

Abstract
Ischemic tolerance can be induced by numerous preconditioning stimuli, including various Toll-like receptor (TLR) ligands. We have shown previously that systemic administration of the TLR4 ligand LPS or the TLR9 ligand unmethylated CpG oligodeoxynucleotide before transient brain ischemia in mice confers substantial protection against ischemic damage. To elucidate the molecular mechanisms of preconditioning, we compared brain genomic profiles in response to preconditioning with these TLR ligands and with preconditioning via exposure to brief ischemia. We found that exposure to the TLR ligands and brief ischemia induced genomic changes in the brain characteristic of a TLR pathway-mediated response. Interestingly, all three preconditioning stimuli resulted in a reprogrammed response to stroke injury that converged on a shared subset of 13 genes not evident in the genomic profile from brains that were subjected to stroke without prior preconditioning. Analysis of the promoter region of these shared genes showed sequences required for interferon regulatory factor (IRF)-mediated transcription. The importance of this IRF gene network was tested using mice deficient in IRF3 or IRF7. Our data show that both transcription factors are required for TLR-mediated preconditioning and neuroprotection. These studies are the first to discover a convergent mechanism of neuroprotection induced by preconditioning—one that potentially results in reprogramming of the TLR-mediated response to stroke and requires the presence of IRF3 and IRF7.
Introduction
Toll-like receptors (TLRs) are sentinels of the innate immune response recognizing both endogenous damage-associated molecular patterns (DAMPs) and exogenous pathogen-associated molecular patterns (PAMPs). Activation of these receptors by cognate ligands can induce NFκB-dependent proinflammatory cytokines and/or interferon regulatory factor (i.e., IRF1/IRF3/IRF7/IRF8) activation of antiviral type I interferons (IFNs). Whereas TLR signaling in response to PAMPs has been studied extensively, the recognition of TLR activation via DAMPs has come to light more recently. One such example is the effect of endogenous DAMPs in a model of brain focal ischemia wherein mice deficient in TLR2 or TLR4 showed significantly less brain damage compared with their wild-type counterparts, highlighting a deleterious role for these receptors in ischemic injury (Cao et al., 2007; Lehnardt et al., 2007; Tang et al., 2007). In humans, TLR2 and TLR4 expression was recently shown to be associated with poor functional outcome in human ischemic stroke patients and to correlate with higher serum levels of proinflammatory cytokines (Brea et al., 2011).
TLRs play a critical role in initiating the inflammatory response to “danger,” and they also play a vital role in autoregulation of these responses. A hallmark of the TLR family is the ability to induce tolerance to subsequent TLR signaling by priming initially with a small amount of the ligand. The priming event leads to suppression and redirection of the subsequent response to stimulation with a secondary TLR ligand (Broad et al., 2007). TLRs are known to play a role in ischemic injury, which suggests that TLR stimulation in the setting of ischemia may be subject to prior tolerance via primary stimulation with a TLR ligand. Indeed, we and others have shown that exogenous administration of ligands for at least three TLRs (TLR2, TLR4, and TLR9) provides protection against a subsequent ischemic event when administered in small doses before stroke (Tasaki et al., 1997; Rosenzweig et al., 2004; Hickey et al., 2007; Hua et al., 2008; Stevens et al., 2008).
Protection against an ischemic injury via administration of a small dose of an otherwise harmful stimulus is not restricted to exogenous TLR ligands. Preconditioning stimuli can also include brief ischemia and mild seizures (for review, see Dirnagl et al., 2009), which are physiological events that could presumably result in the exposure of endogenous TLR ligands (e.g., heat shock proteins, fibronectin). In the case of brief ischemia, Pradillo et al. (2009) showed that neuroprotection was attenuated in TLR4-deficient mice, implicating a role for endogenous TLR ligands in protection against ischemic injury.
One important question that we and others have raised is whether distinct preconditioning stimuli induce a conserved neuroprotective response (da Silva et al., 2004; Karikó et al., 2004; Sergeev et al., 2004; Stenzel-Poore et al., 2007). We previously reported that LPS preconditioning alters the genomic response to ischemia by reprogramming stroke-induced TLR signaling and that such reprogramming occurs through preferential activation of the IRF3 pathway (Marsh et al., 2009a). Here we postulate that other preconditioning stimuli may cause a similar bias toward IRF3 activation. We tested this hypothesis by comparing the genomic profiles of mice preconditioned by the following treatments: TLR4 agonist (LPS), TLR9 agonist (CpG), and brief ischemia. We found that each of the three preconditioning stimuli resulted in TLR signaling in the brain prior to stroke, and each induced a similar IRF-associated response following stroke. These data suggest that the neuroprotective mechanism is shared among these stimuli and may be, on a general level, independent of the nature of the stimulus. In addition, we provide the first evidence that there is a dual requirement for both IRF3 and IRF7 transcription factors in TLR-induced neuroprotection against ischemic injury.
Materials and Methods
Mice.
C57BL/6 mice (male, 8–12 weeks) were purchased from Jackson Laboratories. IRF7−/− mice were provided by Dr. Ian Rifkin (Boston University School of Medicine, Boston, MA). IRF3−/− mice were obtained from RIKEN BioResource Center. Both knock-out strains were back-crossed at least eight generations onto C57BL/6. All mice were housed in an American Association for Laboratory Animal Care-approved facility. Procedures were conducted according to Oregon Health & Science University, Institutional Animal Care and Use Committee, and National Institutes of Health guidelines.
TLR ligands.
TLR4 ligand LPS (Escherichia coli serotype 0111:B4; Cat# L2630, purified by phenol extraction, protein content <3%) was obtained from Sigma-Aldrich. The TLR9 ligand CpG oligodeoxynucleotide (ODN) 1826 (tccatgacgttcctgacgtt), a mouse-specific phosphothioate unmethylated CpG ODN ligand for TLR9, was obtained from InvivoGen. InvivoGen has confirmed the specificity of ODN 1826 for mouse TLR9 by testing against cells transfected with the other TLR family members. In addition, endotoxin levels were determined to be negligible (<0.125 EU/mg). Systemic administration of TLR ligands was via either intraperitoneal injection or subcutaneous injection as noted. We have found that both routes of administration provide significant protection. The TLR ligands were titrated for the optimal neuroprotective dose in the particular strain of mouse being tested to control for variation between product lots. We found consistent gene regulation between different neuroprotective doses of the individual TLR ligands.
Middle cerebral artery occlusion.
Mice were anesthetized and subjected to middle cerebral artery occlusion (MCAO) using the monofilament suture method described previously (Stevens et al., 2002). Briefly, a silicone-coated 7-0 monofilament nylon surgical suture was threaded through the external carotid artery to the internal carotid artery to block the middle cerebral artery; it was maintained intraluminally for either 12 min for ischemic preconditioning or 45 min for injurious ischemia. The suture was then removed to restore blood flow. Mice undergoing ischemic preconditioning followed by injurious ischemia were rested in their home cage for 72 h before receiving the second MCAO (45 min). Cerebral blood flow (CBF) was monitored throughout surgery by laser Doppler flowmetry. The mean CBF value between groups was consistent for each experiment. In addition, any mouse that did not maintain a CBF during occlusion of <20% of baseline was excluded from the study. Body temperature was monitored and maintained at 37°C with a thermostat-controlled heating pad.
Experimental design for gene expression studies.
Groups of C57BL/6 mice (n = 4/treatment/time) received either preconditioning alone, preconditioning plus injurious ischemia (45 min MCAO), or injurious ischemia alone. Preconditioning paradigms included LPS (0.2 mg/kg, i.p.), CpG (0.8 mg/kg, i.p.), saline (i.p.), or short-term MCAO (12 min). For groups receiving preconditioning alone, mice were killed and tissue was collected at 3, 24, and 72 h after preconditioning. In groups receiving preconditioning plus injurious ischemia, MCAO was performed 72 h following the preconditioning stimulus, and mice were killed for tissue collection at either 3 or 24 h after occlusion. Groups receiving injurious ischemia alone received 45 min MCAO with no prior pretreatment, and tissue was collected at 3 and 24 h after occlusion. Tissue was also collected from six untreated mice to include as a baseline control group. When the mice were killed, they were all anesthetized and then perfused with heparinized saline. Under RNase-free conditions, a 1 mm section was removed (4 mm from rostral end) to determine the area of infarct based on 2,3,5-triphenyltetrazolium chloride (TTC) staining. The ipsilateral cortex region from the frontal 4 mm was isolated and snap frozen in liquid nitrogen as described previously (Stenzel-Poore et al., 2003).
Infarct evaluation.
For microarray experiments, a 1 mm coronal midsection was placed in 1.5% TTC in 0.9% PBS and stained at 37°C for 15 min to visualize infarct and confirm preconditioning-induced protection. For all other experiments, the brain was sliced into 7 × 1 mm coronal sections before TTC staining, and the volume of infarct was determined by summing of the area of infarct from individual slices. The infarct size for each image was determined using NIH image analyses. To account for edema within the infarct region, we computed infarct area indirectly as follows: 100 × (contralateral hemisphere area − area of live tissue on ipsilateral hemisphere)/(contralateral hemisphere area) (Swanson et al., 1990). Table 1 shows the infarct sizes 24 h after stroke for each of the groups, confirming that the preconditioning paradigms reflect our previously published results (Stenzel-Poore et al., 2003; Rosenzweig et al., 2007; Stevens et al., 2008).
Table 1.
Percentage of tissue damage 24 h following 45 min MCAO
| Pretreatment | Percentage infarct (mean ± SEM) |
|---|---|
| None | 46 ± 3 |
| Saline | 47 ± 2 |
| CpG (0.8 mg/kg) | 15 ± 2 |
| LPS (0.2 mg/kg) | 22 ± 5 |
| Ischemic (12 min) | 10 ± 4 |
RNA isolation.
Total RNA was isolated using the Qiagen RNeasy Lipid Mini Kit (Qiagen). RNA from individual mice was hybridized to single arrays as described below.
GeneChip expression analyses.
Microarray assays were performed in the Affymetrix Microarray Core of the Oregon Health & Science University Gene Microarray Shared Resource. RNA samples were labeled using the NuGEN Ovation Biotin RNA Amplification and Labeling System_V1. Hybridization was performed as described in the Affymetrix technical manual with modification as recommended for the Ovation labeling protocol (NuGEN Technologies). Labeled cRNA target was quality-checked based on yield and size distribution. Quality-tested samples were hybridized to the MOE430 2.0 array. The array image was processed with Affymetrix GeneChip Operating Software (GCOS). Data were normalized using the Robust Multichip Average method (RMA) (Irizarry et al., 2003). The normalized data were then analyzed using a two-way ANOVA model for each gene, using conditions and time as groups. Post hoc comparisons were made using the untreated mice as a control group. We used the method of Hochberg and Benjamini (1990) to adjust p values for multiple comparisons. Genes were considered significantly regulated if the adjusted p value was <0.01 and the fold change in regulation was greater than or equal to two compared with untreated control mice.
As an internal control, to approximate the number of false-positive/-negative identifications that we may expect to encounter from our analyses, we compared the saline-treated mice with the untreated control group, both before and following injurious ischemia. Before stroke, the vehicle-treated mice (saline injected) would be anticipated to be similar to the untreated mice, and in fact, there were no statistically regulated genes between the two groups at any of the three time points. Following stroke, the saline-pretreated mice should be equivalent to the stroke-alone mice; however, we found only a 65% overlap between the two groups, with the remainder identified as regulated in only one of the stroke paradigms. To increase our stringency in identifying gene regulation associated only with the preconditioning paradigms, we used the union of regulated transcripts identified in the saline and stroke-alone groups to represent the gene regulation induced by stroke (i.e., if a gene was regulated under either of the stroke paradigms, we considered it regulated following stroke).
We have previously reported on the individual analysis of the LPS genomic dataset (Marsh et al., 2009a) and the CpG genomic dataset (Marsh et al., 2009b); however, in these studies, the data have been renormalized to include all preconditioning paradigms. All three preconditioning studies were performed during the same timeframe and microarray hybridizations were performed based on a randomization schedule of the entire dataset.
Kyoto Encyclopedia of Genes and Genomes pathway analysis.
For Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, Affymetrix CEL files were uploaded into GeneSifter (www.genesifter.net) and normalized using RMA. ANOVA was performed for each preconditioning paradigm [LPS, CpG, and ischemic preconditioning (IP)] using the three preconditioning time points (3, 24, and 72 h) and the untreated group. The gene lists for KEGG pathway term comparisons were generated using a twofold threshold compared with untreated and an adjusted p value <0.05.
Transcriptional regulatory network analysis.
Using the web-based program Promoter Analysis and Interaction Network Toolset (PAINT) version 3.5 (http://www.dbi.tju.edu/dbi/tools/paint/), we examined the predicted regulatory elements associated with the unique gene regulation identified by microarray (Vadigepalli et al., 2003). In brief, using PAINT we obtained the 5000 bp upstream sequence for the transcripts represented on the MOE430 Affymetrix gene chip (33,635 transcripts). PAINT identified putative transcription factor-binding sequences [transcriptional regulatory elements (TREs)] in these upstream sequences using the Match algorithm in TRANSFAC PRO database version 10.4. Match Filter was set to Minimize False Positives, TRE core similarity threshold was set at 1.00, and TREs found on complementary strand were included. These Affymetrix transcripts were used as our reference comparison group. In addition, we identified the predicted TREs in our experimental groups using the same criteria. The statistical component of PAINT [false discovery rate (FDR)-adjusted p value set at ≤0.1, which is the most stringent analysis within the software] was used to determine the overrepresented TREs associated with our experimental gene cluster compared with the reference group.
Quantitative real-time PCR.
Total RNA isolated from brain cortex was reverse transcribed using an Omniscript Reverse Transcription kit (Qiagen). Quantitative PCR was performed using the TaqMan Gene Expression Assays (Applied Biosystems) on an ABI-prism 7700. Results were normalized to β-actin expression and analyzed relative to appropriate controls. The relative quantification of the gene of interest was determined using the comparative CT method (2−ΔΔCt).
Results
Preconditioning results in gene regulation in the brain associated with TLR and cytokine signaling pathways
We tested three distinct preconditioning stimuli, LPS, CpG, and brief ischemia, to determine whether the brain's response to subsequent injury exhibited common or distinct pathways of ischemic neuroprotection. The three preconditioning stimuli represent distinct molecular processes of induction given that LPS and CpG act as exogenous activators of TLR4 and TLR9, respectively, whereas brief ischemia may have multiple targets, one of which appears to be endogenous activation of TLR4. We found that soon after treatment (3 h), all three stimuli induced significant gene regulation in the brain. The greatest overlap in expression profiles occurred between the TLR ligands, CpG and LPS. Nearly 98% of the genes regulated by CpG were consistent with the LPS response at 3 h. However, LPS shared only 30% overlap with CpG. This finding may be due to the fact that TLR4 signals via two different adaptor molecules, MyD88 and TRIF, each of which induces independent gene regulation (for review, see Akira and Takeda, 2004). TLR9 signals exclusively through MyD88; thus, the 98% overlap of the genes induced by CpG with genes induced by LPS may represent the shared MyD88 pathway of gene regulation. Ischemic preconditioning shared <21% with either of the TLR preconditioning stimuli at any of the preconditioning time points (Fig. 1A). Although IP does not share a large overlap of gene regulation, comparison of the KEGG pathways (Kanehisa and Goto, 2000; Kanehisa et al., 2006, 2010) associated with the genes regulated in each paradigm before stroke indicated that three pathways were significantly over-represented (Z > 2) in all preconditioning paradigms: the TLR receptor signaling pathway, cytokine–cytokine receptor interaction pathway, and apoptosis (Fig. 1B). The identification of the TLR receptor-signaling pathway supports the concept of a TLR-signaling event in the brain instigated by all three preconditioning stimuli.
Figure 1.

Common signaling pathways identified during preconditioning. A, Venn diagrams comparing the regulated* genes in each of the preconditioning paradigms. B, Z score analysis of KEGG pathway terms associated with genes regulated* in the brain following preconditioning (3, 24, and 72 h). Z scores >2 were considered significant (black line). *p < 0.01; at least twofold regulation compared with nontreated mice.
Preconditioning alters the genomic response to stroke
We have previously shown that both IP and LPS preconditioning result in a reprogrammed genomic response to stroke (Stenzel-Poore et al., 2003; Marsh et al., 2009a). To compare these reprogrammed responses and to determine whether CpG preconditioning resulted in a similar reprogramming of the brain's genomic response to stroke, we examined the unique gene sets induced in response to ischemic injury in the setting of preconditioning that were not evident in stroke alone. Examination of the 3 h following the stroke time point revealed that ∼12% of the genomic response to ischemia in each preconditioning paradigm was not shared with stroke alone (Fig. 2). These early differences are unlikely due to extent of injury because at 3 h after stroke, little if any damage or cellular infiltration is reported in the literature. The induction of new gene regulation in response to stroke supports the idea that preconditioning altered the endogenous response to injury, potentially via redirection of TLR signaling.
Figure 2.
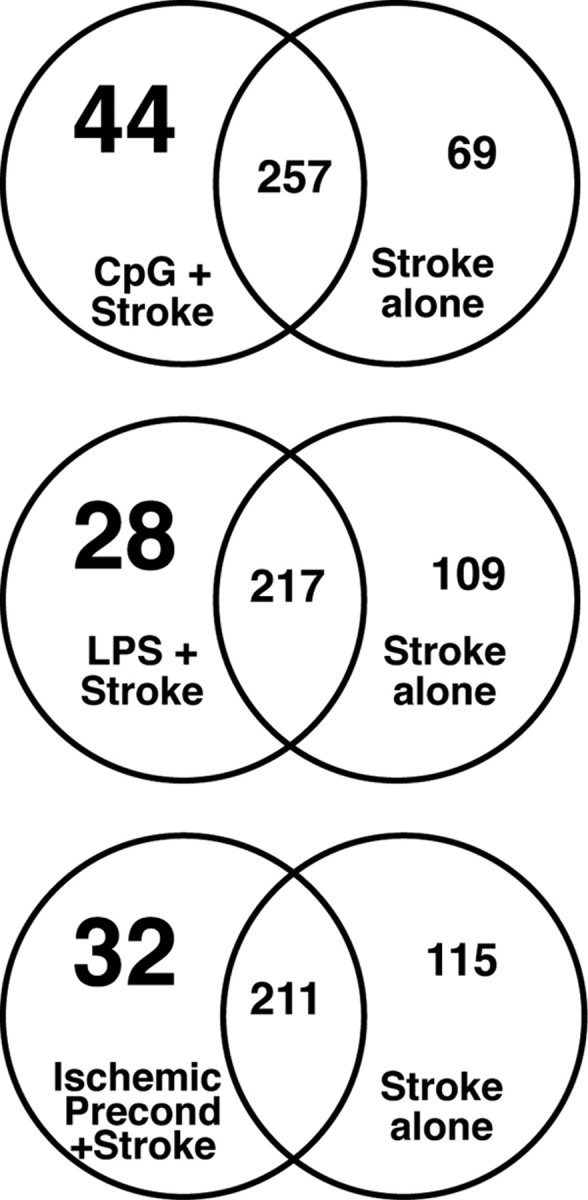
Preconditioning induces unique gene regulation in response to stroke. Venn diagrams of genes regulated at 3 h in response to stroke. Comparisons are made between the preconditioning stimulus + stroke and stroke alone. Numbers in large type represent the reprogrammed response. Stroke alone includes all genes regulated in either saline + stroke or nontreated stroke mice.
Preconditioned mice share a common genomic response to stroke
To determine whether the reprogrammed response to stroke induced in the preconditioned mice results in shared gene regulation, we compared the genes regulated at 3 and 24 h after stroke in each of the preconditioned paradigms that were not evident in stroke alone (mice given no prior preconditioning). We identified three genes at 3 h after stroke (Fig. 3, left panel) that were induced in all three preconditioning paradigms: cytohesion 1 interacting protein (Cytip), scavenger receptor class A (Scara5), and interferon-activatable protein (AI607873). At 24 h after stroke, we identified 13 genes that were upregulated in all three preconditioning paradigms (Fig. 3, right panel; Table 2). The majority of the 13 genes were also induced before stroke in response to preconditioning with LPS and CpG either at 3 or 24 h, but not in response to IP. However, all of the common genes identified at both time points following stroke were at basal levels immediately before stroke (data not shown) and, thus, are induced as new gene regulation in response to ischemic injury. These genes may represent a shared protective response to injury, which could indicate that preconditioning elicits a common mechanism of protection regardless of the nature of the preconditioning stimulus.
Figure 3.

A core set of commonly regulated genes induced following stroke in preconditioned mice. Venn diagrams comparing the reprogrammed genes in each of the preconditioning paradigms at 3 and 24 h after stroke. Reprogrammed genes are those regulated only with preconditioning + stroke and not seen in either of the stroke-alone paradigms.
Table 2.
Thirteen genes regulated in common at 24 h after stroke in mice preconditioned with LPS, CpG, or brief ischemia
| Symbol | Title | LPS + stroke 24 h |
CpG + stroke 24 h |
IP + stroke 24 h |
Strokea 24 h |
||||
|---|---|---|---|---|---|---|---|---|---|
| Fold changeb | FDR-adjusted p value | Fold change | FDR-adjusted p value | Fold change | FDR-adjusted p value | Fold change | FDR-adjusted p value | ||
| Fcgr1 | Fc receptor, IgG, high affinity I | 3.06 | 2.8 × 10−9 | 3.03 | 2.3 × 10−9 | 2.36 | 1.3 × 10−5 | 1.85/1.77 | 0.0004/0.001 |
| HGF | Hepatocyte growth factor | 2.56 | 2.1 × 10−9 | 3.04 | 6.1 × 10−12 | 2.30 | 6.0 × 10−7 | 1.49/1.85 | 0.007/3.2 × 10−5 |
| Ifit1 | IFN-induced protein with tetratricopeptide repeats 1 | 3.04 | 0.0001 | 2.61 | 0.0009 | 2.85 | 0.001 | 1.39/1.28 | 0.32/0.45 |
| Ifit3 | IFN-induced protein with tetratricopeptide repeats 3 | 2.07 | 2.1 × 10−5 | 2.04 | 2.4 × 10−5 | 2.04 | 0.0001 | 1.54/1.40 | 0.01/0.05 |
| Oasl2 | 2′-5′Oligoadenylate synthetase-like 2 | 2.47 | 0.0009 | 2.59 | 0.0004 | 2.55 | 0.002 | 1.24/1.19 | 0.51/0.58 |
| Plscr2 | Phospholipid scramblase 2 | 2.56 | 3.9 × 10−7 | 2.93 | 9.4 × 10−9 | 2.11 | 0.0001 | 1.32/1.45 | 0.15/0.04 |
| Pycard | PYD and CARD domain containing | 2.60 | 8.9 × 10−9 | 2.57 | 8.3 × 10−9 | 2.27 | 3.6 × 10−6 | 1.69/1.68 | 0.0007/0.0009 |
| Pyhin1 | Pyrin and HIN domain family, 1 | 2.66 | 3.9 × 10−6 | 2.52 | 9.4 × 10−6 | 2.17 | 0.0008 | 1.44/1.85 | 0.09/0.003 |
| Viperin | Radical S-adenosyl methionine domain containing 2 | 3.82 | 4.2 × 10−6 | 3.02 | 0.0001 | 2.67 | 0.002 | 1.84/1.75 | 0.03/0.05 |
| Rtp4 | Receptor transporter protein 4 | 2.96 | 1.3 × 10−7 | 2.66 | 1.0 × 10−6 | 2.48 | 3.9 × 10−5 | 1.56/1.52 | 0.02/0.03 |
| Trim30 | Tripartite motif protein 30 | 2.50 | 2.5 × 10−6 | 2.43 | 3.9 × 10−6 | 2.09 | 0.0005 | 1.50/1.53 | 0.03/0.02 |
| Usp18 | Ubiquitin specific peptidase 18 | 2.93 | 0.0001 | 2.68 | 0.0003 | 3.40 | 8.7 × 10−5 | 1.35/1.16 | 0.34/0.6 |
| Zbp1 | Z-DNA binding protein 1 | 2.20 | 1.0 × 10−5 | 2.79 | 2.0 × 10−8 | 2.07 | 0.0002 | 1.57/1.66 | 0.01/0.003 |
aNontreated stroke alone/saline-treated stroke.
bFold change compared with untreated mice. Genes are considered significantly regulated if fold change > 2 and FDR-adjusted p < 0.01.
Type I interferon regulatory elements overrepresented in the shared response to stroke in preconditioned mice
To determine whether a common regulatory event may be associated with these shared genes, we used PAINT version 3.5 (Vadigepalli et al., 2003) to identify TREs overrepresented in the promoter region of the commonly regulated genes (Fig. 3, center overlap). Upstream sequence was identified for each gene using the ENSEMBLE database and Transfac Pro was used to identify predicted TREs. The TREs associated with these genes were compared with those identified in a reference cluster consisting of ∼33,000 transcripts from the Affymetrix MOE430 gene chip to determine which TREs were significantly overrepresented. There were no significantly overrepresented TREs associated with the three commonly regulated genes at 3 h after stroke. However, using the 13 common genes found 24 h after stroke, PAINT identified five TREs as overrepresented: ISRE, IRF8, IRF, Freac3, and IRF7 (Table 3). Four of the five identified TREs are associated with type I interferon signaling. A network depiction of interactions between the overrepresented TREs and the genes shared among the preconditioning paradigms is displayed in Figure 4. Of the 13 genes, only HGF (hepatocyte growth factor) did not have a type I interferon-associated TRE. This analysis suggests that each of the three preconditioning paradigms reprograms the response to stroke, driving a type I interferon response that is not evident in stroke alone.
Table 3.
TREs identified as significantly overrepresented in genes commonly induced 24 h following stroke in preconditioned mice
| TRE | Adjusted p values for overrepresentation comparison |
|---|---|
| ISRE/V$ISRE_01 | < 0.001 |
| IRF8/V$ICSBP_Q6 | 0.002 |
| IRF/V$IRF_Q6 | 0.014 |
| Freac-3/V$FREAC3_01 | 0.037 |
| IRF7/V$IRF7_01 | 0.059 |
Figure 4.

A shared IRF-driven reprogrammed response to stroke in preconditioned mice. PAINT-generated Hypothesis Gene–TRE network showing the relationship of identified TREs to the genes common to all preconditioning paradigms 24 h following stroke. Genes are in blue, and TREs are in red. Fcgr1, Fc receptor, IgG, high affinity I; HGF, hepatocyte growth factor; Ifit1, IFN-induced protein with tetratricopeptide repeats 1; Ifit3, IFN-induced protein with tetratricopeptide repeats 3; Oasl2, 2′-5′oligoadenylate synthetase-like 2; Plscr2, phospholipid scramblase 2; Pycard, PYD and CARD domain containing; Pyhin1, pyrin and HIN domain family, 1; Rtp4, receptor transporter protein 4; Trim30, tripartite motif protein 30; Usp18, ubiquitin specific peptidase 18; Viperin, radical S-adenosyl methionine domain containing 2; Zbp1, Z-DNA binding protein 1.
Preconditioning significantly increases the type I interferon regulatory factor IRF7 following stroke
To determine whether a specific IRF was involved in the shared protective phenotype associated with preconditioning, we first examined the genomic regulation of the known IRFs. We found that IRF7 was significantly induced compared with nontreated mice (p < 0.01) 24 h after stroke in all preconditioning paradigms, while showing no induction following stroke alone (Table 4). Although IRF1 and IRF8 were significantly regulated following stroke in preconditioned mice, they were also induced following stroke in non-preconditioned mice (data not shown), suggesting that expression of these regulators does not distinguish between the protective and injurious genomic response. Each of the other IRFs, including IRF3, showed no differential regulation compared with untreated mice. To confirm the IRF7 induction in the preconditioned mice, we used qRT-PCR to examine message levels of IRF7 in the brain following stroke. We found that IRF7 mRNA was significantly increased 24 h following stroke in all three preconditioning paradigms compared with stroke alone (Fig. 5). Thus, the induction of IRF7 may likely play a key role in mediating the shared protective response to injury in all three preconditioning paradigms.
Table 4.
IRF7 significantly induced in the brain 24 h after stroke only in preconditioned mice
| Fold changea | FDR-adjusted p value | |
|---|---|---|
| LPS + stroke 24 h | 1.69 | 0.0011 |
| CpG + stroke 24 h | 1.77 | 0.0003 |
| IP + stroke 24 h | 1.79 | 0.0012 |
| Stroke 24 hb | 1.11/1.15 | 0.6/0.4 |
aFold change compared with untreated mice.
bNontreated stroke alone/saline-treated stroke.
Figure 5.

IRF7 is upregulated following MCAO in preconditioned mouse brain. IRF7 gene expression is significantly induced 24 h following MCAO in mice preconditioned with LPS, CpG, or IP. Fold change is represented compared with IRF7 expression in non-preconditioned mice 24 h following MCAO (ischemia alone). One-way ANOVA, Bonferroni post hoc, **p < 0.01, ***p < 0.001 compared with ischemia alone.
Loss of IRF3 or IRF7 abrogates preconditioning-induced neuroprotection
The fact that IRF7 induction was common to each preconditioning stimulus, taken together with our previous report that LPS preconditioning requires IRF3 (Marsh et al., 2009a), led us to postulate that the preconditioning paradigms shared a common dependence on IRF transcription factors. To determine whether IRF3 and/or IRF7 were required for neuroprotection induced by the three preconditioning paradigms, we preconditioned mice deficient in either IRF3 or IRF7. IRF7−/− or wild-type mice were preconditioned with LPS (1 mg/kg, s.c.), CpG (1.6 mg/kg, s.c.), or IP (12 min MCAO) 72 h before 45 min MCAO. We had previously established that IRF3−/− mice failed to be protected with LPS (Marsh et al., 2009a); thus, we tested whether preconditioning with CpG or IP induced neuroprotection in IRF3−/− mice. As expected, each preconditioning stimulus significantly reduced infarct size in the control wild-type mice (Figs. 6A,B, 7). In contrast, IRF7−/− mice showed no reduction in infarct size when treated with either LPS or CpG compared with saline-treated mice (36.88 ± 4.3%, 39.76 ± 3.0%, respectively, vs saline-treated 38.49 ± 1.5%) (Fig. 6A). The IRF3−/− mice also failed to show a reduction in infarct size when preconditioned with CpG (36.32 ± 2.3% vs 36.90 ± 2.5%) (Fig. 6B). These data demonstrate that IRF3 and IRF7 are required for the protective effects of both LPS and CpG preconditioning. In contrast, ischemic preconditioning of IRF3 and IRF7 knock-out mice caused a modest reduction in infarct size (IRF3: 24.06 ± 2.0% vs stroke alone 39.35 ± 1.8%; IRF7: 32.51 ± 3.2% vs stroke alone 46.06 ± 2.6%) (Fig. 7), although the extent of reduction was not as marked as that found in wild-type mice preconditioned with brief ischemia. The reduction in infarct size observed in the IRF3-deficient and IRF7-deficient mice preconditioned with brief ischemia was 40% and 30%, respectively, which is significantly attenuated when compared with the extent of protection in the wild-type mice (78% reduction in injury) (Fig. 7). These results demonstrate that LPS and CpG preconditioning require the presence of IRF3 and IRF7 for neuroprotection, whereas ischemic preconditioning exhibits partial dependence on these transcription factors for neuroprotection.
Figure 6.

IRF7 and IRF3 are required for TLR4 and TLR9 preconditioning-induced neuroprotection. A, Wild-type (Wt; n = 8–13) or IRF7 knock-out (KO; n = 8–14) mice were pretreated with LPS (1.0 mg/kg), CpG (1.6 mg/kg), or saline 72 h before 45 min MCAO. B, Wild-type (n = 8–10) or IRF3 knock-out (n = 7–8) mice were pretreated with CpG (1.6 mg/kg) or saline 72 h before 45 min MCAO. Infarct volumes were measure 24 h following surgery using TTC staining. Data shown are group means + SEM; two-way ANOVA, Bonferroni post hoc, ***p < 0.001 compared with saline-treated mice for respective genotype.
Figure 7.

Partial requirement for IRF7 and IRF3 in ischemic preconditioning-induced neuroprotection. Wild-type (Wt; n = 7–8) and IRF7 (n = 9–10) or IRF3 (n = 7–8) knock-out (KO) mice were either preconditioned with a 12 min MCAO 72 h before 45 min MCAO or exposed to 45 min MCAO without preconditioning. Infarct volumes were measure 24 h following surgery using TTC staining. Data shown are group means + SEM; ***p < 0.001 compared with stroke-alone mice for respective genotype; #p < 0.05 and ##p < 0.001 compared with wild-type IP. There were no significant differences between wild-type and IRF3 or IRF7 knock-out mice with MCAO alone.
Discussion
Recent findings that stroke injury is mediated partially through TLR-induced inflammation (Cao et al., 2007; Lehnardt et al., 2007; Tang et al., 2007) have furthered our speculation that preconditioning paradigms share a common TLR-mediated mechanism of protection. We postulated that if the mechanism of preconditioning is similar to the phenomenon of tolerance described for TLR family members, then an initial TLR-signaling event would be evident in the brain following the preconditioning stimulus and a reprogrammed TLR response should be apparent following stroke. We examined the genomic profiles in response to preconditioning to determine whether TLR signaling occurred in the brain parenchyma before the subsequent ischemic injury. Although both LPS and CpG are systemically administered, we found gene changes in the brain as early as 3 h, and in both cases, the regulated genes contained an overrepresentation of genes associated with the TLR-signaling pathway (LPS: Z = 9.98; CpG: Z = 6.98). It is not clear whether LPS and CpG cross the blood–brain barrier to directly signal the brain or whether signaling occurs through the endothelium to release endogenous mediators into the brain parenchyma.
IP also induced an overabundance of TLR-signaling pathway-associated genes (Z = 4.36) before stroke. This suggests that, similar to CpG and LPS, IP induces TLR signaling in the brain presumably via potential endogenous TLR ligands such as heat shock proteins, which are induced in response to IP [Hsp70-1 (45-fold), Hsp70-3 (38-fold), Hsp73 (6-fold), Hsp40 (3-fold), Hsp25 (4-fold), Hsp110 (2-fold)]. The importance of TLR signaling to IP has been previously reported whereby mice deficient in TLR4 showed less neuroprotection from IP in a model of brain focal ischemia (Pradillo et al., 2009). Interestingly, the protective phenotype was not completely abrogated, suggesting that although TLR4 signaling played a role in neuroprotection, additional mechanisms of protection are also initiated with IP. Our genomic data support a multicomponent effect associated with IP through the large number of genes induced before stroke that are unique to IP (Fig. 1A). In addition, pathway analysis identified several pathways significantly over-represented (Z ≥ 2) in IP that were not apparent in LPS or CpG preconditioning, including ECM–receptor interaction pathways and complement and coagulation cascades (Fig. 1B).
Our analysis following stroke indicates that all three preconditioning paradigms reprogrammed the response to the ischemic injury, leading to new gene regulation not induced in non-preconditioned mice. This reprogramming of the response to stroke resulted in a core set of 13 genes shared among all three preconditioning paradigms at 24 h. In line with our postulate that these preconditioning stimuli share a common mechanism of TLR reprogramming, the transcriptional analysis of these 13 genes identified type I interferon regulatory sequences (ISRE, IRF7, IRF8, and IRF) as overrepresented transcriptional regulatory elements associated with these genes. These regulatory factors were not identified in the three genes shared at 3 h after stroke; however, one of these genes codes an interferon-activatable protein, further supporting an IRF-mediated response to stroke shared between the preconditioning paradigms. This is consistent with our previously published data showing that both LPS and CpG preconditioning result in a reprogrammed signaling event in response to stroke that culminates in type I interferon-induced gene regulation (Marsh et al., 2009a, 2009b). The appearance of a consensus set of genes, whose transcriptional regulation is dominated by IFN regulatory elements, parallels the process seen in TLR tolerance, which has been shown to be linked to TRIF–IRF-mediated gene regulation (Biswas et al., 2007; Broad et al., 2007).
IRF3 and IRF7 are key modulators of type I interferon gene regulation and have been shown to induce both redundant as well as distinct transcriptional responses (Sato et al., 2000). IRF3 is constitutively expressed (Sato et al., 2000), whereas IRF7 is induced via both IFN-dependent (Sato et al., 1998) and IFN-independent mechanisms (Ning et al., 2005). We found that whereas IRF3 was not regulated at the transcriptional level in our system, IRF7 mRNA was significantly increased in the brains of preconditioned mice following stroke. Mice deficient in IRF3 or IRF7 showed complete abrogation of the protective effect of LPS and CpG preconditioning, whereas IP showed an attenuation of the protective effect. The failure to completely reverse the protective effect of IP in these mice implies that IP, unlike TLR4 and TLR9 preconditioning, is only partially dependent on IRF signaling. These findings support the notion that CpG, LPS, and IP mediate neuroprotection through a shared IRF3/IRF7-dependent mechanism. This mechanism has a dominant role in protection induced by CpG and LPS but only serves as a component of the protective cascade induced by IP. In addition, the dependence on IRF3 further parallels the mechanism of TLR tolerance in which IRF3-deficient mice show a significant reduction in tolerance-induced suppression of proinflammatory cytokines (Biswas et al., 2007).
Our data showing a loss of preconditioning in both IRF3-deficient and IRF7-deficient mice suggest that both IRF3 and IRF7 are required for induction of neuroprotection and that the absence of either transcription factor is sufficient to block the protective affect. This dual requirement reflects the complicated interplay between these two transcription factors, which have been shown to form heterodimers, regulate each other, and induce synergistic gene regulation (Au et al., 2001; Yang et al., 2004; Ning et al., 2005). In macrophages exposed to Sendai virus, a virus that can signal via TLRs, Ning et al. (2005) showed that IRF7 expression was induced in an IRF3-dependent manner, independent of IFN signaling. They showed that cells expressing a dominant-negative form of IRF3 failed to increase virus-induced IRF7, and that the pathway of activation occurred via an IRF3/IRF7 complex that was not dependent on Janus kinase/signal transducers and activators of transcription signaling (Ning et al., 2005). In our system, we found an increase in IRF7 following stroke only in preconditioned mice. This increase in IRF7 may depend on the availability of IRF3; thus, if IRF7 is required for neuroprotection, mice deficient in either IRF3 or IRF7 would fail to be protected. Of particular interest to our model are findings showing that IRF7 can directly interact with MyD88 to redirect TLR signaling away from NFκB activation toward activation of IFN genes. Honda et al. (2004) postulated that TLRs modulate their signaling of NFκB-regulated and IRF-regulated genes via a transductional–transcriptional processor involving a MyD88/IRAK/TRAF6 complex that can be dynamically redirected by the addition of IRF7. That study supports our model of a reprogrammed TLR response to stroke in preconditioned mice that could ultimately be triggered by increased IRF7 such as we found in preconditioned mice. The subsequent suppression of NFκB signaling would contribute to the neuroprotective response as has been reported for inhibition of NFκB in the setting of stroke (for review, see Ridder and Schwaninger, 2009). In addition, the switch in the signaling cascade would culminate in the induction of IFN genes, as we have reported here. We are currently pursuing the effect of IRF7 expression on NFκB activation in our preconditioning model.
An alternative explanation for the involvement of both IRF3 and IRF7 may be through their synergistic effect on the regulation of IFNβ (Yang et al., 2004). We and others have found that IFNβ is neuroprotective against stroke injury (Liu et al., 2002; Veldhuis et al., 2003a,b; Marsh et al., 2009a); thus, the absence of IRF3 or IRF7 may result in failure to reach threshold levels needed for IFNβ-induced protection. In addition, the protective effect of IFNβ has been linked to the stabilization of the blood–brain barrier (Minagar et al., 2003; Veldhuis et al., 2003b; Kraus et al., 2008), an interface that involves endothelial cells. Recently, Buss et al. (2010) reported that siRNA inhibition of either IRF3 or IRF7 was sufficient to suppress the induction of IFNβ in endothelial cells exposed to Chlamydophila pneumoniae. Thus, the induction of IFNβ by IRF3/IRF7 in endothelial cells may stabilize the blood–brain barrier and reduce ischemic injury following preconditioning.
In summary, we have provided the first evidence of a shared mechanism that underlies tolerance to ischemia. This shared mechanism manifests as genomic reprogramming of TLR signaling following preconditioning with TLR ligands CpG or LPS or brief exposure to ischemia. We have shown that for each of the three preconditioning agents tested, the genomic profile reflects a primary TLR-signaling event followed by a potential redirection of the stroke-induced TLR-signaling cascade toward an IRF-dominant response. The critical importance of IRF-mediated signaling to the neuroprotective process is shown clearly by the fact that mice lacking IRF3 or IRF7 fail to be protected or exhibit much reduced protection when treated with CpG, LPS, or brief ischemia. This implicates IRF3 and IRF7 as key mediators in the induction of an endogenously neuroprotective genomic program. Understanding this and other mechanisms of protection induced via preconditioning will further our ability to initiate and complement these pathways to minimize the damage associated with stroke.
Footnotes
This work was supported by the NIH–NINDS Grants NS050567, NS062381, and NS035965. We thank Dr. Nikola Lessov, Valerie Conrad, Amy Packard, Eric Tobar, and Delfina Homen for excellent technical support and Solange Mongoue-Tchokote and Dr. Motomi Mori of the Oregon Health & Science University Biostatistics Core for statistical analysis of the microarray data. Microarray assays were performed in the Oregon Health & Science University Gene Microarray Shared Resource.
M.P.S.-P. and S.L.S. have financial interest in Neuroprotect, Inc. This potential conflict of interest has been reviewed and managed by Oregon Health & Science University.
References
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Au WC, Yeow WS, Pitha PM. Analysis of functional domains of interferon regulatory factor 7 and its association with IRF-3. Virology. 2001;280:273–282. doi: 10.1006/viro.2000.0782. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Bist P, Dhillon MK, Kajiji T, Del Fresno C, Yamamoto M, Lopez-Collazo E, Akira S, Tergaonkar V. Role for MyD88-independent, TRIF pathway in lipid A/TLR4-induced endotoxin tolerance. J Immunol. 2007;179:4083–4092. doi: 10.4049/jimmunol.179.6.4083. [DOI] [PubMed] [Google Scholar]
- Brea D, Blanco M, Ramos-Cabrer P, Moldes O, Arias S, Perez-Mato M, Leira R, Sobrino T, Castillo J. Toll-like receptors 2 and 4 in ischemic stroke: outcome and therapeutic values. J Cereb Blood Flow Metab. 2011 doi: 10.1038/jcbfm.2010.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad A, Kirby JA, Jones DE. Toll-like receptor interactions: tolerance of MyD88-dependent cytokines but enhancement of MyD88-independent interferon-beta production. Immunology. 2007;120:103–111. doi: 10.1111/j.1365-2567.2006.02485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss C, Opitz B, Hocke AC, Lippmann J, van Laak V, Hippenstiel S, Krüll M, Suttorp N, Eitel J. Essential role of mitochondrial antiviral signaling, IFN regulatory factor (IRF)3, and IRF7 in Chlamydophila pneumoniae-mediated IFN-beta response and control of bacterial replication in human endothelial cells. J Immunol. 2010;184:3072–3078. doi: 10.4049/jimmunol.0902947. [DOI] [PubMed] [Google Scholar]
- Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Commun. 2007;353:509–514. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- da Silva R, Lucchinetti E, Pasch T, Schaub MC, Zaugg M. Ischemic but not pharmacological preconditioning elicits a gene expression profile similar to unprotected myocardium. Physiol Genomics. 2004;20:117–130. doi: 10.1152/physiolgenomics.00166.2004. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8:398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey EJ, You X, Kaimaktchiev V, Stenzel-Poore M, Ungerleider RM. Lipopolysaccharide preconditioning induces robust protection against brain injury resulting from deep hypothermic circulatory arrest. J Thorac Cardiovasc Surg. 2007;133:1588–1596. doi: 10.1016/j.jtcvs.2006.12.056. [DOI] [PubMed] [Google Scholar]
- Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9:811–818. doi: 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh WC, Taniguchi T. Role of a transductional–transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci U S A. 2004;101:15416–15421. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua F, Ma J, Ha T, Kelley J, Williams DL, Kao RL, Kalbfleisch JH, Browder IW, Li C. Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J Neuroimmunol. 2008;199:75–82. doi: 10.1016/j.jneuroim.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karikó K, Weissman D, Welsh FA. Inhibition of Toll-like receptor and cytokine signaling—a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab. 2004;24:1288–1304. doi: 10.1097/01.WCB.0000145666.68576.71. [DOI] [PubMed] [Google Scholar]
- Kraus J, Voigt K, Schuller AM, Scholz M, Kim KS, Schilling M, Schäbitz WR, Oschmann P, Engelhardt B. Interferon-beta stabilizes barrier characteristics of the blood–brain barrier in four different species in vitro. Mult Scler. 2008;14:843–852. doi: 10.1177/1352458508088940. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Lehmann S, Kaul D, Tschimmel K, Hoffmann O, Cho S, Krueger C, Nitsch R, Meisel A, Weber JR. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Liu H, Xin L, Chan BP, Teoh R, Tang BL, Tan YH. Interferon beta administration confers a beneficial outcome in a rabbit model of thromboembolic cerebral ischemia. Neurosci Lett. 2002;327:146–148. doi: 10.1016/s0304-3940(02)00371-3. [DOI] [PubMed] [Google Scholar]
- Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY, Harrington CA, Stenzel-Poore MP. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci. 2009a;29:9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh BJ, Stevens SL, Hunter B, Stenzel-Poore MP. Inflammation and the emerging role of the Toll-like receptor system in acute brain ischemia. Stroke. 2009b;40:S34–S37. doi: 10.1161/STROKEAHA.108.534917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minagar A, Long A, Ma T, Jackson TH, Kelley RE, Ostanin DV, Sasaki M, Warren AC, Jawahar A, Cappell B, Alexander JS. Interferon (IFN)-beta 1a and IFN-beta 1b block IFN-gamma-induced disintegration of endothelial junction integrity and barrier. Endothelium. 2003;10:299–307. doi: 10.1080/10623320390272299. [DOI] [PubMed] [Google Scholar]
- Ning S, Huye LE, Pagano JS. Regulation of the transcriptional activity of the IRF7 promoter by a pathway independent of interferon signaling. J Biol Chem. 2005;280:12262–12270. doi: 10.1074/jbc.M404260200. [DOI] [PubMed] [Google Scholar]
- Pradillo JM, Fernández-López D, García-Yébenes I, Sobrado M, Hurtado O, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. J Neurochem. 2009;109:287–294. doi: 10.1111/j.1471-4159.2009.05972.x. [DOI] [PubMed] [Google Scholar]
- Ridder DA, Schwaninger M. NF-kappaB signaling in cerebral ischemia. Neuroscience. 2009;158:995–1006. doi: 10.1016/j.neuroscience.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning prevents the cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35:2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. [DOI] [PubMed] [Google Scholar]
- Rosenzweig HL, Minami M, Lessov NS, Coste SC, Stevens SL, Henshall DC, Meller R, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning protects against the cytotoxic effects of TNFa after stroke: a novel role for TNFa in LPS-ischemic tolerance. J Cereb Blood Flow Metab. 2007;27:1663–1674. doi: 10.1038/sj.jcbfm.9600464. [DOI] [PubMed] [Google Scholar]
- Sato M, Tanaka N, Hata N, Oda E, Taniguchi T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett. 1998;425:112–116. doi: 10.1016/s0014-5793(98)00210-5. [DOI] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- Sergeev P, da Silva R, Lucchinetti E, Zaugg K, Pasch T, Schaub MC, Zaugg M. Trigger-dependent gene expression profiles in cardiac preconditioning: evidence for distinct genetic programs in ischemic and anesthetic preconditioning. Anesthesiology. 2004;100:474–488. doi: 10.1097/00000542-200403000-00005. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, Chu X, Simon RP. Effect of ischemic preconditioning on genomic response to cerebral ischemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet. 2003;362:1028–1037. doi: 10.1016/S0140-6736(03)14412-1. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, King JS, Simon RP. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke. 2007;38:680–685. doi: 10.1161/01.STR.0000251444.56487.4c. [DOI] [PubMed] [Google Scholar]
- Stevens SL, Bao J, Hollis J, Lessov NS, Clark WM, Stenzel-Poore MP. The use of flow cytometry to evaluate temporal changes in inflammation. Brain Res. 2002;932:110–119. doi: 10.1016/s0006-8993(02)02292-8. [DOI] [PubMed] [Google Scholar]
- Stevens SL, Ciesielski TM, Marsh BJ, Yang T, Homen DS, Boule JL, Lessov NS, Simon RP, Stenzel-Poore MP. Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab. 2008;28:1040–1047. doi: 10.1038/sj.jcbfm.9600606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki K, Ruetzler CA, Ohtsuki T, Martin D, Nawashiro H, Hallenbeck JM. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997;748:267–270. doi: 10.1016/s0006-8993(96)01383-2. [DOI] [PubMed] [Google Scholar]
- Vadigepalli R, Chakravarthula P, Zak DE, Schwaber JS, Gonye GE. PAINT: a promoter analysis and interaction network generation tool for gene regulatory network identification. Omics. 2003;7:235–252. doi: 10.1089/153623103322452378. [DOI] [PubMed] [Google Scholar]
- Veldhuis WB, Derksen JW, Floris S, Van Der Meide PH, De Vries HE, Schepers J, Vos IM, Dijkstra CD, Kappelle LJ, Nicolay K, Bär PR. Interferon-beta blocks infiltration of inflammatory cells and reduces infarct volume after ischemic stroke in the rat. J Cereb Blood Flow Metab. 2003a;23:1029–1039. doi: 10.1097/01.WCB.0000080703.47016.B6. [DOI] [PubMed] [Google Scholar]
- Veldhuis WB, Floris S, van der Meide PH, Vos IM, de Vries HE, Dijkstra CD, Bär PR, Nicolay K. Interferon-beta prevents cytokine-induced neutrophil infiltration and attenuates blood–brain barrier disruption. J Cereb Blood Flow Metab. 2003b;23:1060–1069. doi: 10.1097/01.WCB.0000080701.47016.24. [DOI] [PubMed] [Google Scholar]
- Yang H, Ma G, Lin CH, Orr M, Wathelet MG. Mechanism for transcriptional synergy between interferon regulatory factor (IRF)-3 and IRF-7 in activation of the interferon-beta gene promoter. Eur J Biochem. 2004;271:3693–3703. doi: 10.1111/j.1432-1033.2004.04310.x. [DOI] [PubMed] [Google Scholar]