

Summary
The extraordinary sensitivity and specificity of T cells for their cognate antigen make them a highly attractive cancer therapeutic. However, the rarity of tumor-reactive T cells in cancer patients, the difficulty isolating them in sufficient numbers for adoptive immunotherapy, and the unpredictable persistence of transferred cells have been significant obstacles to broad application. Technologies that enable genetic modification of T cells have been refined and are being used to redirect the specificity of T cells to tumor antigens. An issue the field is now grappling with is how the diverse phenotypic and functional heterogeneity in T cells that could potentially be genetically modified can be capitalized upon to enhance the efficacy, safety, and reproducibility of cancer immunotherapy.
Introduction
Gene transfer to redirect the specificity of any human T lymphocyte is a rapidly developing area in cancer immunotherapy. This approach relies on introducing genes into T cells that encode T cell receptors (TCR) specific for a tumor associated antigen, or chimeric antigen receptors (CAR) that typically consist of a single chain Fv constructed from a monoclonal antibody specific for a tumor cell surface molecule and linked to one or more T cell signaling moieties [1–3]. The initial clinical applications of adoptive immunotherapy with genetically retargeted T cells have primarily employed unselected T cells from the patient’s peripheral blood for gene transfer, which ignores the considerable phenotypic diversity of T lymphocytes that have been programmed for distinct functions by previous experience [4–7]. Here, we will review the strategies being used to engineer therapeutic T cells, and discuss how the phenotypic and functional diversity of human CD8+ T cells may provide opportunities to enhance cancer immunotherapy with genetically modified cells.
Genetic modification of T cells to confer tumor specificity
The potential for adoptively transferred T cells to eradicate human malignancies is illustrated by the graft versus leukemia effect of allogeneic T cells administered as part of a stem cell transplant [8], and by the dramatic tumor regressions that can occur in melanoma patients who receive autologous T cells derived from the tumor infiltrate and expanded ex vivo prior to transfer [9]. Although culture methods have improved, it is still not possible to rapidly derive tumor-reactive T cells from the blood or tumor infiltrates of most patients with cancer. Even when T cells are isolated from the endogenous repertoire, expanded, and adoptively transferred, their ability to persist and mediate antitumor activity in vivo has been unpredictable [10]. An approach to overcome the low frequency of tumor-reactive T cells in patients and potentially ensure predictable behavior after adoptive transfer is to redirect the specificity of T cells using gene insertion (Figure 1). Vector systems to deliver transgenes into primary human T cells have been developed and the advantages and disadvantages of the various gene delivery methods have been reviewed [11,12].
Figure 1. Redirecting T cell specificity by insertion of genes encoding a TCR or CAR.
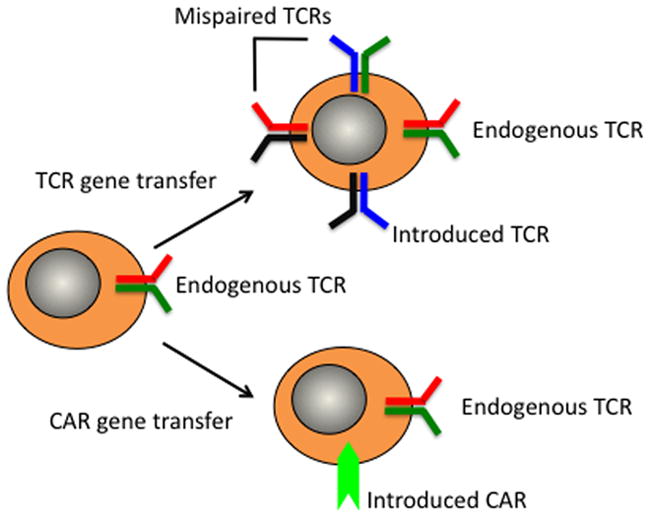
Peripheral blood T cells (or selected subsets) are activated and exposed to a gene delivery vector to introduce a tumor targeting T cell receptor (TCR) or chimeric antigen receptor (CAR). In the case of TCR introduction, the possibility of mispairing with endogenous α and β chains leads to the potential for four receptors to be expressed on the transduced cell.
T cell receptor gene transfer
Tumor antigens are predominantly derived from non-mutated self-proteins that are either cell lineage specific, aberrantly expressed, or overexpressed in tumor cells; and most T cells that express TCRs with high affinity for such self-determinants are eliminated by thymic selection. Although rare, moderate and high avidity T cells specific for self-antigens can occasionally be cloned from patients or healthy individuals. These T cells provide a source of TCR α and β genes that can be used de novo or after mutation to increase affinity, as “off the shelf” reagents that confer tumor reactivity to T cells from any patient with the appropriate HLA restricting allele. TCR genes have been cloned that redirect specificity to several tumor antigens including MART-1, gp-100, WT-1, NY-ESO-1, and CEA [13–17].
A problem with TCR gene transfer is mispairing of the introduced TCR chains with the endogenous TCR chains, creating potentially deleterious reactivity with normal host tissues [18]. Such off-target toxicity has not been definitively observed in the small clinical trials that have been performed thus far [19,20], however studies in animal models have clearly demonstrated the potential for toxicity resulting from acquired self reactivity as a consequence of mispairing in TCR gene-modified T cells [21]. The potential for toxicity can be minimized by modifications to the design of TCR transgenes that promote appropriate pairing of the introduced chains [18], and by the selection of defined T cells as recipients of TCR gene transfer, such as TCRγδ cells that lack endogenous αβ chains, or virus-specific T cells that utilize a very limited endogenous TCR repertoire [21,22].
Chimeric antigen receptor gene transfer
Redirecting T cells to recognize tumor antigens through TCR gene transfer is inherently constrained by the requirement for MHC restricted peptide presentation by tumor cells. Chimeric non-MHC restricted artificial receptors that recognize tumor cell surface molecules have been developed to overcome this limitation. A CAR is typically comprised of a fusion gene that encodes monoclonal antibody-derived single chain variable fragments (scFv), consisting of heavy (VH) and light (VL) chains joined by a flexible linker, and then fused through a transmembrane domain to cytoplasmic signaling moieties consisting of CD3ζ alone, or CD3ζ combined with activation domains from costimulatory molecules [2]. As with TCRs, CARs have been constructed for many tumor-associated molecules including CD19, CD20, EGFR, Her2neu, GD2, PSMA, CAIX and ROR1 [23–29].
Heterogeneity of peripheral blood CD8+ T cells
Several clinical trials in which polyclonal T cells are obtained from the peripheral blood, genetically modified with either a TCR or a CAR, expanded, and re-infused into the patient are in progress. Impressive antitumor activity has been observed in some trials, but inconsistent T cell persistence and serious toxicities that are often not fully explained have also been reported [17,30,31]. A factor that is emerging as an important variable is the potential for vast differences in the composition of T cell products if unselected T cells are used for genetic modification. For brevity, this review will focus on heterogeneity in human CD8+ T cell subsets and the implications for deriving genetically modified T cells for adoptive therapy, however the issues discussed are equally germane for redirecting CD4+ T cells for tumor reactivity.
A variety of phenotypic and functional subsets of CD8+ T cells are present in peripheral blood, and this heterogeneity ensures appropriate responses to neo-antigens and to antigens to which the host has been previously exposed (Figure 2). CD8+ T cells are broadly divided into CD45RA+ antigen inexperienced naïve T cells (TN) that contain the greatest diversity of endogenous T cell receptors [32], and express CD62L+ and CCR7+ to enable their transit through lymph nodes where they survey for foreign antigens; and CD45RO+ memory T cells that have clonally expanded in response to prior antigen encounter, and can be subdivided into CD62L+ central memory (TCM) and effector memory (TEM) subsets [33]. In humans, the CD45RO TCM and TEM subsets have recently been shown to contain a major population of distinct CD161hi, IL-18Rαhi T cells that remain to be fully characterized but differ significantly in functional properties from their CD161lo counterparts [35–37]. A subset of memory T cells with a CD44loCD62LhiSca-1hiCD122hi bcl-2hi phenotype that is intermediate between that naïve and memory cells was identified in a murine model of graft versus host disease (GVHD) and suggested to represent a “memory stem cell” based on the ability to self-renew, and give rise to effector (TE), and TCM and TEM subsets [34]. A counterpart for this memory CD8+ T cell has not been reported in viral infections in mice or in humans, and whether specialized CD8+ T cells with stem cell qualities develop after antigen exposure remains unproven.
Figure 2. Surface markers and functional attributes of major subsets of CD8+ T cells in peripheral blood.
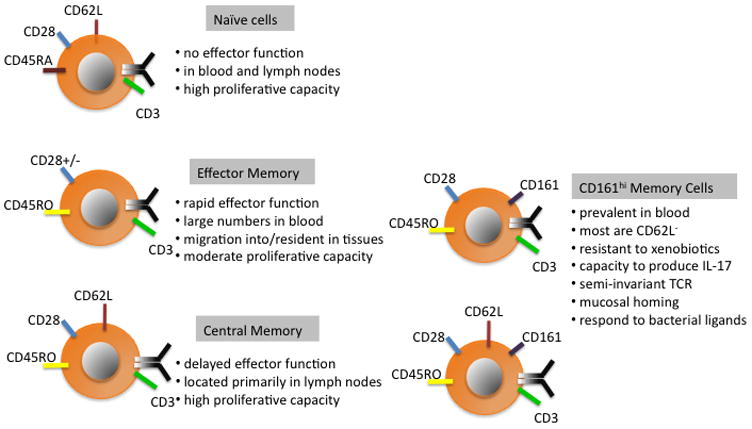
CD8+ T cells can be identified as naïve or memory based on the acquisition of the CD45RO isoform on memory cells. Memory cells can then be broadly subdivided into CD161hi and CD161lo subsets. Within the CD161lo pool of memory cells, the expression of CD62L distinguishes functionally disparate central (TCM) and effector memory (TEM) subsets, although it should be noted that there are cells with an overlapping phenotype. The CD161hi memory cells are predominantly CD62L− and differ in specificity and function from CD161lo memory cells. The functional properties of the small subset of CD62L+CD161hi cells have not been separately evaluated.
An important consideration for genetic modification of T cells is that the proportion of each of the TN, and CD161hi and CD161lo TEM and TCM subsets in the blood of normal individuals and cancer patients can vary dramatically depending on age, pathogen exposure, and prior chemotherapy [5,35,37]. This heterogeneity poses a potential variable that could impact safety and efficacy of cancer immunotherapy if polyclonal unselected cells derived from the blood are genetically modified with TCR or CAR genes. Indeed, recent data suggests that depending on the clinical situation it may prove valuable or even essential to select T cells for therapy from a defined subset, or of singular antigen specificity.
Properties of effector cells derived from CD8+ TCM and TEM
The longevity of T cell memory is a cardinal feature of adaptive immunity, and the cell intrinsic and extrinsic mechanisms that determine memory cell differentiation and survival continue to be intensively studied [38,39]. Adoptive transfer studies in humans demonstrated that virus-specific TE cells could be isolated and expanded from the memory pool of immune hematopoietic stem cell donors, expanded in vitro as clones or polyclonal populations and transferred to transplant recipients to restore durable immunity [40,41]. However, whether TE cells from the TCM or TEM subsets contributed more or less to reconstitution was unknown.
Studies using gene-marked virus-specific TE cells in non-human primates have now demonstrated that TE cells derived from the CD62L+ TCM subset rather than the TEM subset possess a markedly superior capacity to survive in vivo, revert to a memory phenotype, and establish long-lived immunity that is capable of responding to antigen challenge [42,43]. In these studies, the transferred TE cells were clonally derived and had uniformly upregulated granzyme B and perforin, and downregulated CD62L, CCR-7, CD127 and CD28. Despite the similarities in differentiation markers at the time of infusion, only TE cells derived from TCM reacquired memory markers and established reservoirs of CD62L+ CCR-7+ CD28+ TCM that largely resided in lymph nodes, and CD62L− TEM in the blood and bone marrow. This result suggested that TCM-derived TE retain a cell-intrinsic capacity to revert to a quiescent memory cell through cloning, effector differentiation, and long-term culture [42]. A similar analysis of polyclonal human TCM- and TEM-derived TE cells has been performed using NOD/SCID/γc−/− (NSG) mice as recipients of the transferred T cells. Here again, TCM-derived TE exhibited vastly superior engraftment and antitumor activity [44]. Moreover, analysis of the TCR Vβ repertoire of persisting human T cells in the NSG mice suggest that longevity is a general property of TCM-derived TE, and not confined to a rare subset of cells [44].
Gene transfer into virus-specific memory T cells
While animal model data has identified superior in vivo persistence of TCM-derived TE, it is premature to conclude that virus-specific or polyclonal CD8+ TCM should be selected for introducing tumor-targeting receptors for adoptive therapy in all circumstances. However, this strategy may improve safety and be necessary in some clinical settings. For example, genetic modification of unselected T cells from an allogeneic stem cell donor with a TCR or CAR to direct specificity to leukemia and augment the graft versus leukemia effect after allogeneic stem cell transplant is likely to be complicated by GVHD mediated by T cells in the product that have an alloreactive endogenous TCR. Donor CMV and EBV-specific T cells do not cause GVHD in the allogeneic transplant setting, thus selecting such virus-specific cells from TCM to genetically modify with a CAR or TCR to treat leukemia should improve safety. As previously discussed, selecting virus-specific T cells for TCR gene transfer will also limit the endogenous TCR repertoire available for cross-pairing of the introduced TCR. Furthermore, stimulation through the endogenous virus-specific TCR during episodes of viral reactivation or by vaccination could promote selective activation, survival and expansion of the transferred cells, and potentially overcome tolerance mechanisms [45].
Modifying virus-specific memory cells rather than unselected T cells may also have advantages for autologous cell therapy. In a clinical trial performed by Brenner et al, eleven neuroblastoma patients were treated with cell products that contained both EBV-specific T cells and anti-CD3 activated T cells that were retrovirally transduced with a GD2-specific CAR [46]. Sequence differences in the vectors used to transduce EBV-specific or αCD3-stimulated T cell preparations enabled measurement of persistence of the two products, which suggested that EBV-specific CAR-transduced T cells were better equipped to survive after transfer.
CD161hi CD8+ memory cells
Several recent papers have shed light on the function of a specialized subset of CD45RO+ CD8+ memory cells characterized by high levels of CD161 expression. In the course of studies analyzing chemotherapy resistance of human T cells, we identified CD8+ T cells that rapidly effluxed fluorescent dyes based on high levels of ABC transporter activity and found that these effluxing cells were distinguished by the expression of high levels of CD161 and IL-18Rα [35]. CD161hi CD8+ T cells represent a significant fraction of the total circulating CD8+ memory pool in normal donors, and are characterized by uniform expression of CD28 and CD27, and high levels of bcl-2 and bcl-xl. In a series of elegant papers, Lantz and colleagues have shown that CD161hi CD8+ T cells comprise an innate-like subset that expresses a semi-invariant TCR Vα7.2-Jα33 that confers specificity for undefined bacterial ligands presented by the MHC class Ib MR1 molecule [37,47,48]. CD161hi cells also express tissue homing integrins and preferentially localize to liver and gastrointestinal tract [37]. Analysis of the functional properties of this subset of CD8+ T cells remains incomplete but in addition to IFN-γ, CD161hi CD8+ T cells produce IL17 after PMA/ionomycin stimulation [35–37]. Because these T cells have enhanced resistance to certain chemotherapy drugs, they may be further enriched in the blood of cancer patients. Thus, if peripheral blood T cells from cancer patients are genetically modified in bulk, the expression of tumor-targeting receptor in these peculiar but prevalent memory T cells is likely to occur to some degree, with uncertain clinical consequences. Infiltration of normal colon with CD8+ T cells and serious colitis was observed in 3/3 patients treated with autologous T cells modified with a CEA-specific TCR for metastatic colorectal cancer [17]. This was presumed to be due to recognition of CEA on normal colonic cells by the transferred gene modified cells raising the interesting possibility that the preferential gut-homing properties of certain subsets of CD8+ T cells might contribute to toxicity.
CD8+ naïve T cells
As noted above, TN have clear disadvantages for immunotherapy in the allogeneic stem cell transplant setting because this subset has been shown to be primarily responsible for GVHD [49]. However, TN exhibit many traits that could be advantageous when genetically modifying autologous T cells for adoptive therapy. Elegant cell transfer studies in mice have indicated that a single TN can give rise to TE, TCM, and TEM cells, illustrating both the proliferative and differentiation potential of this subset [50]. In a murine TCR transgenic model, superior tumor elimination was demonstrated after transfer of engineered TE derived from TN compared to those derived from TCM and TEM, and this was linked to less upregulation of KLRG-1, which is a marker of a terminal effector cell [51]. The properties of human TE cells derived from TN, TCM, and TEM and modified with a tumor-specific TCR have thus far only been compared in vitro. These studies demonstrated that TN cells were easily transduced, and the derived TE cells exhibited greater proliferation without acquiring markers of late effector differentiation, and retained longer telomeres -- traits that would be preferred in therapeutic T cell products [52]. However, in the absence of in vivo experiments, it remains uncertain whether TN that are expanded and differentiated in vitro will have the cell-intrinsic properties that have been convincingly demonstrated to confer long-term survival of adoptively transferred CMV and EBV-specific TE cells. The answer to this critical question will require carefully designed clinical adoptive transfer studies.
A potential advantage of TN is their plasticity, which could enable modifications of culture conditions to direct cell fate decisions. For example, an in vitro counterpart of the memory stem cell identified in murine GVHD studies has been generated by chemical augmentation of Wnt signaling during in vitro priming of murine TCR transgenic CD8+ T cells, which led to upregulation of Tcf-1 and Lef-1 and arrested the acquisition of effector function [53]. After adoptive transfer, these T cells exhibited enhanced in vivo recall responses and antitumor activity in murine models compared with TCM and TEM. In vivo pharmacologic manipulation of mTOR activation and fatty acid metabolism has also been demonstrated to augment the generation of memory T cells during priming in animal models [54,55]. These results suggest that altering signaling pathways involved in directing cell fate decisions might be used in vitro during genetic modification to derive T cells with selected qualities that promote in vivo persistence and function.
Conclusions
The past decade has seen an apparent coming of age of T cell adoptive immunotherapy for cancer, due in large part to the success in melanoma where the use of lymphodepleting therapy before T cell transfer led to improvements in the in vivo persistence of transferred cells and therapeutic efficacy, and to advances in T cell genetic engineering. Our understanding of the fundamental properties of heterogeneous subsets of T cells is also evolving and suggests that selection of defined populations for gene modification may be necessary to capitalize fully on the opportunities for adoptive immunotherapy for cancer. Clinical investigation of distinct cell populations will require the development of improved cell sorting and selection technologies that enable cell manipulation without compromising function, and current approaches have serious limitations in this regard. It is also imperative that future clinical trials incorporate detailed characterization of the cell products that are infused and sophisticated monitoring of their fate, migration and function in vivo, to provide insight into the basis for therapeutic success and toxicity, and direct subsequent investigation.
Acknowledgments
This work was supported by National Institutes of Health grants CA114536, CA18029, CA136551, AI053193, AI086683, and P50CA138293.
Footnotes
Conflict of Interest Statement: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schumacher TN. T-cell-receptor gene therapy. Nat Rev Immunol. 2002;2:512–519. doi: 10.1038/nri841. [DOI] [PubMed] [Google Scholar]
- 2.Eshhar Z. Adoptive cancer immunotherapy using genetically engineered designer T-cells: First steps into the clinic. Curr Opin Mol Ther. 2010;12:55–63. [PubMed] [Google Scholar]
- 3.Bendle GM, Haanen JB, Schumacher TN. Preclinical development of T cell receptor gene therapy. Curr Opin Immunol. 2009;21:209–214. doi: 10.1016/j.coi.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. 2007;27:393–405. doi: 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angelosanto JM, Wherry EJ. Transcription factor regulation of CD8+ T-cell memory and exhaustion. Immunol Rev. 2010;236:167–175. doi: 10.1111/j.1600-065X.2010.00927.x. [DOI] [PubMed] [Google Scholar]
- 6.Rutishauser RL, Kaech SM. Generating diversity: transcriptional regulation of effector and memory CD8 T-cell differentiation. Immunol Rev. 2010;235:219–233. doi: 10.1111/j.0105-2896.2010.00901.x. [DOI] [PubMed] [Google Scholar]
- 7.Beuneu H, Lemaitre F, Deguine J, Moreau HD, Bouvier I, Garcia Z, Albert ML, Bousso P. Visualizing the functional diversification of CD8+ T cell responses in lymph nodes. Immunity. 2010;33:412–423. doi: 10.1016/j.immuni.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 8.Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, Rimm AA, Ringden O, Rozman C, Speck B, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75:555–562. [PubMed] [Google Scholar]
- *9.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. This study evaluated different lymphodepleting regimens prior to adoptively transferring unmodified tumor infiltrating lymphocytes and demonstrated that the antitumor activity and T cell persistence was improved with intensive lymphodepletion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, Powell DJ, Jr, Rosenberg SA. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breckpot K, Emeagi PU, Thielemans K. Lentiviral vectors for anti-tumor immunotherapy. Curr Gene Ther. 2008;8:438–448. doi: 10.2174/156652308786848058. [DOI] [PubMed] [Google Scholar]
- 12.Hackett PB, Largaespada DA, Cooper LJ. A transposon and transposase system for human application. Mol Ther. 2010;18:674–683. doi: 10.1038/mt.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson LA, Heemskerk B, Powell DJ, Jr, Cohen CJ, Morgan RA, Dudley ME, Robbins PF, Rosenberg SA. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol. 2006;177:6548–6559. doi: 10.4049/jimmunol.177.9.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xue SA, Gao L, Hart D, Gillmore R, Qasim W, Thrasher A, Apperley J, Engels B, Uckert W, Morris E, et al. Elimination of human leukemia cells in NOD/SCID mice by WT1-TCR gene-transduced human T cells. Blood. 2005;106:3062–3067. doi: 10.1182/blood-2005-01-0146. [DOI] [PubMed] [Google Scholar]
- 15.Wargo JA, Robbins PF, Li Y, Zhao Y, El-Gamil M, Caragacianu D, Zheng Z, Hong JA, Downey S, Schrump DS, et al. Recognition of NY-ESO-1+ tumor cells by engineered lymphocytes is enhanced by improved vector design and epigenetic modulation of tumor antigen expression. Cancer Immunol Immunother. 2009;58:383–394. doi: 10.1007/s00262-008-0562-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chinnasamy N, Wargo JA, Yu Z, Rao M, Frankel TL, Riley JP, Hong JJ, Parkhurst MR, Feldman SA, Schrump DS, et al. A TCR Targeting the HLA-A*0201-Restricted Epitope of MAGE-A3 Recognizes Multiple Epitopes of the MAGE-A Antigen Superfamily in Several Types of Cancer. J Immunol. 2010 doi: 10.4049/jimmunol.1001775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *17.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, et al. T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Mol Ther. 2010 doi: 10.1038/mt.2010.272. The authors describe three patients who developed colitis after adoptive therapy with T cells modified to express a carcinoembryonic antigen (CEA)-specific TCR by gene transfer. The toxicity was presumed to result from on target recognition of normal colonic mucosa mediated by CEA-specific CD8+ T cells that infiltrated the colon after infusion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Govers C, Sebestyen Z, Coccoris M, Willemsen RA, Debets R. T cell receptor gene therapy: strategies for optimizing transgenic TCR pairing. Trends Mol Med. 2010;16:77–87. doi: 10.1016/j.molmed.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 19.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenberg SA. Of mice, not men: no evidence for graft-versus-host disease in humans receiving T-cell receptor-transduced autologous T cells. Mol Ther. 2010;18:1744–1745. doi: 10.1038/mt.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **21.Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, Kaiser AD, Pouw N, Debets R, Kieback E, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 2010;16:565–570. doi: 10.1038/nm.2128. 561p following 570. The authors provide definitive evidence in a murine model for the development of undesired reactivity for self antigens as a consequence of the mispairing of introduced TCR chains with endogenous TCR chains in TCR gene modified polyclonal T cells and demonstrate the efficacy of various strategies to minimize mispairing for preventing autoreactivity. [DOI] [PubMed] [Google Scholar]
- 22.van der Veken LT, Coccoris M, Swart E, Falkenburg JH, Schumacher TN, Heemskerk MH. Alpha beta T cell receptor transfer to gamma delta T cells generates functional effector cells without mixed TCR dimers in vivo. J Immunol. 2009;182:164–170. doi: 10.4049/jimmunol.182.1.164. [DOI] [PubMed] [Google Scholar]
- 23.Ohno M, Natsume A, Ichiro Iwami K, Iwamizu H, Noritake K, Ito D, Toi Y, Ito M, Motomura K, Yoshida J, et al. Retrovirally engineered T-cell-based immunotherapy targeting type III variant epidermal growth factor receptor, a glioma-associated antigen. Cancer Sci. 2010;101:2518–2524. doi: 10.1111/j.1349-7006.2010.01734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 25.Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, Wright C, Popplewell L, Raubitschek A, Forman SJ, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101:1637–1644. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 26.Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini MT, Nishida T, Yamamoto TN, Bleakley M, Turtle CJ, Chang WC, Greisman HA, et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood. 2010;116:4532–4541. doi: 10.1182/blood-2010-05-283309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen M, Tan G, Forman S, Wu AM, Raubitschek A. CD20 is a molecular target for scFvFc:zeta receptor redirected T cells: implications for cellular immunotherapy of CD20+ malignancy. Biol Blood Marrow Transplant. 1998;4:75–83. doi: 10.1053/bbmt.1998.v4.pm9763110. [DOI] [PubMed] [Google Scholar]
- 28.Gade TP, Hassen W, Santos E, Gunset G, Saudemont A, Gong MC, Brentjens R, Zhong XS, Stephan M, Stefanski J, et al. Targeted elimination of prostate cancer by genetically directed human T lymphocytes. Cancer Res. 2005;65:9080–9088. doi: 10.1158/0008-5472.CAN-05-0436. [DOI] [PubMed] [Google Scholar]
- 29.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol. 2002;20:70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 30.Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010;18:666–668. doi: 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **31.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. The authors describe a patient who was treated with a high dose of autologous T cells modified to express an ERBB2-specific chimeric antigen receptor and developed a rapid onset and fatal pulmonary toxicity, presumably due to recognition of normal pulmonary cells that expressed ERBB2. This illustrates the need for dose escalation and careful monitoring of patients that receive T cells capable of reacting with normal as well as malignant cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, Riddell SR, Warren EH, Carlson CS. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114:4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanzavecchia A, Sallusto F. Dynamics of T lymphocyte responses: intermediates, effectors, and memory cells. Science. 2000;290:92–97. doi: 10.1126/science.290.5489.92. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat Med. 2005;11:1299–1305. doi: 10.1038/nm1326. [DOI] [PubMed] [Google Scholar]
- *35.Turtle CJ, Swanson HM, Fujii N, Estey EH, Riddell SR. A distinct subset of self-renewing human memory CD8+ T cells survives cytotoxic chemotherapy. Immunity. 2009;31:834–844. doi: 10.1016/j.immuni.2009.09.015. The authors use drug efflux assays to characterize T cells that have the capacity to resist chemotherapy and identify and characterize a distinct and prevalent subset of human memory CD8+ T cells that express high levels of CD161 and are enriched in patients undergoing chemotherapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Billerbeck E, Kang YH, Walker L, Lockstone H, Grafmueller S, Fleming V, Flint J, Willberg CB, Bengsch B, Seigel B, et al. Analysis of CD161 expression on human CD8+ T cells defines a distinct functional subset with tissue-homing properties. Proc Natl Acad Sci U S A. 2010;107:3006–3011. doi: 10.1073/pnas.0914839107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **37.Dusseaux M, Martin E, Serriari N, Peguillet I, Premel V, Louis D, Milder M, Le Bourhis L, Soudais C, Treiner E, et al. Human MAIT cells are xenobiotic resistant, tissue-targeted, CD161hi IL-17 secreting T cells. Blood. 2010 doi: 10.1182/blood-2010-08-303339. This study confirms the quiescense and chemotherapy resistance of CD161hi CD8+ memory T cells and demonstrates that this subset of cells are identical to previously described mucosal associated invariant T (MAIT) cells that are transcriptionally programmed for IL-17 production and largely specific for bacterial ligands presented by the class Ib MR-1 molecule. The authors speculate that the resistance mechansms may be necessary this subset of gut-homing T cells to survive xenobiotics secreted by gut bacteria. [DOI] [PubMed] [Google Scholar]
- 38.Cui W, Kaech SM. Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol Rev. 2010;236:151–166. doi: 10.1111/j.1600-065X.2010.00926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hammarlund E, Lewis MW, Hansen SG, Strelow LI, Nelson JA, Sexton GJ, Hanifin JM, Slifka MK. Duration of antiviral immunity after smallpox vaccination. Nat Med. 2003;9:1131–1137. doi: 10.1038/nm917. [DOI] [PubMed] [Google Scholar]
- 40.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 41.Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, Bollard CM, Liu H, Wu MF, Rochester RJ, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115:925–935. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *42.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. This study is the first in primates to analyze whether the fate of clonally derived effector CD8+ T cells is determined by the phenotype of the precursor memory cell from which they are derived. The use of gene marking enabled detailed and prolonged tracking of survival and migration, and demonstrated that effector cells derived from central rather than effector memory cells could survive long term and revert to functional memory cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berger C, Berger M, Anderson D, Riddell SR. A non-human primate model for analysis of safety, persistence, and function of adoptively transferred T cells. J Med Primatol. 2010 doi: 10.1111/j.1600-0684.2010.00451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X, Berger C, Wong CW, Forman SJ, Riddell SR, Jensen MC. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood. 2010 doi: 10.1182/blood-2010-10-310599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teague RM, Greenberg PD, Fowler C, Huang MZ, Tan X, Morimoto J, Dossett ML, Huseby ES, Ohlen C. Peripheral CD8+ T cell tolerance to self-proteins is regulated proximally at the T cell receptor. Immunity. 2008;28:662–674. doi: 10.1016/j.immuni.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *46.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. This clinical study compared in the same patient the in vivo survival of genetically redirected antitumor T cells derived from polyclonal unselected cells or from virus-specific memory cells, and demonstrated improved persistence and antiumor activity of gene-modified memory cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, Affaticati P, Gilfillan S, Lantz O. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature. 2003;422:164–169. doi: 10.1038/nature01433. [DOI] [PubMed] [Google Scholar]
- 48.Le Bourhis L, Martin E, Peguillet I, Guihot A, Froux N, Core M, Levy E, Dusseaux M, Meyssonnier V, Premel V, et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol. 2010;11:701–708. doi: 10.1038/ni.1890. [DOI] [PubMed] [Google Scholar]
- 49.Shlomchik WD. Graft-versus-host disease. Nat Rev Immunol. 2007;7:340–352. doi: 10.1038/nri2000. [DOI] [PubMed] [Google Scholar]
- 50.Stemberger C, Huster KM, Koffler M, Anderl F, Schiemann M, Wagner H, Busch DH. A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity. 2007;27:985–997. doi: 10.1016/j.immuni.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 51.Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, Sanchez-Perez L, Muranski P, Kern SJ, Logun C, et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A. 2009;106:17469–17474. doi: 10.1073/pnas.0907448106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, Klebanoff CA, Johnson LA, Kerkar SP, Yang S, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2010 doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **53.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15:808–813. doi: 10.1038/nm.1982. The authors examine whether altering signaling through the Wnt/beta-catenin pathway, which promotes hematopoietic stem cell renewal can be used to derive T cells from the naïve T cell pool that have enhanced memory characteristics. They demonstrate that chemical activation of Wnt signaling during in vitro priming alters cell phenotype and arrests effector differentiation resulting in T cells that are more effective in adoptive transfer models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]