

Abstract
A random sequence survey of the genome of Trypanosoma cruzi, the agent of Chagas disease, was performed and 11,459 genomic sequences were obtained, resulting in ∼4.3 Mb of readable sequences or ∼10% of the parasite haploid genome. The estimated total GC content was 50.9%, with a high representation of A and T di- and trinucleotide repeats. Out of the estimated 5000 parasite genes, 947 putative new genes were identified. Another 1723 sequences corresponded to genes detected previously in T. cruzi through expression sequence tag analysis. 7735 sequences had no matches in the database, but the presence of open reading frames that passed Fickett's test suggests that some might contain coding DNA. The survey was highly redundant, with ∼35% of the sequences included in a few large sequence families. Some of them code for protein families present in dozens of copies, including proteins essential for parasite survival and retrotransposons. Other sequence families include repetitive DNA present in thousands of copies per haploid genome. Some families in the latter group are new, parasite-specific, repetitive DNAs. These results suggest that T. cruzi could constitute an interesting model to analyze gene and genome evolution due to its plasticity in terms of sequence amplification and divergence. Additional information can be found at http://www.iib.unsam.edu.ar/tcruzi.gss.html.
[The sequence data described in this paper have been submitted to the dbGSS database under the following GenBank accession nos.: AQ443439–AQ443513, AQ443743–AQ445667, AQ902981–AQ911366, AZ049857–AZ051184, and AZ302116–AZ302563.]
Protozoan parasites from the order Kinetoplastida are the causative agents of widespread diseases in humans as well as of considerable economic loss through infection of domestic animals and wildlife. Among them are Trypanosoma cruzi and Trypanosoma brucei, the agents of Chagas disease in the Americas and sleeping sickness in Africa, respectively, and Leishmania spp., causing a variety of pathologies in humans. In addition to their medical importance, these parasites were the source of discoveries of fundamental cellular and molecular phenomena such as RNA editing (Stuart et al. 1997; Estevez and Simpson 1999), mRNA trans-splicing (Nilsen 1995; Lee and Van der Ploeg 1997), glycosylphosphatidyl–inositol anchoring of proteins (Krakow et al. 1986), and antigenic variation (Rudenko et al. 1998), among others. Kinetoplastids are also interesting evolutionarily because they represent one of the earliest eukaryotic organisms that diverged from the ancestor of the main eukaryotic branch.
Initial studies on the genome structure of these organisms were hampered by the difficulty of applying classical genetic tools. As is the case with yeast, chromosomes do not condense during mitosis and cannot be visualized directly with color stains. In spite of these drawbacks several recent studies have revealed a highly plastic genome with an unusual gene organization (Zingales et al. 1997; Network 1998; Ersfeld et al. 1999). In the case of T. cruzi it has been shown that there is a large chromosomal size variation between strains that can also be observed between pairs of homologous chromosomes (Henriksson et al. 1995).
Many genes in kinetoplastids, including housekeeping genes, are present in multiple copies, either clustered in tandems or distributed in different chromosomes (Campetella et al. 1992b; El-Sayed and Donelson 1997). Recently, the complete sequence of chromosome 1 from L. major Friedlin (Myler et al. 1999) and a partial sequence of chromosome 3 from T. cruzi (Andersson et al. 1998) were obtained, showing in both cases a similar organization with two clusters of genes per chromosome transcribed in opposite directions. These studies also showed that the gene density is high in the regions where genes are clustered, with ∼1 gene every 3.6 Kb in both cases. These and other unusual characteristics in genome structure and organization make these parasites an interesting field of study to further understand eukaryote gene and genome evolution.
As part of the parasite genome projects launched by the Tropical Disease Program of the World Health Organization, we and others have performed expressed sequence tag (EST) analysis of the T. cruzi genome as a means for rapid gene finding (Verdún et al. 1998; Porcel et al. 2000). As a first step toward obtaining the complete sequence of the clone selected for the T. cruzi genome project (CL-Brener clone) we have now made a random genome survey of ≈10% (4.3 Mb) of the haploid genome of the parasite. The results obtained allowed us to make a general characterization of the T. cruzi genome, identify putative new genes, and define large gene families and repetitive sequence families present in the parasite genome, including a novel repetitive element and uncharacterized abundant sequences.
RESULTS AND DISCUSSION
Overall Structure of the T. cruzi Genome
A random genomic library of T. cruzi DNA was constructed and used to produce 11,459 reads with an average length of 374 bp after vector removal and quality clipping. These GSS (Genomic Sequence Survey) sequences represent 4.3 Mb of readable sequences or ∼10% of the parasite haploid genome, which is ∼40 Mb (Frohme et al. 1998). The total GC content of the sequence produced was 50.9%, a value that is slightly larger than the one obtained from estimations made on 8796 T. cruzi ESTs (49.6 %), and the one obtained for 93.4 Kb from chromosome 3 (48.5%) (Andersson et al. 1998). The fractional GC content for all the GSSs varied from 0.18–0.71, with ∼69% of the sequences falling in the 0.47–0.57 range.
The frequency of di- and trinucleotide repeats calculated for all the GSS sequences (11,459 GSS) is shown in Figure 1. T. cruzi has levels of CpG, TpG, and CpA dinucleotides which are near their expected values, unlike mammalian or vertebrate genomes that show a suppression of the CpG dinucleotide and increased levels of the TpG and CpA dinucleotides (Regev et al. 1998). The TpA dinucleotide, however, is suppressed in T. cruzi, with an observed/expected ratio of 0.54. This dinucleotide is also suppressed in mammalian genomes (Smith and Waterman 1983), and in Fugu the pufferfish (Elgar et al. 1999), although the reason for this is not known. Also, the abundance of (A)n and (T)n di- and trinucleotides is remarkable. The frequency of these di- and trinucleotides is greater than the observed frequency for the CpA dinucleotide, the most common dinucleotide in vertebrates. When the same analysis was done for the partial sequence of chromosome 3, we obtained similar values, except for the (A)n and (T)n di- and tri-nucleotides. These also represent the most frequent species, but their observed/expected ratios are higher. In our survey the (A)n dinucleotide had a 0.13 obs/exp ratio, whereas the partial sequence of chromosome 3 had a 0.4 ratio. For the (A)n trinucleotide these values are higher: a 0.67 ratio in our survey and a 1.43 ratio in the partial sequence of chromosome 3.
Figure 1.

Frequency of di- and tri-nucleotide repeats in the Trypanosoma cruzi genome. The total of 11,459 sequences were used to search for the occurrence of all possible words of length 2 (A) and 3 (B) on both strands of the sequences using COMPSEQ. The expected frequency of each word is based on the assumption that all words have the same probability of occurrence. Di- and tri-nucleotide frequencies are expressed as Observed (Obs)/Expected (Exp) − 1, so that negative values correspond to suppressed di- and tri-nucleotides and positive values correspond to di- and tri-nucleotides with frequencies over that expected. Because the search is done on both strands, only one reverse complementary di- or tri-nucleotide of a pair is shown.
Coding Content of the GSS Sequences
We compared the sequences in our data set to a protein nonredundant database using BLASTX (Altschul et al. 1997). From this analysis, 3724 GSS (32.5%) showed significant (E<1e-5) similarities to sequences present in the public database (GenBank release 115) (Table 1); 2778 GSS (24.2%) matched T. cruzi sequences while 947 GSS (8.3%) matched non-T. cruzi sequences. 67.5% did not match any sequence in the database. When we compared each GSS to the dbEST database, only 1723 (15%) of the GSSs gave significant matches and 96% of these were matches to T. cruzi ESTs. The low hit rate against ESTs could be explained in part by the low sequence coverage attained in the EST sequencing, which only covered one of the four main life cycle stages of the parasite. Thus, the coding content of our survey could be considered to be 15% based on the number of matches against dbEST or 32.5% based on the number of matches against proteins in nonredundant databases. However, this does not include sequences absent from the databases.
Table 1.
BLASTX Matches to Nonredundant Databases
| No. of GSS | % of GSS | No. ESTs | % of ESTs | |
|---|---|---|---|---|
| Total | 11,459 | 100 | 8,796 | 100 |
| Database matches: | ||||
| Total | 3,724 | 32.5 | 2,552 | 29 |
| T. cruzi | 2,778 | 24.2 | 861 | 9.8 |
| Other trypanosomatids | 331 | 2.9 | 262 | 3 |
| Other organisms | 616 | 5.4 | 1,429 | 16.2 |
| No database match | 7,735 | 67.5 | 6,244 | 71 |
All of the GSSs presented in this paper and all the T. cruzi ESTs present in the dbEST division of GenBank were used to search the NCBI nonredundant database (GenBank Release 115). Matches with an E value ≤ 1e-5 were considered positive.
To detect putative coding sequences using a different criteria, we first searched our sequences for open reading frames (ORFs) > 300 bp (defined as sequences without a stop codon and no requirement for start codons). We then evaluated which of these resulting sequences could be regarded as coding using the testcode algorithm developed by Fickett (1982), which measures the positional randomness of a sequence, and is independent of the reading frame. About 83% of the sequences (9520 GSSs) had ORFs with the mentioned requirements, and 23% of these (2222 GSSs) were found to be potentially coding by the testcode algorithm. Using this figure as the minimum number of coding sequences in our survey, we can estimate the number of genes for T. cruzi to be about 5000 per haploid genome (considering an average gene size of 1500 bp and a haploid genome size of 40 Mbp).
Identification of Putative New Genes
Based on the BLASTX analysis, which is summarized in Table 1, we detected 947 putative new genes, which are those positive matches against a non-T. cruzi protein in the nonredundant database. The best 50 matches are shown in Table 2 (detailed information on T. cruzi GSS sequences can be found at http://www.iib.unsam.edu.ar/tcruzi/gss.html).
Table 2.
Identification of New T. cruzi Genes
| dbGSSa | Descriptionb | Score | Expect |
|---|---|---|---|
| 11462 | ref-NP_011059.1-GLC7-protein phosphatase type I [Saccharomyces cereviseae] | 474 | 0.00E+00 |
| 10993 | sp-P22679-elongation factor TU (EF-TU) [Mycoplasma hominis] | 312 | 0.00E+00 |
| 00184 | sp-P46794-cystathionine beta-synthase [Dictyostelium discoldeum] | 303 | 0.00E+00 |
| 11438 | sp-O76767-lumen protein retaining receptor [Drosophila melanogaster] | 208 | 0.00E+00 |
| 11472 | emb-CAB56598.1-alpha dynein heavy chain [Chlamydomonas reinhardtii] | 202 | 0.00E+00 |
| 11026 | gi-2425121-Spalten [Dictyostelium discoldeum] | 116 | 0.00E+00 |
| 12036 | gb-AAF19802.1-N-myristoyl transferase [Brassica oleracea] | 107 | 0.00E+00 |
| 01761 | gl-3004644-trypanothione synthetase [Crithidia fasciculata] | 428 | 5.00E-42 |
| 11137 | gb-AAB67249.1-T-complex protein 1, Beta subunit [Homo sapiens] | 424 | 9.00E-42 |
| 11193 | ref-NP_014850.1-RET1-second-largest subunit of RNA polymerase III [Saccharomyces cereviseae] | 422 | 1.00E-41 |
| 11590 | gb-AAF08387.1-26S proteasome regulatory complex subunit p48A [Drosophila melanogaster] | 419 | 2.00E-41 |
| 11120 | emb-CAA65384-malate dehydrogenase [Mesembryanthemum crystallinum] | 371 | 2.00E-35 |
| 11285 | gi-1931649-DNA helicaso isolog [Arabidopsis thaliana] | 353 | 2.00E-33 |
| 11563 | ref-NP_013458.1-transaldolase [Saccharomyces cerevisiae] | 348 | 7.00E-33 |
| 09938 | gi-2246458-S-adenosyl-methionine-sterol-C-methyltransferase [Ricinus communis] | 348 | 8.00E-33 |
| 0705 | pir-T1017324-sterol C-methyltransferase-castor bean [Ricinus communis] | 348 | 9.00E-33 |
| 11338 | gb-AAC02737.1-3-hydroxyisobutyryl-coenzyme A hydrolase [Arabidopsis thaliana] | 341 | 5.00E-32 |
| 11467 | gb-AAF04493.1-acetyl-CoA carboxylase 1 [Toxoplasma gondii] | 330 | 7.00E-31 |
| 10956 | dbj-BAA84364.1-DEIH-box RNA/DNA helicase [Arabidopsis thaliana] | 328 | 2.00E-30 |
| 11575 | gb-AAC73040.1-putative AAA-type ATPase [Arabidopsis thaliana] | 323 | 5.00E-30 |
| 11516 | sp-P05439-ATP synthase alpha chain [Rhodobacter blasticus] | 319 | 2.00E-29 |
| 11606 | sp-P51044-citrate synthase, mitochondrial precursor [Aspergillus niger] | 321 | 2.00E-28 |
| 11417 | gb-AAF21464.1-proline oxidase 2 [Homo sapiens] | 310 | 2.00E-28 |
| 11502 | gi-2654103-MAPKK kinase [Neurospora crassa] | 304 | 9.00E-28 |
| 11523 | gb-AAD26855.1-phenylalanyl tRNA synthetase beta subunit [Mus musculus] | 301 | 2.00E-27 |
| 11568 | gi-4101722-histone deacetylase mHDA1 [Mus musculus] | 301 | 2.00E-27 |
| 11480 | gi-2462752-phosphatidylinositol 3-kinase [Arabidopsis thaliana] | 299 | 4.00E-27 |
| 11463 | sp-P32826-serine carboxypeptidase precursor [Arabidopsis thaliana] | 299 | 4.00E-27 |
| 10965 | gi-687208-dynein heavy chain isotype 5C [Tripneustes gratilla] | 289 | 5.00E-26 |
| 11328 | pir-A56220-protein kinase aurora-fruit fly [Drosophila melanogaster] | 287 | 1.00E-25 |
| 11446 | sp-O15228-dihydroxyacetone phosphate acyltransferase (DAP-AT) [Homo sapiens] | 280 | 7.00E-25 |
| 11433 | gb-AAF11511.1-acetyl-CoA acetyltransferase [Deinococcus radiodurans] | 279 | 9.00E-25 |
| 11601 | sp-P30575-enolase 1 (2-phosphoglycerate dehydratase) [Candida albicans] | 278 | 1.00E-24 |
| 01810 | sp-O94476-eukaryotic translation initiation factor 6 (EIF-6) [Schizosaccharomyces pombe] | 275 | 3.00E-24 |
| 0740 | gb-AAF62506.1-ribosomal protein LS [Trypanoplasma borreli] | 273 | 5.00E-24 |
| 11274 | pir-S70896-aminomethyltransferase [Saccharomyces cerevisiae] | 271 | 5.00E-24 |
| 11279 | gi-780410-helicase [African swine fever virus] | 272 | 6.00E-24 |
| 11882 | sp-Q07405-ATP synthase alpha chain [Myxococcus xanthus] | 269 | 1.00E-23 |
| 11432 | emb-CAB40791.1-centrin [Euplotes octocarinatus] | 265 | 3.00E-23 |
| 11654 | gi-1872473-delta-24-sterol methyltransferase [Triticum aestivum] | 258 | 7.00E-23 |
| 01015 | pir-A56492-protein kinase ERK2 [Dictyostelium discoideum] | 280 | 8.00E-23 |
| 11584 | ref-NP_005678.1-phenylalanyl-tRNA synthetase beta-subunit [Homo sapiens] | 259 | 1.00E-22 |
| 11434 | sp-O05593-methionyl-tma synthetase [Mycobacterium tuberculosis] | 254 | 6.00E-22 |
| 11038 | gi-1354084-axonemal dynein light chain p33 (Strongylocentrotus purpuratus] | 251 | 2.00E-21 |
| 11440 | gi-2665637-mismatch repair protein MSH6 [Mus musculus] | 248 | 4.00E-21 |
| 11248 | gb-AAF22155.1-ARD-1 N-acetyltransferase homologue [Mus musculus] | 244 | 1.00E-20 |
| 11852 | gb-AAC32590.1-sperm flagellar protein Repro-SA-1 [Homo sapiens] | 239 | 5.00E-20 |
| 01825 | dbj-BAA20996-kinesin-like protein [Caenorhabditis elegans] | 239 | 6.00E-20 |
| 11210 | pir-A35630-regulatory protein algR3 [Pseudomonas aeruginosa] | 237 | 7.00E-20 |
GSS sequences were used to search NCBI's non-redundant database using BLASTX. The first 50 GSSs out of the 947 GSSs showing matches against non-T. cruzi sequences are listed. Detailed information about the homologies found for GSSs can be found at http://www.iib.unsam.edu.ar/genomelab/tcruzi/gss.html.
GSS names in dbGSS are the numbers given here preceded by GSSTc (e.g., GSSTc11210).
Descriptions are taken directly from the BLAST reports.
Among the new genes found there is a GSS (GSSTc12036) with similarity to N-myristoyl transferases (Nmt). Nmt is only found in eukaryotic cells and transfers fatty acid myristate from myristoyl-CoA to the amino-terminal glycine of substrate proteins (Russell Johnson et al. 1994). Genetic and biochemical studies have established Nmt as a target for the development of a new class of fungicidal drugs, and the structure of Nmt from two lower eukaryotes, namely Saccharomyces and Candida has been solved (Weston et al. 1998; Bhatnagar et al. 1999). In trypanosomes protein N-myristoylation has not yet been demonstrated, whereas it has been shown that these parasites can do S-myristoylation of proteins (Armah and Mensa-Wilmot 1999). Other interesting findings were two GSS homologous to proteins involved in chromatin remodeling. GSSTc788 is homologous to the Drosophila ISWI protein, which is part of several ATP-dependent chromatin remodeling complexes such as NURF (Nucleosome remodeling factor), CHRAC (chromatin accessibility complex) and ACF (ATP-utilizing chromatin assembly and remodeling factor) (Muchardt and Yaniv 1999), whereas GSSTc11568 is homologous to several histone deacetylases. Chromatin remodeling is a mechanism of transcriptional regulation that has been demonstrated in many eukaryotes including yeast, which as trypanosomes does not show chromosome condensation during its cell cycle. Another GSS (GSSTc12012) had homology with a T. brucei VSG expression site-associated protein precursor (ESAG-2), which is a member of a large gene family that includes nonfunctional genes (Kooter et al. 1988). Also, several GSSs with homology to proteins having RNA binding domains were identified, including one clone (GSSTc11533) that showed homology to the RNA binding domain present in the developmentally regulated proteins p37 and p34 from T. brucei (Zhang et al. 1998).
Identification of Large Gene Families
To identify large gene families, GSS sequences were clustered using the PHRAP program to assemble contigs. Using this method we were able to group 7883 reads in 2091 contigs; the other 3576 were singlets (reads having no nonvector match to any other read). This means that our survey contains 5667 unique sequences, according to our clustering method, and thus a redundancy of at least ∼50%. Further clustering is possible, however, because the results from BLASTX contained several GSS that belong to different contigs showing matches to the same sequences in the database. To estimate the total number of GSS that belong to a given gene family, we used the consensus sequence from each contig to search a local T. cruzi GSS database using BLASTN. Based on this analysis we were able to delineate abundant sequences in our survey, which are the main contributors to the observed redundancy. The most abundant gene families in the T. cruzi genome are summarized in Table 3.
Table 3.
Large Gene Families in T. cruzi
| A Gene Families | No. of GSSs | Estimated No. of Copies | Relative % | No. of ESTs | Reference |
|---|---|---|---|---|---|
| dgf-1 | 494 | 154 | 4.3 | 6 | (Wincker et al. 1992) |
| trans-sialidase | 427 | 632 | 3.7 | 10 | (Parodi et al. 1992) |
| L1 non-LTR retrotransposon | 214 | 149 | 1.9 | 5 | (Martín et al. 1995) |
| Mucin | 122 | 710 | 1.1 | 5 | (Di Noia et al. 1995) |
| Cysteine proteinase (Cruzipain) | 39 | 91 | 0.3 | 7 | (Campetella et al. 1992a) |
| predicted ORF (gi3053534), chromosome 3 | 38 | 103 | 0.3 | 4 | (Andersson et al. 1998) |
| gp63 | 34 | 70 | 0.3 | 9 | AF110951, unpubl. |
| Histone H4 | 29 | 337 | 0.2 | 10 | (Soto et al. 1997) |
| Casein kinase homolog | 23 | 81 | 0.2 | 7 | AF089709, unpubl. |
| Adenylyl cyclase | 19 | 18 | 0.2 | 0 | (Taylor et al. 1999) |
| Hsp70 | 18 | 25 | 0.2 | 48 | (Requena et al. 1988) |
| Histone H2A | 17 | 145 | 0.1 | 106 | (Puerta et al. 1994) |
| Helicase | 14 | 24 | 0.1 | 15 | |
| Hsp90 | 11 | 18 | 0.1 | 7 | (Mottram et al. 1989) |
| Total | 1499 | 14.5 | |||
| B Repetitive DNA Families | No. of GSSs | Estimated No.of Copies | Relative % | No. of ESTs | Reference |
|---|---|---|---|---|---|
| minichromosomal 195 bp repeat | 854 | 15287 | 7.45 | ND | (Gonzalez et al. 1984) |
| TcIRE (I) | 266 | 1664 | 2.3 | 8 | this work |
| VIPER | 174 | 257 | 1.5 | ND | (Vázquez et al. 2000) |
| C6 interspersed element | 230 | 560 | 2.0 | ND | (Araya et al. 1997) |
| SIRE | 201 | 3011 | 1.8 | ND | (Vázquez et al. 2000) |
| telomere associated sequences | 131 | 1963 | 1.1 | ND | |
| TcIRE (II) | 47 | 2310 | 0.4 | 2 | this work |
| TRBSEQA | 31 | 105 | 0.3 | ND | (Requena et al. 1992) |
| HCR6 | 10 | 57 | 0.1 | ND | (de Mendonça-Lima and Traub-Cseko 1991) |
| Spliced Leader gene | 12 | 69 | 0.1 | ND | |
| Total | 2133 | 18.6 |
| C Unknown Families | No. of GSSs | Estimated No.of Copies | Relative % | No. of ESTs | Consensus Size (bp) |
|---|---|---|---|---|---|
| Cluster 2009 | 19 | 54 | 0.1 | 0 | 1220 |
| Cluster 2047 | 85 | 136 | 0.7 | 1 | 2170 |
| Cluster 2015 | 53 | 96 | 0.5 | 0 | 1917 |
| Cluster 1994 | 25 | 82 | 0.2 | 1 | 1056 |
| Cluster 2056 | 22 | 30 | 0.2 | 0 | 2571 |
| Cluster 2019 | 21 | 102 | 0.2 | 3 | 1051 |
| Cluster 2027 | 12 | 58 | 0.1 | 0 | 718 |
| Cluster 1986 | 10 | 48 | 0.1 | 0 | 728 |
| Total | 247 | 2.1 |
GSS sequences were clustered and their similarities against sequences in nonredundant databases were determined. Total number of GSS for each family was determined as described in the text. Note, however, that this approach can lead to a GSS belonging to more than one family. To calculate the number of copies, the value of the gene size (GS) used was the length of the coding sequence (excluding UTRs) of a representative member from each family; in the case of genes with different sizes, an average was used. When only partial sequences were available, copy numbers were not determined as this could lead to overestimation of the figures. To determine the number of ESTs for a given gene family, the consensus sequence or a sequence of a representative member was used to do a BLASTN search against the 8796 T. cruzi ESTs. Matches with E < 1e-40 were considered positive. In the case of unpublished sequences that are available from nucleotide databases, the GenBank accession number is given. (A) Gene families (protein coding). (B) Repetitive DNA families (likely to be noncoding). (C) Uncharacterized sequences described in this work. Information about these unknown families (consensus sequence and individual GSSs included in the contig) can be found at http://www.iib.unsam.edu.ar/genomelab/tcruzi/gss.html.
Among the largest families identified is the superfamily of T. cruzi antigens, also known as trans-sialidase-like molecules (632 copies per haploid genome). Their members have a number of different activities, most of them involved in the host–parasite interaction (Frasch 2000). Other sequences already known to conform large gene families in T. cruzi identified through this screening were the cysteine proteinase cruzipain (Campetella et al. 1992a), dgf-1, a large protein of unknown function (Wincker et al. 1992), and the parasite mucins (Di Noia et al. 1998). In all of these cases, the estimated number of copies agreed well with experimental data, showing that our sampling can be considered a good representation of the whole genome. A considerable number of GSS gave matches to proteins known to be part of retrotransposons: reverse transcriptase, apurinic/apyrimidinic endonuclease (AP-nuclease) and homologs of the gag protein present in retroviral and LTR-retrotransposons. These proteins most probably correspond to the L1Tc non-LTR retrotransposon (Martín et al. 1995; Olivares et al. 1997), the most abundant retrotransposable element found in our survey (250 GSS). However, they could also come from other elements since 40 GSS with homology to the T. brucei INGI retrotransposon and 11 GSS with homology to an Anopheles retrotransposon were detected when we performed a search in a database of repetitive elements (see below).
Some GSS contigs failed to show similarity to any known protein in the nonredundant databases (BLASTX), and also failed to show homology to any known DNA sequence (BLASTN) in the nonredundant database. To see if these unknown sequences were part of uncharacterized large gene families, we used the consensus sequence from each of these contigs to search a database made from all the 2091 consensus sequences of the contigs. We then grouped together contigs showing significant (E < 1e-10) homology as reported by BLASTN. None of the homologous contigs found, nor any GSS that formed those contigs, had significant matches in the databases. Out of 1050 contigs analyzed (2562 GSSs, 22.3% of the survey), the first 10 representing the most abundant unknown sequences in the T. cruzi genome are included in Table 3B.
Identification of Repetitive DNA Families
The program REPEATMASKER was used to search for simple repeats (1–6 bp), low-complexity regions, and known repetitive DNA. Using the database of simple repeats from RepBase (Genetic Information Research Institute, http://www.girinst.org/, Release 5.02) the program found 1556 GSS (13.5%) containing 1091 matches to 62 different simple repeats comprising 46,965 bp (0.74%) and 1001 matches to low-complexity regions comprising 56,455 bp (0.89%). The most abundant simple repeats are shown in Figure 2A. As was observed in the analysis of di- and trinucleotide repeats, the most abundant simple repeat in T. cruzi is (T)n and its complement. Interestingly, (TA)n and (TAA)n appear to be the second two most abundant repeats in our survey. In agreement, the most abundant low-complexity regions, as reported by REPEATMASKER are AT-rich, T-rich and A-rich regions, which together represent >85% of the low complexity regions in Figure 2B. Taken together, this analysis and the data on frequencies of di- and tri-nucleotide repeats suggest that, although suppressed in the whole survey, (TA)n and (TAA)n are abundant in a subset of the GSS. This subset most probably includes GSS corresponding to intergenic regions that are rich in A and T in T. cruzi.
Figure 2.

Microsatellite repeats and low-complexity regions in the Trypanosoma cruzi genome. Simple repeats and low-complexity regions were searched for in the T. cruzi GSS database using REPEATMASKER as described in the text. (A) The 20 most abundant microsatellite repeats in the survey are shown. The minimum value of n is the one that gives a Smith-Waterman (SW) score ⩾180, which is the cutoff to consider a match as positive. This value varied from 18 for a single nucleotide repeat to 3 for a hexanucleotide repeat. Each named microsatellite in the graph includes all combinations thereof; so (A)n also includes its complement (T)n, and (ATG)n also includes (CAT)n, (ATC)n, (TCA)n, (TGA)n and (GAT)n. (B) Low-complexity regions were searched for as described in the text. The length of the regions detected varied from 16 bp to 308 bp.
To search for repetitive elements, we used the invertebrate database of RepBase that, although not specific for trypanosomatids, contains repetitive elements from these organisms. The program found 3523 matches to 13 different elements comprising 629,831 bp (14.7%) in 3290 GSS (29%). The great majority of these elements were from T. cruzi or T. brucei with the exception of a repetition from T. borreli (TBRP1) which had five matches and a retrotransposon from Anopheles gambiae (RT1) which had 11 matches. As shown in Table 3C, the most abundant element in the T. cruzi genome is the 195-bp minichromosomal repetitive element (González et al. 1984). Also included in Table 3C are repetitive elements that were described in T. cruzi but were absent from RepBase. This is the case of telomere-associated sequences, TRBSEQA (Requena et al. 1992), HCR6 (de Mendonça-Lima and Traub-Cseko 1991), and the spliced leader gene for which we detected 27, 31, 9, and 12 GSS, respectively, using BLASTN (E < 1e-40). We did not find any match to primate or mammalian repetitive elements when we searched the respective RepBase libraries.
TcIRE, A New T. cruzi Repetitive Element
A new repetitive element apparently dispersed in the T. cruzi genome was found during the analysis of the GSS contigs. This element, which was named TcIRE (for T. cruzi Interspersed Repeated Element) was found in 44 contigs (266 GSS) representing almost 3% of the genome with 1664 copies per haploid genome. The structure of TcIRE, depicted in Figure 3, shows a central conserved core flanked by less conserved regions. The BLASTN analysis showed that the 3′ region of TcIRE is similar in some cases to the last 70 bp of the 3′-UTRs of some mucin genes. This results in two different groups of TcIRE elements that differ in their 3′ regions. As can be seen in the alignment shown in Figure 3B, the two groups start to differ after the conserved region. The 5′ region, although less conserved, appears to be shared by both groups.
Figure 3.

Structure of TcIRE. (A) General scheme of the structure of TcIRE. (B) Five GSS containing a copy of TcIRE were aligned using CLUSTALW with the corresponding region of the 25-Kb cosmid sequenced by Gao et al. 1999 (GenBank accession no. AB017765) and the last portion of the 3′ UTR from the Emuce-31l3 mucin gene. The rest of the mucin gene does not show any homology with the sequences aligned and was cut off for the sake of clarity. Coloring is based on BLOSUM 62 scores: 3.0, black; 1.5, gray; 0.5, light gray. Similar residues are colored as the most conserved one. Arrows indicate the two oligonucleotides used to generate the probe for the Southern blot analysis. Arrowheads indicate the site of divergence between the two groups of TcIRE sequences. The top three sequences, including AB017765, are representative of one group of sequences, denoted Group I, whereas the other sequences, including the last portion of the 3′-UTR region from the Emuce-31l3 mucin gene are representative of another group, denoted Group II.
One copy of TcIRE is present in an intergenic region of a recently sequenced T. cruzi cosmid containing enzymes for the synthesis of pyrimidines (Gao et al. 1999). TcIRE is located between the OMPDCase–OPRTase gene and a surface antigen gene from the transsialidase superfamily. This intergenic region is ∼4.5 Kb long, and the TcIRE copy is located past the center (∼13,480–13,960 bp) of this region. For means of comparison, we found only three GSS showing significant homology with the OMPDCase–OPRTase gene. Southern blots from several strains and clones of T. cruzi and from Leishmania were probed with a 300-bp fragment amplified from clone GSSTc11311 corresponding to the most conserved region of TcIRE. This analysis showed that TcIRE is present in high copy numbers in all the strains and clones tested (Fig. 4). Conversely, it seems to be absent in Leishmania, as we did not get any hybridization signal even under low-stringency conditions (data not shown). Hybridization of chromosomes separated by PFGE using the same probe showed that TcIRE might be present in several chromosomes, although the region of hybridization corresponds to poorly resolved chromosomes of high molecular weight. When the TcIRE consensus sequence was used to search the T. cruzi EST database using BLASTN, it showed significant (E < 1e-40) homology with eight ESTs. None of these ESTs, however, had homologs in nonredundant databases. Furthermore, no significant matches were obtained when these ESTs or the TcIRE consensus sequence were used to search the Pfam database of multiple alignments using HMMER.
Figure 4.

(A) Genomic DNA was prepared as described in Methods, digested with PstI and run in a 0.7% agarose gel at 3 V/cm. and transferred to a nylon membrane. (B) PFGE blots were prepared and processed as described (Henriksson et al. 1995). Chromosomal DNA markers were the CHEF DNA size markers, 0.2–2.2 Mbp (BioRad). Both nylon membranes (A,B) were hybridized with a radioactively labeled 300-bp fragment amplified by PCR using the oligonucleotides TcIRE-fwd and TcIRE-rev as shown in Figure 3 and described in Methods. Parasites, strains, and clones used in A or B are: Leishmania mexicana (Lm); Crithidia fasciculata (Cf); Trypanosoma cruzi strains and clones Tul0, Tul2, Corpus christi (Cc), Y, Perú (P), Sonya (S), CL-Brener (CL), and Sylvio (Sv).
When the same fragment from clone GSSTc11311 was used to probe a Northern blot made from epimastigote total RNA, no signal was found (data not shown), which suggests that the ESTs detected might come from a genomic contamination of the original library used to sequence the ESTs. This data suggest that TcIRE does not code for a protein product and can be considered part of the T. cruzi repetitive DNA.
METHODS
Genomic Library
A random genomic library from T. cruzi strain CL Brener was constructed in the plasmid vector pBS(−) (Stratagene). DNA was prepared by using the proteinase K–phenol extraction method (Sambrook et al. 1989) and mechanically sheared by using a nebulizer. After treatment with Bal31 nuclease, phenol extraction, and ethanol precipitation, the DNA was blunt-ended with T4 DNA polymerase. Fragments were size-fractionated by agarose gel electrophoresis and the range between 1.4–2.1 Kb were recovered and cloned into the dephosphorylated HincII site of the vector.
Nucleotide Sequencing
Fresh plated colonies were grown at 37°C in Terrific broth containing 100 μg/mL ampicillin in 96 deep-well plates at 350 rpm. The bacterial cultures were grown in two steps, first in 0.2 mL for 14–16 h; afterward 0.8 mL of medium was added and grown overnight. The template DNA for the sequencing reaction was prepared by a modified alkaline lysis method (Sambrook et al. 1989) using a 96-well format, followed by a purification step with Wizard Midipreps DNA Purification Resin (Promega) and 96-well MultiScreen plates with glass fiber membrane (Millipore).
The amount of isolated DNA template was estimated on 1.0% agarose gel by comparison to serial dilutions of pBluescript II KS(+) (Stratagene). Sequencing reactions were performed in a Genius (Techne) or an Uno II (Biometra) thermal cycler by using BigDye Terminator Cycle Sequencing Ready Reaction kits, with AmpliTaq DNA Polymerase (FS enzyme) (PE Biosystems) following the protocols supplied by the manufacturer, and analyzed in an ABI prism 377 (PE Biosystems). Single-pass sequencing was performed on each template using T7 or T3 primer. Bases were called by PHRED, and edited to remove vector sequence (as detected by CROSSMATCH) from the 5′ end and unreliable data from the 3′ end of sequences. Sequences longer than 100 bases were further analyzed.
Sequence Analysis
The sequences were compared against the NCBI nonredundant protein or nucleotide databases by using the program BLASTX or BLASTN respectively (Altschul et al. 1997) on the BLAST network service at the National Center for Biotechnology Information (NCBI). Blast searches against in-house databases were run locally using the BLAST suite of programs as distributed by the NCBI in a PC computer running Linux. Sequences used to search the databases were either consensus sequences or individual GSS sequences as explained in the text.
For contig assembly, sequences were assembled with PHRAP; the assemblies were visually inspected with CONSED. PHRED, PHRAP, CROSS-MATCH, and CONSED are courtesy of B. Ewing, P. Green, and D. Gordon (University of Washington, Seattle). The T. cruzi GSS database used to run local BLAST searches was compiled from the 11,459 GSS after base-calling and vector masking with the mentioned programs. The frequency of di- and trinucleotides was calculated using COMPSEQ (EMBOSS, European Molecular Biology Open Software Suite; The Sanger Centre, UK) and searching for all possible words of length 2 and 3, respectively. The expected values are calculated on the assumption that all words have the same probability of occurrence. The fractional GC content of sequences was calculated with the program GEECEE (EMBOSS). The number of ORFs was calculated using GETORF (EMBOSS). Fickett's test (testcode) was implemented as a Perl program, based on the original algorithm (Fickett 1982), and used to calculate a testcode value for each sequence. Microsatellite repeats, low-complexity regions and repetitive elements were searched for using the program REPEATMASKER (courtesy of A. Smit and P. Green, University of Washington, Seattle) over the entire GSS database using different repeat libraries that are part of RepBase (release 5.02). The Pfam database (Bateman et al. 2000) was searched using HMMPFAM, from the HMMER package (courtesy of S. Eddy, Washington University, St. Louis).
Estimation of the Copy Number and the Relative Abundance
Copy number (CN) was estimated directly from the number of GSS (GSS) belonging to a given gene family. The estimation also included the size (GS) of the gene/sequence element to account for the fact that bigger sequences should be more represented in a random sequence survey than smaller ones at the same copy number. For this calculation the haploid genome size (HGS) used was 4 × 107 bp (Frohme et al. 1998) and the total number of GSS (TGSS) was 11,459:
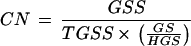 |
The estimation of the relative abundance (R) was done using the estimated number of copies as follows:
 |
Southern Blot and Pulse Field Gel Electrophoresis
DNA was purified using the conventional proteinase K–phenol extraction method. 4 μg from each sample was digested with PstI (New England BioLabs) and separated on a 0.7% agarose gel. Southern blot transfer and analysis was performed following standard procedures (Sambrook et al. 1989). For the PFGE blots, DNA was prepared and processed as described (Henriksson et al. 1995). The probe was radioactively labeled with [α-32P]dCTP (NEN Life Science Products, Inc.) by a PCR-based method using DNA from clone GSSTc11311 as template and the following oligonucleotides: TcIRE-rev, 5′-cgccaacacaactgatacg-3′, and TcIRE-fwd, 5′-gcaaaagagagcaccaac-3′. After hybridization the nylon filters were washed under stringent conditions (0.1× SSC, 0.1 % SDS, at 62°C).
Acknowledgments
We thank J.J. Cazzulo for critical reading of the manuscript. We thank Diego Rey Serantes, Fernanda Peri and Rodrigo Pavón for technical assistance. This work was funded by grants from the UNDP/World Bank/WHO Special Program for Research and Training in Tropical Diseases; SAREC, the Research Department of the Swedish International Development Agency (SIDA); Agencia Nacional de Promoción Científica y Tecnológica, Argentina and Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina. The work of A.C.C.F. was partially supported by an International Research Scholar award from the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL dsanchez@iib.unsam.edu.ar; FAX 54-11-4752-9639.
Article published online before print: Genome Res., 10.1101/gr.146300.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.146300.
REFERENCES
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson B, Aslund L, Tammi M, Tran A, Hoheisel JD, Pettersson U. Complete sequence of a 93.4-kb contig from chromosome 3 of Trypanosoma cruzi containing a strand-switch region. Genome Res. 1998;8:809–816. doi: 10.1101/gr.8.8.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya J, Cano MI, Gomes HB, Novak EM, Requena JM, Alonso C, Levin MJ, Guevara P, Ramirez JL, Da Silveira JF. Characterization of an interspersed repetitive DNA element in the genome of Trypanosoma cruzi. Parasitology. 1997;115:563–570. doi: 10.1017/s0031182097001674. [DOI] [PubMed] [Google Scholar]
- Armah DA, Mensa-Wilmot K. S-myristoylation of a glycosylphosphatidylinositol-specific phospholipase C in Trypanosoma brucei. J Biol Chem. 1999;274:5931–5938. doi: 10.1074/jbc.274.9.5931. [DOI] [PubMed] [Google Scholar]
- Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer ELL. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar RS, Futterer K, Waksman G, Gordon JI. The structure of myristoyl-CoA:protein N-myristoyltransferase. Biochim Biophys Acta. 1999;1441:162–172. doi: 10.1016/s1388-1981(99)00155-9. [DOI] [PubMed] [Google Scholar]
- Campetella O, Henriksson J, Aslund L, Frasch ACC, Pettersson U, Cazzulo JJ. The major cysteine proteinase (cruzipain) from Trypanosoma cruzi is encoded by multiple polymorphic tandemly organized genes located on different chromosomes. Mol Biochem Parasitol. 1992a;50:225–234. doi: 10.1016/0166-6851(92)90219-a. [DOI] [PubMed] [Google Scholar]
- Campetella O, Sánchez DO, Cazzulo JJ, Frasch ACC. A superfamily of Trypanosoma cruzi surface antigens. Parasitol Today. 1992b;8:378–381. doi: 10.1016/0169-4758(92)90175-2. [DOI] [PubMed] [Google Scholar]
- de Mendonça-Lima L, Traub-Cseko YM. A new repetitive DNA sequence from Trypanosoma cruzi. Mem Inst Oswaldo Cruz. 1991;86:475. doi: 10.1590/s0074-02761991000400020. [DOI] [PubMed] [Google Scholar]
- Di Noia JM, D'Orso I, Åslund L, Sánchez DO, Frasch ACC. The Trypanosoma cruzi mucin family is transcribed from hundreds of genes having hypervariable regions. J Biol Chem. 1998;273:10843–10850. doi: 10.1074/jbc.273.18.10843. [DOI] [PubMed] [Google Scholar]
- El-Sayed NMA, Donelson JE. A survey of the Trypanosoma brucei rhodesiense genome using shotgun sequencing. Mol Biochem Parasitol. 1997;84:167–178. doi: 10.1016/s0166-6851(96)02792-2. [DOI] [PubMed] [Google Scholar]
- Elgar G, Clark MS, Meek S, Smith S, Warner S, Edwards YJK, Bouchireb N, Cottage A, Yeo GSH, Umrania Y, et al. Generation and analysis of 25 Mb of genomic DNA from the pufferfish Fugu rubripes by sequence scanning. Genome Res. 1999;9:960–971. doi: 10.1101/gr.9.10.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersfeld K, Melville SE, Gull K. Nuclear and genome organization of Trypanosoma brucei. Parasitol Today. 1999;15:58–63. doi: 10.1016/s0169-4758(98)01378-7. [DOI] [PubMed] [Google Scholar]
- Estevez AM, Simpson L. Uridine insertion/deletion RNA editing in trypanosome mitochondria. Gene. 1999;29:247–260. doi: 10.1016/s0378-1119(99)00437-0. [DOI] [PubMed] [Google Scholar]
- Fickett JW. Recognition of protein coding regions in DNA sequences. Nucleic Acids Res. 1982;10:5303–5318. doi: 10.1093/nar/10.17.5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasch ACC. Functional diversity in members of the trans-sialidase and mucin families in Trypanosoma cruzi. Parasitol Today. 2000;16:282–286. doi: 10.1016/s0169-4758(00)01698-7. [DOI] [PubMed] [Google Scholar]
- Frohme M, Hanke J, Åslund L, Pettersson U, Hoheisel JD. Selective generation of chromosomal cosmid libraries within the Trypanosoma cruzi genome project. Electrophoresis. 1998;19:478–481. doi: 10.1002/elps.1150190404. [DOI] [PubMed] [Google Scholar]
- Gao G, Nara T, Nakajima-Shimada J, Aoki T. Novel organization and sequences of five genes encoding all six enzymes for de novo pyrimidine biosynthesis in Trypanosoma cruzi. J Mol Biol. 1999;285:149–161. doi: 10.1006/jmbi.1998.2293. [DOI] [PubMed] [Google Scholar]
- González A, Prediger E, Huecas ME, Nogueira N, Lizardi PM. Minichromosomal repetitive DNA in Trypanosoma cruzi: Its use in a high-sensitivity parasite detection assay. Proc Natl Acad Sci. 1984;81:3356–3360. doi: 10.1073/pnas.81.11.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson J, Porcel B, Rydåker M, Ruiz A, Cazzulo JJ, Frasch ACC, Petersson U. Chromosome specific markers reveal conserved linkage groups in spite of extensive chromosomal size variation in Trypanosoma cruzi. Mol Biochem Parasitol. 1995;37:64–73. doi: 10.1016/0166-6851(95)00096-j. [DOI] [PubMed] [Google Scholar]
- Kooter JM, Winter AJ, de Oliveira C, Wagter R, Borst P. Boundaries of telomere conversion in Trypanosoma brucei. Gene. 1988;69:1–11. doi: 10.1016/0378-1119(88)90372-1. [DOI] [PubMed] [Google Scholar]
- Krakow JL, Hereld D, Bangs JD, Hart GW, Englund PT. Identification of a glycolipid precursor of the Trypanosoma brucei variant surface glycoprotein. J Biol Chem. 1986;261:12147–12153. [PubMed] [Google Scholar]
- Lee MG, Van der Ploeg LH. Transcription of protein-coding genes in trypanosomes by RNA polymerase I. Annu Rev Microbiol. 1997;51:463–489. doi: 10.1146/annurev.micro.51.1.463. [DOI] [PubMed] [Google Scholar]
- Martín F, Marañón C, Olivares M, Alonso C, López MC. Characterization of a non-long terminal repeat retrotransposon cDNA (L1Tc) from Trypanosoma cruzi: Homology of the first ORF with the ape family of DNA repair enzymes. J Mol Biol. 1995;247:49–59. doi: 10.1006/jmbi.1994.0121. [DOI] [PubMed] [Google Scholar]
- Mottram JC, Murphy WJ, Agabian N. A transcriptional analysis of the Trypanosoma brucei hsp83 gene cluster. Mol Biochem Parasitol. 1989;37:115–127. doi: 10.1016/0166-6851(89)90108-4. [DOI] [PubMed] [Google Scholar]
- Muchardt C, Yaniv M. ATP-dependent chromatin remodelling: SWI/SNF and Co. are on the job. J Mol Biol. 1999;293:187–198. doi: 10.1006/jmbi.1999.2999. [DOI] [PubMed] [Google Scholar]
- Myler PJ, Audleman L, deVos T, Hixson G, Kiser P, Lemley C, Magness C, Rickel E, Sisk E, Sunkin S, et al. Leishmania major Friedlin chromosome 1 has an unusual distribution of protein-coding genes. Proc Natl Acad Sci. 1999;96:2902–2906. doi: 10.1073/pnas.96.6.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network TLG. The complete chromosomal organization of the reference strain of the Leishmania genome project, L. major Friedlin. Parasitol Today. 1998;14:301–303. doi: 10.1016/s0169-4758(98)01275-7. [DOI] [PubMed] [Google Scholar]
- Nilsen TW. Trans-splicing: An update. Mol Biochem Parasitol. 1995;73:1–6. doi: 10.1016/0166-6851(94)00107-x. [DOI] [PubMed] [Google Scholar]
- Olivares M, Alonso C, Lopez MC. The open reading frame 1 of the L1Tc retrotransposon of Trypanosoma cruzi codes for a protein with apurinic-apyrimidinic nuclease activity. J Biol Chem. 1997;272:25224–25228. doi: 10.1074/jbc.272.40.25224. [DOI] [PubMed] [Google Scholar]
- Parodi AJ, Pollevick GD, Mautner M, Buschiazzo A, Sanchez DO, Frasch ACC. Identification of the gene(s) coding for the trans-sialidase of Trypanosoma cruzi. EMBO J. 1992;11:1705–1710. doi: 10.1002/j.1460-2075.1992.tb05221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcel BM, Tran AN, Tammi M, Nyarady Z, Rydaker M, Urmenyi TP, Rondinelli E, Pettersson U, Andersson B, Aslund L. Gene survey of the pathogenic protozoan Trypanosome cruzi. Genome Res. 2000;10:1103–1107. doi: 10.1101/gr.10.8.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puerta C, Martin J, Alonso C, Lopez MC. Isolation and characterization of the gene encoding histone H2A from Trypanosoma cruzi. Mol Biochem Parasitol. 1994;64:1–10. doi: 10.1016/0166-6851(94)90129-5. [DOI] [PubMed] [Google Scholar]
- Regev A, Lamb MJ, Jablonka E. The role of DNA methylation in invertebrates: Developmental regulation or genome defense? Mol Biol Evol. 1998;15:880–891. [Google Scholar]
- Requena JM, Lopez MC, Jimenez-Ruiz A, de la Torre JC, Alonso C. A head-to-tail tandem organization of hsp70 genes in Trypanosoma cruzi. Nucleic Acids Res. 1988;16:1393–1406. doi: 10.1093/nar/16.4.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Requena JM, Jimenez-Ruiz A, Soto M, Lopez MC, Alonso C. Characterization of a highly repeated interspersed DNA sequence of Trypanosoma cruzi: Its potential use in diagnosis and strain classification. Mol Biochem Parasitol. 1992;51:271–280. doi: 10.1016/0166-6851(92)90077-w. [DOI] [PubMed] [Google Scholar]
- Rudenko G, Cross M, Borst P. Changing the end: Antigenic variation orchestrated at the telomeres of African trypanosomes. Trends Microbiol. 1998;6:113–116. doi: 10.1016/s0966-842x(97)01200-6. [DOI] [PubMed] [Google Scholar]
- Russell Johnson D, Bhatnagar RS, Knoll LJ, Gordon JI. Genetic and biochemical studies of protein N-myristoylation. Annu Rev Biochem. 1994;63:869–914. doi: 10.1146/annurev.bi.63.070194.004253. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular sequences. J Mol Biol. 1983;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Soto M, Quijada L, Alonso C, Requena JM. Molecular cloning and analysis of expression of the Leishmania infantum histone H4 genes. Mol Biochem Parasitol. 1997;90:439–447. doi: 10.1016/s0166-6851(97)00178-3. [DOI] [PubMed] [Google Scholar]
- Stuart K, Allen TE, Kable ML, Lawson S. Kinetoplastid RNA editing: Complexes and catalysts. Curr Opin Chem Biol. 1997;1:340–346. doi: 10.1016/s1367-5931(97)80071-3. [DOI] [PubMed] [Google Scholar]
- Taylor MC, Muhia DK, Baker DA, Mondragon A, Schaap PB, Kelly JM. Trypanosoma cruzi adenylyl cyclase is encoded by a complex multigene family. Mol Biochem Parasitol. 1999;104:205–217. doi: 10.1016/s0166-6851(99)00154-1. [DOI] [PubMed] [Google Scholar]
- Verdún RE, Di Paolo N, Urmenyi RP, Rondinelli E, Frasch ACC, Sanchez DO. Gene discovery through expressed sequence tag sequencing in Trypanosoma cruzi. Infect Immun. 1998;66:5393–5398. doi: 10.1128/iai.66.11.5393-5398.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston SA, Camble R, Colls J, Rosenbrock G, Taylor I, Egerton M, Tucker AD, Tunnicliffe A, Mistry A, Mancia F, et al. Crystal structure of the anti-fungal target N-myristoyl transferase. Nature Struct Biol. 1998;5:213–221. doi: 10.1038/nsb0398-213. [DOI] [PubMed] [Google Scholar]
- Wincker P, Murto-Dovales AC, Goldenberg S. Nucleotide sequence of a representative member of a Trypanosoma cruzi dispersed gene family. Mol Biochem Parasitol. 1992;55:217–220. doi: 10.1016/0166-6851(92)90142-7. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ruyechan W, Williams N. Developmental regulation of two nuclear RNA binding proteins, p34 and p37, from Trypanosoma brucei. Mol Biochem Parasitol. 1998;92:79–88. doi: 10.1016/s0166-6851(97)00228-4. [DOI] [PubMed] [Google Scholar]
- Zingales B, Rondinelli E, Degrave W, da Silveira JF, Levin M, Le Paslier D, Modabber F, Dobrokhotov B, Swindle J, Kelly JM, et al. The Trypanosoma cruzi genome initiative. Parasitol Today. 1997;13:16–22. [Google Scholar]