

Abstract
The radical intermediates generated during the catalytic cycles of adenosylcobalamin-dependent enzymes occur in pairs. The positions of radicals residing on the cofactor, substrate or protein, relative to the position of the low-spin Co2+ from the cob(II)alamin intermediate, can be extracted from electron paramagnetic resonance (EPR) spectra of the spin-coupled pairs. Examples of radical–Co2+ pairs that span a range of interspin distances from 3 to 13 Å have been presented. Interspin distances greater than 5 Å require motion of one or more of the participating species. EPR spectroscopy provides a convenient means to determine the structures of these transient intermediates.
Introduction
The list of enzymes that use free radical intermediates as part of their catalytic cycles continues to expand [1]. Initiation of these radical-generating reactions typically involves homolytic cleavage of a bond (such as the cobalt–carbon bond in adenosylcobalamin [AdoCbl]-dependent enzymes), electron transfer to or from a metal-locofactor, or one-electron oxidation by an oxidant such as oxygen [2].
In AdoCbl-dependent reactions, two radicals are created in the initiation step — the 5′-deoxyadenosyl radical and low-spin Co2+in the cob(II)alamin portion of the original AdoCbl cofactor. During the catalytic cycle of such enzymes, the organic radical migrates from the adenosyl moiety to substrate, to product and back to the adenosyl moiety [3]. Recombination of the paramagnetic 5′-deoxyadenosyl radical and cob(II)alamin to give diamagnetic AdoCbl, and release of product complete the catalytic cycle (Figure 1).
Figure 1.

Schematic outline of the catalytic cycle of AdoCbl-dependent enzymes. RH and R′H represent the substrate and product, respectively; B12r stands for cob(II)alamin.
Magnetic interactions between the paramagnetic species create mutual perturbations in the electron paramagnetic resonance (EPR) spectra of the component radicals. These electron spin–spin interactions contain valuable information on the relative positions of the interacting radicals (see Figure 2). Knowledge of the relative positions of the paramagnetic centers is useful for understanding the multiple migrations of the organic radical center during the catalytic cycle [3], as well as revealing how unwanted side reactions, such as suicidal electron transfers, are minimized [4•]. The paramagnetic states of these systems are typically transient, such that they are usually accessed spectroscopically by freeze trapping samples during steady-state or pre-steady-state turnover conditions. Thus, EPR spectroscopy constitutes a logical tool for the structural examination of these species. Research on these systems has experienced resurgence of late, partly because of the increased availability of enzymes through the use of cloning technology and partly because of the phenomenal increases in computational speed and convenience, which allow more rigorous analysis of the EPR spectra. This review will discuss recent progress in determining the positions of radical intermediates in the active sites of AdoCbl-dependent enzymes. More exhaustive treatments of electron spin–spin interactions are covered in monographs and review articles [5–8,9•].
Figure 2.

Schematic diagram showing the location of the g-axis system of Co2+ within the cob(II)alamin molecular frame and its relation to the interspin vector connecting it to the spin-bearing atom of the substrate-derived radical. The interspin distance, r, and the angle, ζ, between the interspin vector and the g||-axis of C2+ are shown.
Electron spin–spin interactions
The two magnetic interactions that operate between paramagnetic centers are the through-space dipole–dipole interaction, and an exchange interaction that depends on orbital overlap and spin polarization effects. The former is an anisotropic interaction that follows a 1/r3 dependence on the spacing between the interacting centers. The latter is usually considered an isotropic interaction, which falls off approximately exponentially with the distance between the partners. At distances greater than approximately 9 Å, the exchange interaction creates a doublet splitting in the EPR spectrum of each partner. At closer distances, the exchange interaction mixes the two spin systems, such that their g-values become averaged and eventually converge to a triplet state at interspin separations of <7 Å. The dipole–dipole interaction is the most useful in terms of structural analysis because of its explicit dependence on the distance between the two spins. Thus, if one is able to determine the magnitude of the dipole–dipole splitting in the spectrum, one can determine the separation of the two interacting spins.
The dipole–dipole interaction lifts the degeneracy of the spin states in the absence of an external magnetic field and is a source of zero-field splitting. In the most general formulation, the zero-field splitting is described as a traceless tensor with an axial, D, and a rhombic, E, term. In the commonly used point-dipole approximation, E ≡ 0. The principal axis of the zero-field splitting normally contains the interspin vector. In simulations, Euler rotations are required to relate the axis system of the zero-field splitting tensor to a reference system, such as the g-axis of Co2+. Examples of the dependence of the EPR spectra on the distance between Co2+ and the organic radical are shown in Figure 3. In cases in which one or both of the spins have intrinsic anisotropy in their g or hyperfine (A) terms, the position of the interspin vector in the molecular axis system, defined by these intrinsic anisotropies, can be evaluated from the spectrum [5]. In regard to the AdoCbl-dependent enzymes, wherein the low-spin Co2+ of cob(II)alamin is one of the paramagnetic species, the g and 59 Co A terms are anisotropic, and either could be exploited to determine the position of the interspin vector in a molecule fixed axis system. However, as the g and A tensors are collinear, information from these interactions is redundant. Examples of the dependence of the EPR spectra on the angle between the interspin vector and the g||-axis of Co2+ are shown in Figure 4.
Figure 3.

Spectral simulations showing the effect on EPR spectra of varying the distance between paramagnetic centers. In the simulations, the g-values for Co2+ are g⊥ = 2.25 and g|| = 2, and g = 2 for the substrate-derived radical. The 59Co nuclear hyperfine splittings are A⊥ = 5 G and A|| = 115 G. The zero-field splitting parameter, D, was varied according to the equation D = Do/r3, where r (Å) is the interspin distance and Do = 2.785 × 104 GÅ3. The rhombic zero-field splitting parameter, E, was set to 0 G. The angle between the interspin vector and the g||-axis of Co2+ was set to 0°. The exchange coupling constant, J, was varied according to the empirical equation J = Joexp(−r), where Jo was chosen to be 7.5 × 104 G, making its variation with distance consistent with what is typically found in AdoCbl-dependent systems.
Figure 4.

Spectral simulations showing the effect on EPR spectra of varying the angle between the interspin vector connecting the paramagnetic centers and the g||-axis of the Co2+. In the simulations, the g-values for Co2+are g⊥ = 2.25 and g|| = 2, and g = 2 for the substrate-derived radical. The 59Co nuclear hyperfine splittings are A⊥ = 5 G and A|| =115 G. The zero-field splitting parameters, D and E, were set to 125 G and 25 G, respectively, and the exchange coupling constant, J, was held fixed at 1750 G. The values for the zero-field splitting parameters and the exchange coupling constant are consistent with an interspin distance of approximately 6 Å.
Experimental EPR spectra are normally taken from frozen solutions, in which there is a random orientation of the molecules in the laboratory frame of reference. Hence, powder averages obtained by numerical integrations are required to simulate the spectra [10]. EPR spectra are normally interpreted with the aid of a spin Hamiltonian containing the g and A tensors of the individual radicals, as well as the exchange and dipole–dipole interaction terms of the spin–spin coupling. Good approximations for the g and A tensors of cob(II)alamin bound to the enzyme of interest are frequently available, such that the spin–spin interaction terms and Euler angles relating the interspin vector to an appropriate molecule fixed axis (e.g. the g-axis of Co2+) are the major unknowns in the analysis.
Weakly coupled spin systems
Two examples of weakly coupled spin systems have been described for cob(II)alamin and radicals generated by the suicide inactivation of ethanolamine ammonia-lyase (EAL) by hydroxyethylhydrazine [10] and of diol dehydrase (DDH) by glycolaldehyde [11]. In both cases, the signals from the low-spin Co2+ and the partner radical were split by a combination of exchange and dipole–dipole coupling. Furthermore, the hydrazine cation radical in the inactivation of EAL and the cis-ethanesemidione radical in the inactivation of DDH were positioned 13 Å and 11 Å (Figure 5), respectively, from Co2+ and approximately along the g||-axis of the ion (i.e. directly above the plane of the corrin ring). In each case, the identity of the companion radical was revealed by isotopic substitutions within the original suicide inactivator.
Figure 5.

Schematic representation of the disposition of the cis-ethanesemidione radical and Co2+ of cob(II)alamin in the active site of DDH. The radical is generated from the suicide inactivator glycolaldehyde. The position of the radical relative to Co2+ was determined from the electron spin–spin splitting in the EPR spectra of the coupled pair [11].
The spin–spin interaction parameters in EPR spectra of intermediates observed during turnover of S-2-aminopropanol and of ethanolamine by EAL differ from those in spectra of the hydrazine cation radical–cob(II)alamin pair, mainly because of a larger exchange coupling in the former cases [12–14]. The distance between Co2+ and the organic radicals is 10–12 Å (Figure 6). As shown in Figure 6, rotation of the deoxyribosyl moiety about the glycosidic bond effectively moves C5′ between a position near the Co2+ and a position within van der Waals contact of the substrate. This motion accounts for the 11 Å separation between the substrate radical and Co2+. In the case of the steady-state radical formed with ethanolamine, the radical is reported to lie 40° off the g||-axis of Co2+[13]. The recent results with S-2-aminopropanol are in good agreement with an earlier analysis [15].
Figure 6.

Schematic representation of the position of the substrate radical derived from hydrogen-atom abstraction from S-2-aminopropanol in the reaction catalyzed by EAL. The position of the radical relative to the g-axis system of Co2+ was determined from analysis of the EPR spectrum of the spin-coupled pair [12]. The position of the 5′-carbon of the 5′-deoxyadenosyl moiety was determined from electron nuclear double resonance experiments with [U-13C] ribosyl AdoCbl [32]. The arrows indicate how rotation about the glycosidic bond of the adenosyl moiety (A) would allow movement of the 5′-carbon of deoxyadenosine between the position near the substrate and its origin in a bond with Co2+. Adapted with permission from [12]. Copyright 2002 American Chemical Society.
Strongly coupled spin systems
AdoCbl-dependent ribonucleotide reductase provided the first example of a strongly coupled radical–cob(II)-alamin spin system [16]. The novel EPR spectrum, which appeared in samples prepared by rapid-mix freeze quenching of reaction mixtures, had g-values that were between those of low-spin Co2+ and an organic radical. Moreover, the 59 Co hyperfine splitting was approximately half the value typically observed in EPR spectra of cob(II)alamin. Subsequently, the spectrum was shown to be due to a ‘hybrid’ triplet spin system comprising the low-spin Co2+ of cob(II)alamin and a thiyl radical from Cys408 of the protein [17]. The complicated EPR spectrum was subsequently reproduced in simulations; the dipole–dipole splitting parameters were found to be consistent with a 5–7 Å separation between Co2+ and the thiyl radical, and a 25° angle (ζ) between the interspin vector and the g|| axis of Co2+[18].
EPR spectra indicative of strongly coupled spin systems have been reported for other AdoCbl-dependent enzymes, including glutamate mutase [19], methyleneglutamate mutase [20] and methylmalonyl-CoA mutase [21–23]. Triplet-state EPR patterns have been analyzed for the glutamate mutase intermediate [24]; the analysis indicates a cobalt–radical separation of 6–7 Å and a ζ of 50°. These values were in excellent agreement with the positions of substrate and cofactor determined in a subsequent X-ray crystal structure [25]. Triplet EPR spectra have recently been reported for an alternative substrate, L-2-hydroxyglutarate [26], and for a suicide inactivator, 2-methyleneglutarate [27], of glutamate mutase.
Very strongly coupled spin systems
Radicals produced from substrates or substrate analogs approach to within approximately 6 Å of the Co2+ in cob(II)alamin as a lower limit. The precursor to these radicals, the 5′-deoxyadenosyl radical, is an unstable primary alkyl radical — the short life-time of which has prevented its direct spectroscopic observation. Magnus-son and Frey [28] have prepared an analog of AdoCbl, 3′,4′-anhydroadenosylcobalamin (anhydro-AdoCbl), which gives a more stable allylic radical upon homolysis of the cobalt–carbon bond. Anhydro-AdoCbl undergoes cobalt–carbon bond cleavage upon binding to DDH in the presence or absence of substrate. The analog supports a measurable amount (0.02%) of catalytic turnover, in addition to undergoing a slow suicidal electron transfer reaction [29••]. EPR spectra of enzyme–cofactor complexes with or without substrate are consistent with a rhombic triplet-state species. Spectra of both complexes show a prominent half-field transition that is a hallmark of strongly coupled triplet spin systems. Changes in the line-widths of transitions in the EPR spectra resulting from 13C and 2H isotopic substitutions in the anhydroribosyl moiety identified one of the triplet spin partners as the anhydroadenosyl radical. The presence of 59Co hyperfine splitting, as well as the apparent g-values of the signals, identified low-spin Co2+ of cob(II)alamin as the other spin in the hybrid triplet system. Spectra of the complex in the presence of substrate have been simulated and the magnitudes of the parameters in the D tensors indicate that the radical is <4 Å from Co2+. The close spacing of the unpaired electrons, together with the spin delocalization within the allylic radical, requires a higher level of treatment than the point-dipole approximation that is commonly used in the analysis of dipole–dipole coupling in radical pairs with larger interspin separations. A radical–cob(II)alamin geometry that is consistent with the parameters in the D tensor and with the X-ray coordinates of DDH complexes with an analog of AdoCbl (adeninyl-pentylcobalamin) [30] is shown in Figure 7 (SO Mansoorabadi, OTh Magnusson, RR Poyner, PA Frey, GH Reed, unpublished data).
Figure 7.
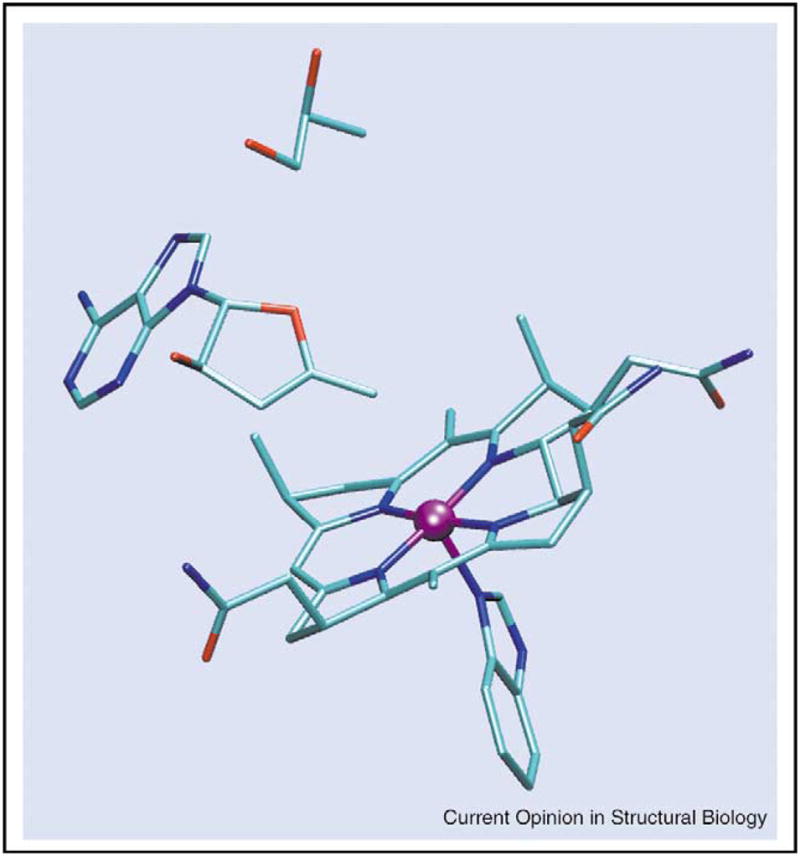
Representation of the position of the anhydroadenosyl radical relative to cob(II)alamin in the active site of DDH in the presence of 1,2-propanediol. The geometry reproduces the elements of the D tensor obtained by simulation of the triplet EPR powder pattern (SO Mansoorabadi, OTh Magnusson, RR Poyner, PA Frey, GH Reed, unpublished data). The initial position of the adenine ring was obtained from the X-ray coordinates of the complex of DDH with adeninylpentylcobalamin [30]. The distance between the 5′-carbon of the radical and Co2+is ~3 Å. The substrate, 1,2-propanediol, is shown. A truncated version of cob(II)alamin is shown.
Conclusions
Structural information on AdoCbl-dependent enzymes from X-ray crystallography and EPR spectroscopy has provided clues into how the radical intermediates inter-convert during the catalytic cycle [3]. Recognition of the possibility of ‘suicidal’ electron transfer between the radicals and cob(II)alamin underscores the importance of interspin distance and orbital overlap (exchange interaction) in this process [4•]. The positions of organic radical intermediates with respect to cob(II)alamin can be extracted from EPR spectra of the complexes. Increases in computational power allow the application of sophisticated methods of global minimization [31] to the analysis of complicated EPR patterns. Thus, one can anticipate more accurate and rigorous analyses of the EPR spectra, and the acquisition of more detailed information on the geometries of interacting paramagnetic centers in the active sites of enzymes.
Acknowledgments
We thank V Bandarian, RR Poyner, A Abend, OTh Magnusson and PA Frey for their collaborations on work cited from our laboratory. We are grateful to the National Institutes of Health (grant GM 35752) for the support of research from this laboratory. SOM is supported by National Institutes of Health Predoctoral Training Grant T32 GM08293 in Molecular Biophysics.
Abbreviations
- AdoCbl
adenosylcobalamin
- DDH
diol dehydrase
- EAL
ethanolamine ammonia-lyase
- EPR
electron paramagnetic resonance
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Frey PA. Radical mechanisms in enzymatic catalysis. Annu Rev Biochem. 2001;70:121–148. doi: 10.1146/annurev.biochem.70.1.121. [DOI] [PubMed] [Google Scholar]
- 2.Frey PA, Reed GH. Radical mechanisms in adenosylmethionine-and adenosylcobalamin-dependent enzymatic reactions. Arch Biochem Biophys. 2000;382:6–14. doi: 10.1006/abbi.2000.2010. [DOI] [PubMed] [Google Scholar]
- 3.Banerjee R. Radical peregrinations catalyzed by coenzyme B12-dependent enzymes. Biochemistry. 2001;40:6191–6198. doi: 10.1021/bi0104423. [DOI] [PubMed] [Google Scholar]
- 4•.Tang K-H, Frey PA. Electron transfer in the substrate-dependent suicide inactivation of lysine 5,6-aminomutase. Biochemistry. 2001;40:5190–5199. doi: 10.1021/bi010157j. This paper describes suicide inactivation of lysine 5,6-aminomutase as a result of electron transfer from Co2+ to the substrate radical, leaving the inactive cob(III)alamin. [DOI] [PubMed] [Google Scholar]
- 5.Eaton GR, Eaton SS. Resolved electron-electron spin-spin splittings in EPR spectra. Miol Magn Reson. 1989;8:650. [Google Scholar]
- 6.Bencini A, Gatteschi D. EPR of Exchange Coupled Systems. New York: Springer-Verlag; 1990. [Google Scholar]
- 7.Gerfen GJ. EPR spectroscopy of B12-dependent enzymes. In: Banerjee R, editor. Chemistry and Biochemistry of B12. New York: Wiley-Interscience; 1999. pp. 165–195. [Google Scholar]
- 8.Pilbrow JR. EPR of B12-dependent enzyme reactions and related systems. In: Dolphin D, editor. Vitamin B12. Vol. 1. New York: John Wiley & Sons; 1982. pp. 431–463. [Google Scholar]
- 9•.Mansoorabadi SO, Reed GH. Effects of electron spin delocalization and non-collinearity of interaction terms in EPR of triplet powder patterns. In: Telser J, editor. Paramagnetic Resonance of Metallobiomolecules. Washington DC: American Chemical Society; 2003. pp. 82–96. This review deals with two complications that arise in the analysis of triplet EPR spectra. Expressions are given that allow the simulation of the triplet EPR powder pattern spectra. Simulations that illustrate the influences of electron spin delocalization and nonalignment of interaction terms are presented. [Google Scholar]
- 10.Bandarian V, Reed GH. Hydrazine cation radical in the active site of ethanolamine ammonia-lyase: mechanism-based inactivation by hydroxyethylhydrazine. Biochemistry. 1999;38:12394–12402. doi: 10.1021/bi990620g. [DOI] [PubMed] [Google Scholar]
- 11.Abend A, Bandarian V, Reed GH, Frey PA. Identification of cis-ethanesemidione as the organic radical derived from glycolaldehyde in the suicide inactivation of dioldehydrase and of ethanolamine ammonia-lyase. Biochemistry. 2000;39:6250–6257. doi: 10.1021/bi992963k. [DOI] [PubMed] [Google Scholar]
- 12.Bandarian V, Reed GH. Analysis of the electron paramagnetic resonance spectrum of a radical intermediate in the coenzyme B12 dependent ethanolamine ammonia-lyase catalyzed reaction with S-2-aminopropanol. Biochemistry. 2002;41:8580–8599. doi: 10.1021/bi0201217. [DOI] [PubMed] [Google Scholar]
- 13.Ke SC. Spin-spin interaction in ethanolamine deaminase. Biochim Biophys Acta. 2003;1620:267–272. doi: 10.1016/s0304-4165(03)00006-0. [DOI] [PubMed] [Google Scholar]
- 14.Warncke K, Schmidt JC, Ke S-C. Identification of a rearranged-substrate, product radical intermediate and contribution of a product radical trap in vitamin B12 coenzyme dependent ethanolamine deaminase catalysis. J Am Chem Soc. 1999;121:10522–10528. [Google Scholar]
- 15.Boas JF, Hicks PR, Pilbrow JR, Smith TS. Interpretation of electron spin resonance spectra due to some B12-dependent enzyme reactions. J Chem Soc Faraday Trans. 1978;74:417–430. [Google Scholar]
- 16.Orme-Johnson WH, Beinert H, Blakley RL. Carbamides and ribonucleotide reduction XII. The electron paramagnetic resonance spectrum of ‘active coenzyme B12’. J Biol Chem. 1974;249:2338–2343. [PubMed] [Google Scholar]
- 17.Licht S, Gerfen GJ, Stubbe J. Thiyl radicals in ribonucleotide reductases. Science. 1996;271:477–481. doi: 10.1126/science.271.5248.477. [DOI] [PubMed] [Google Scholar]
- 18.Gerfen GJ, Licht S, Willems JP, Hoffman BM, Stubbe J. Electron paramagnetic resonance investigations of a kinetically competent intermediate formed in ribonucleotide reduction - evidence for thiyl radical-cob(II)alamin interaction. J Am Chem Soc. 1996;118:8192–8197. [Google Scholar]
- 19.Zelder O, Beatrix B, Leutbecher U, Buckel W. Characterization of the coenzyme-B12-dependent glutamate mutase from Clostridium chochlearium produced in Escherichia coli. Eur J Biochem. 1994;226:577–585. doi: 10.1111/j.1432-1033.1994.tb20083.x. [DOI] [PubMed] [Google Scholar]
- 20.Michel C, Albracht SPJ, Buckel W. Adenosylcobalamin and cob(II)alamin as prosthetic groups of 2-methyleneglutarate mutase from Clostridium barkeri. Eur J Biochem. 1992;205:767–773. doi: 10.1111/j.1432-1033.1992.tb16841.x. [DOI] [PubMed] [Google Scholar]
- 21.Keep NH, Smithe GA, Evans MC, Kiakun GP, Leadlay PF. The synthetic substrate succinyl(carbadethia)-CoA generates cob(II)alamin on adenosylcobalamin-dependent methylmalonyl-CoA mutase. Biochem J. 1993;295:387–392. doi: 10.1042/bj2950387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, Abend A, Kunz M, Such P, Retey J. Electron paramagnetic resonance studies of the methylmalonyl-CoA mutase reaction. Evidence for radical intermediates using natural and artificial substrates as well as the competitive inhibitor 3-carboxypropyl-CoA. Eur J Biochem. 1994;225:891–896. doi: 10.1111/j.1432-1033.1994.0891b.x. [DOI] [PubMed] [Google Scholar]
- 23.Padmakumar R, Banerjee R. Evidence from electron paramagnetic resonance spectroscopy of the participation of radical intermediates in the reaction catalyzed by methylmalonyl-coenzyme A mutase. J Biol Chem. 1995;270:9295–9300. doi: 10.1074/jbc.270.16.9295. [DOI] [PubMed] [Google Scholar]
- 24.Bothe H, Darley DJ, Albracht SP, Gerfen GJ, Golding BT, Buckel W. Identification of the 4-glutamyl radical as an intermediate in the carbon skeleton rearrangement catalyzed by coenzyme B12-dependent glutamate mutase from Clostridium cochlearium. Biochemistry. 1998;37:4105–4113. doi: 10.1021/bi971393q. [DOI] [PubMed] [Google Scholar]
- 25.Reitzer R, Gruber K, Jogl G, Wagner UG, Bothe H, Buckel W, Kratky C. Glutamate mutase from Clostridium cochlearium: the structure of a coenzyme B12-dependent enzyme provides new mechanistic insights. Structure Fold Des. 1999;7:891–902. doi: 10.1016/s0969-2126(99)80116-6. [DOI] [PubMed] [Google Scholar]
- 26.Roymoulik I, Moon N, Dunham WR, Ballou DP, Marsh ENG. Rearrangement of L-2-hydroxyglutarate to l-threo-3-methylmalate catalyzed by adenosylcobalamin-dependent glutamate mutase. Biochemistry. 2000;39:10340–10346. doi: 10.1021/bi000121b. [DOI] [PubMed] [Google Scholar]
- 27.Huhta MS, Ciceri D, Golding BT, Marsh ENG. A novel reaction between adenosylcobalamin and 2-methyleneglutarate catalyzed by glutamate mutase. Biochemistry. 2002;41:3200–3206. doi: 10.1021/bi011965d. [DOI] [PubMed] [Google Scholar]
- 28.Magnusson OT, Frey PA. Synthesis and characterization of 3′,4′-anhydroadenosylcobalamin: a coenzyme B12 analogue with unusual properties. J Am Chem Soc. 2000;122:8807–8813. [Google Scholar]
- 29••.Magnusson OT, Frey PA. Interactions of diol dehydrase and 3′,4′-anhydroadenosylcobalamin: suicide inactivation by electron transfer. Biochemistry. 2002;41:1695–1702. doi: 10.1021/bi011947w. The interaction of an AdoCbl analog, which gives an allylic radical upon cobalt–carbon bond homolysis, with DDH is described. Homolytic cleavage of the cobalt–carbon bond in the absence of substrate is observed. [DOI] [PubMed] [Google Scholar]
- 30.Masuda J, Shibata N, Morimoto Y, Toraya T, Yasuoka N. How a protein generates a catalytic radical from coenzyme B12: X-ray structure of a diol-dehydratase -adeninylpentylcobalamin complex. Structure Fold Des. 2000;8:775–788. doi: 10.1016/s0969-2126(00)00164-7. [DOI] [PubMed] [Google Scholar]
- 31.Press WH, Flannery BP, Teukolsky SA, Vetterling WT. Numerical Recipes. New York: Cambridge University Press; 1989. [Google Scholar]
- 32.LoBrutto R, Bandarian V, Magnusson OT, Chen X, Schramm VL, Reed GH. 5′-deoxyadenosine contacts the substrate radical intermediate in the active site of ethanolamine ammonia lyase. Biochemistry. 2001;40:9–14. doi: 10.1021/bi001865s. [DOI] [PubMed] [Google Scholar]