalysis in enolase. Biochemistry. 2003;42:8298–8306. doi: 10.1021/bi0346345. [DOI] [PubMed] [Google Scholar]
- 6.Reed GH, Poyner RR, Larsen TM, Wedekind JE, Rayment I. Structural and mechanistic studies of enolase. Curr Opin Struct Biol. 1996;6:736–743. doi: 10.1016/s0959-440x(96)80002-9. [DOI] [PubMed] [Google Scholar]
- 7.Hardy LW, Pacitti DF, Nalivaika E. Use of a purified heterodimer to test negative cooperativity as the basis of substrate inactivation of Escherichia coli thymidylate synthase (Asn177→Asp) Structure. 1994;2:833–838. doi: 10.1016/s0969-2126(94)00084-0. [DOI] [PubMed] [Google Scholar]
- 8.Johnson JL, Lasagna MD, Reinhart GD. Influence of a sulfhydryl cross-link across the allosteric-site interface of E. coli phosphofructokinase. Protein Sci. 2001;10:2186–2194. doi: 10.1110/ps.02401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wedekind JE, Reed GH, Rayment I. Octahedral coordination at the high-affinity metal site in enolase: crystallographic analysis of the mgii–enzyme complex from yeast at 1.9 Å resolution. Biochemistry. 1995;34:4325–4330. doi: 10.1021/bi00013a022. [DOI] [PubMed] [Google Scholar]
- 10.Boel G, Pichereau V, Mijakovic I, Maze A, Poncet S, Gillet S, et al. Is 2-phosphoglycerate-dependent automodification of bacterial enolases implicated in their export? J Mol Biol. 2004;337:485–496. doi: 10.1016/j.jmb.2003.12.082. [DOI] [PubMed] [Google Scholar]
- 11.Sheldrick G, Scnheider T. SHELX: high resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 12.Burbaum JJ, Knowles JR. Internal thermodynamics of enzymes determined by equilibrium quench: values of Kint for enolase and creatine kinase. Biochemistry. 1989;28:9306–9317. doi: 10.1021/bi00450a010. [DOI] [PubMed] [Google Scholar]
- 13.Brewer JM. Characterization of the subunit dissociation of yeast enolase. In: Catsimpoolas N, editor. Physical Aspects of Protein Interactions. Elsevier; North-Holland, Amsterdam: 1978. pp. 57–78. [Google Scholar]
- 14.Chai G, Brewer JM, Lovelace LL, Aoki T, Minor W, Lebioda L. Expression, purification and the 1.8 Å resolution crystal structure of human neuron specific enolase. J Mol Biol. 2004;341:1015–1021. doi: 10.1016/j.jmb.2004.05.068. [DOI] [PubMed] [Google Scholar]
- 15.Segel IH. Enzyme Kinetics. Wiley; New York: 1993. [Google Scholar]
- 16.Fersht A. Structure and Mechanism in Protein Science. W. H. Freeman Co; New York: 1999. [Google Scholar]
- 17.Holleman WH. The use of absorption optics to measure dissociation of yeast enolase into enzymatically active monomers. Biochim Biophys Acta. 1973;327:176–185. doi: 10.1016/0005-2744(73)90115-0. [DOI] [PubMed] [Google Scholar]
- 18.Wedekind JE, Poyner RR, Reed GH, Rayment I. Chelation of serine 39 to Mg2+ latches a gate at the active site of enolase: structure of the bis(Mg2+) complex of yeast enolase and the intermediate analog phosphonoacetohydroxamate at 2.1-Å resolution. Biochemistry. 1994;33:9333–9342. doi: 10.1021/bi00197a038. [DOI] [PubMed] [Google Scholar]
- 19.Poyner RP, Larsen TM, Wong S-W, Reed GH. Functional and structural changes due to a serine to alanine mutation in the active-site flap of enolase. Arch Biochem Biophys. 2002;401:155–163. doi: 10.1016/S0003-9861(02)00024-3. [DOI] [PubMed] [Google Scholar]
- 20.Zhang E, Brewer JM, Minor W, Carreira LA, Lebioda L. Mechanism of enolase: the crystal structure of asymmetric dimer enolase-2-phospho-D-glycerate/enolase-phosphoenolpyruvate at 2.0 Å resolution. Biochemistry. 1997;36:12526–12534. doi: 10.1021/bi9712450. [DOI] [PubMed] [Google Scholar]
- 21.Freitag S, Trong IL, Klumb L, Stayton PS, Stenkamp RE. Structural studies of the steptavidin binding loop. Protein Sci. 1997;6:1157–1166. doi: 10.1002/pro.5560060604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee H, Reyes VM, Kraut J. Crystal structure of Escherichia coli dihydrofolate reductase complexed with 5-formyltetrahydrofolate (folinic acid) in two space groups: evidence for enolization of pteridine O4. Biochemistry. 1966;35:7012–7020. doi: 10.1021/bi960028g. [DOI] [PubMed] [Google Scholar]
- 23.Eyal E, Gerzon S, Potapov V, Edelman M, Sobolev V. The limit of accuracy of protein modeling: influence of crystal packing on protein structure. J Mol Biol. 2005;351:431–442. doi: 10.1016/j.jmb.2005.05.066. [DOI] [PubMed] [Google Scholar]
- 24.Cody V, Luft JR, Pangborn W, Gangjee A. Analysis of three crystal structure determinations of a 5-methyl-6-N-methylamino pyrido-pyrimidine antifolate complex with human dihydrofolate reductase. Acta Crystallog sect D. 2003;59:1603–1609. doi: 10.1107/s0907444903014963. [DOI] [PubMed] [Google Scholar]
- 25.Sims PA, Reed GH. Method for the enzymatic synthesis of 2-phospho-D-glycerate from adenosine 5′-triphosphate and D-glycerate via D-glycerate-2-kinase. J Mol Catal B. 2005;32:77–81. [Google Scholar]
- 26.Warburg O, Christian W. Isolierung and kristallisation des garungsferments enolase. Biochem Z. 1941;310:384–421. [Google Scholar]
- 27.Chin CC, Brewer JM, Eckard E, Wold F. The amino acid sequence of yeast enolase. Preparation and characterization of peptides produced by chemical and enzymatic fragmentation. J Biol Chem. 1981;256:1370–1376. [PubMed] [Google Scholar]
- 28.Holland MJ, Holland JP, Thill GP, Jackson KA. The primary structures of two yeast enolase genes. Homology between the 5′ noncoding flanking regions of yeast enolase and glyceraldehyde-3-phosphate dehydrogenase genes. J Biol Chem. 1981;256:1385–1395. [PubMed] [Google Scholar]
- 29.Poyner RR, Cleland WW, Reed GH. Role of metal ions in catalysis by enolase: an ordered kinetic mechanism for a single substrate enzyme. Biochemistry. 2001;40:8009–8017. doi: 10.1021/bi0103922. [DOI] [PubMed] [Google Scholar]
- 30.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallog. 1997;30:1022–1025. [Google Scholar]
- 31.Roussel A, Cambillau C. Silicon Graphics Geometry Partners Directory. Silicon Graphics; Mountain View, CA: 1991. Turbo-Frodo. [Google Scholar]