

Abstract
A saccharide-terminated Generation 3 (G3) polyamidoamine (PAMAM) dendrimer was synthesized as a drug carrier. Utilizing this dendritic platform, we have successfully synthesized polyvalent conjugates (G3-MTX) containing the drug methotrexate (MTX). Surface plasmon resonance (SPR) results showed that G3-MTX presented 3 orders of magnitude enhancement in binding avidity to folate-binding protein (FBP) as compared to the free folic acid (FA). Flow cytometric and confocal microscopic analysis showed that conjugate (G3-MTX-FI) containing imaging agent fluorescein-5(6)-carboxamidohexanoic acid (FI) was internalized into folate receptor (FR)-expressing KB cells in dose-dependent and receptor-mediated fashion. The G3-MTX induced a dose-dependent cytotoxicity in the KB cells. Therefore, the polyvalent G3-MTX may have potential as an anti-cancer nanodevice for the specific targeting and killing of FR-expressing tumor cells.
Keywords: Targeting anticancer drug, polyamidoamine (PAMAM) dendrimer, methotrexate, polyvalent conjugate, folate receptor
1. Introduction
Over the past two decades, various dendrimers have been synthesized and developed as nanocarriers for the tissue-specific delivery of molecules such as drugs and imaging agents.1–9 The desirable properties of dendrimers, such as aqueous solubility, biocompatibility, and chemically linkable surface functional groups, have made them one of the most studied nanoparticle for use as molecular delivery platforms. PAMAM dendrimer is one of the most extensively investigated dendrimers and has received much attention recently for biomedical applications, especially as a drug carrier.10–13 PAMAM dendrimers are highly branched and narrowly dispersed synthetic macromolecules with well-defined chemical structures. They can be easily modified and conjugated with multiple functionalities such as drugs, targeting ligands, and imaging agents. They are water soluble and essentially non-immunogenic molecules which can be rapidly cleared from the blood through the kidneys (if the size < 5 nm).14 Although the amine-terminated PAMAM dendrimers are cytotoxic, once the dendrimer is decorated with neutral functional groups and the excess amine groups are capped (e.g., by acetylation), they show low or no cytotoxicity.15–17
Over the past few years, we have utilized PAMAM dendrimers as carriers to develop tumor-targeted nanoparticles with folic acid (FA) as the targeting ligand and methotrexate (MTX) as the drug.11,18 A surface plasmon resonance study showed that the nanoparticles with multiple FAs on the surface exhibited a binding avidity improvement of up to 5 orders of magnitude to folate-binding protein (FBP) compared to that of a single FA.10 This allowed the nanoparticles to be targeted to epithelial cancer cells that over-express FR and showed a tumoricidal effect with an improved chemotherapeutic index versus that of free methotrexate.11,18 Despite our success in developing the FA- and MTX-conjugated biologically functional nanomaterial, there are still many challenges before this technology can move forward into the clinic. Major difficulties are the inconsistencies of the starting dendrimer material as well as the variability inherent in the multistep chemistry used for synthesis. The production of the PAMAM dendrimer requires a lengthy synthesis and purification process. Higher generation dendrimers are difficult to be reproduced due to side reactions and formation of defective structures. The covalent attachment of bioactive molecules to the dendrimer often generates heterogeneous populations, which further exacerbate the inconsistency and irreproducibility of the final product, especially during scale-up synthesis of the conjugate.19
One way to produce a more uniform and consistent dendrimer-drug conjugate is to use low generation PAMAM dendrimers, to minimize the number of synthetic steps, and to attach drugs using a simple conjugation chemistry.20 Nonetheless, low generation dendrimers have fewer functional groups on the surface, and their conjugates with lipophilic drugs are usually poorly water-soluble. The enhancement of water solubility of dendrimer can be achieved by introduction of a water-soluble group to the outer shell of the dendrimer.1,3,8 To achieve this goal, poly(ethylene glycol) chains21–24 and saccharides25–28 have been previously explored. In this paper, we report a saccharide-terminated G3 PAMAM dendrimer in which amino termini are modified by D-glucoheptono-1,4-lactone, a saccharide lactone. Each saccharide contains multi-hydroxyl groups which will significantly improve the water solubility of the dendrimer. Also, these hydroxyl groups are available for conjugation of drugs, targeting ligands, and imaging agents. Based on this strategy, we have synthesized a simple MTX-conjugated polyvalent G3-PAMAM dendrimer conjugate (G3-MTX) and demonstrated its in vitro cytotoxicity in the FR-expressing KB cell line. We also synthesized a fluorescein-5(6)-carboxamidohexanoic acid (FI)-containing conjugate (G3-MTX-FI) and demonstrated its cellular internalization in the FR-expressing KB cell line. We believe this simple polyvalent G3-MTX may have potential as a potent FR-targeted cancer therapeutic.
2. Results and Discussion
2.1 Synthesis
G3-OH 3 was synthesized from G3-NH2 1 and D-glucoheptono-1,4-lactone 2 (Scheme 1) with a very high yield (96%). Each amino terminal group reacted with only one D-glucoheptono-1,4-lactone because the newly formed amide is inert to the other lactone group. This reaction won’t cause an intra- or inter-molecular cross-linking reaction. Theoretically there are 6 hydroxyl groups on each terminal group and 192 total hydroxyl groups on the surface of G3-OH 3. The terminal hydroxyl groups of G3-OH 3 can be easily conjugated with drug (MTX), and imaging agent (FI) through ester bonds. As shown in Scheme 1, all conjugates were synthesized using the Mukaiyama reagent, 2-chloro-1-methylpyridinium iodide and 4-(dimethylamino)pyridine, as coupling agents with moderate to high yields (57%–89%). It was observed that the numbers of MTX attached on the surface of the G3-OH 3 increased by increasing the feeding ratio of MTX versus G3-OH 3 in the feed (Table 1). The average numbers of MTX and FI attached to the G3 dendrimer have been calculated based on MALDI-TOF, GPC, and 1H NMR results. All the products are highly water-soluble.
Scheme 1.

Synthesis of G3-OH 3, G3-MTX 4, G3-FI 5, and G3-FI-MTX 6.
Reagents and conditions: (a) DMSO, rt, overnight; (b) MTX, 2-chloro-1-methylpyridinium iodide, 4-(dimethylamino)pyridine, DMSO, rt, overnight; (c) FI, 2-chloro-1-methylpyridinium iodide, 4-(dimethylamino)pyridine, DMSO, rt, overnight; (d) G3-MTX, FI, 2-chloro-1-methylpyridinium iodide, 4-(dimethylamino)pyridine, DMSO, rt, overnight.
Table 1.
Reaction feed molar ratio, average molecular weight measured by MALDI-TOF, and average number of MTX and FI attached on G3-OH
| Name | Reaction feed molar ratio of MTX/G3-OH | Molar Mass | Number of MTX | Number of FI |
|---|---|---|---|---|
| G3-NH2 1 | - | 7106 | - | - |
| G3-OH 3 | - | 12864 | - | - |
| G3-MTX 4 | 5:1 | 14342 | 3.4 | - |
| G3-MTX 4 | 8:1 | 15131 | 5.2 | - |
| G3-MTX 4 | 10:1 | 15780 | 6.6 | - |
| G3-MTX 4 | 20:1 | 17197 | 9.9 | - |
| G3-FI 5 | - | 13976 | - | 2.4 |
| G3-FI-MTX 6 | - | 15033 | 2.4 | 2.4 |
2.2 1H NMR Spectrometry
1H NMR was used to confirm the surface modification of the dendrimers (Figure 1). Compared with G3-NH2 1, NMR spectrum of G3-OH 3 clearly shows new peaks at 8.09, 7.97, and 7.82 ppm which are attributed to newly formed amide bonds. New peaks at 5.56, 4.86, and multiple peaks between 4.48 and 3.30 ppm are from the D-glucoheptononyl moiety. As compared with the spectrum of G3-OH 3, the spectra of G3-MTX 4, G3-FI 5, and G3-FI-MTX 6 all show additional new peaks. The pteridine H-7 protons of the conjugated MTX moiety appear at 8.57 ppm, which is a sharp and well separated single peak. Multiple peaks of protons of methylene groups of FI appear between 1.23 to 2.08 ppm, and aromatic protons of FI appear between 7.27 to 8.69 ppm.
Figure 1.

Comparison of the 1H NMR spectra of G3 dendrimers and conjugates.
2.3 MALDI-TOF Mass Spectrometry
The average molecular weights of G3 dendrimers and conjugates have been measured by MALDI-TOF. The molecular weight increase for each species along the synthetic pathway is shown in Figure 2, which demonstrates that the D-glucoheptononyl group attachment to the G3-NH2 1 and conjugation of MTX and FI to the G3-OH 3 have occurred. The molecular weight gain of the G3-OH 3 from the G3-NH2 1 is due to the attachment of the D-glucoheptononyl groups. The average number of D-glucoheptononyl groups attached to the G3-NH2 1 can be calculated by the difference of average molecular weight between the G3-OH 3 and G3-NH2 1 divided by the molar mass of the D-glucoheptononyl group. The molecular weight increase from the G3-OH 3 to the G3-MTX 4, G3-FI 5, or G3-FI-MTX 6 is due to the addition of MTX and FI. The average number of MTX and FI attached to the conjugates can be calculated by the mass increase of the conjugates divided by the molar mass of the incoming substituent. The average numbers of attached functional molecules have been listed in Table 1.
Figure 2.

MALDI-TOF mass spectra of G3 dendrimers and conjugates.
2.4 High Performance Liquid Chromatography (HPLC)
HPLC was used to evaluate the purity and attachment homogeneity of the conjugates. Small molecule impurities such as unreacted MTX, FI, coupling reagent, and by-products are very easily differentiated from the dendrimers and conjugates by HPLC due to the significant difference in retention time. Figure 3 shows representative chromatograms of the conjugates G3-MTX 4 under UV 285 nm. The single symmetric peak of the conjugates suggests that the MTX has been nearly homogeneously attached to the G3-OH 3, which is in agreement with the GPC results shown below
Figure 3.

HPLC chromatogram of G3-MTX 4 under UV 285 nm.
2.5 Gel Permeation Chromatography (GPC)
GPC was used to evaluate the molar mass and molar mass distribution of the saccharide-terminated PAMAM dendrimer and conjugates. Figure 4 shows that molar mass distribution of the G3-OH 3 and G3-MTX 4 versus retention time. All dendrimer and conjugate samples exhibit just a single peak. The polydispersity indexes (PDI: Mw/Mn) of the G3-OH 3 and G3-MTX 4 are close to 1. This indicates that the mass distribution of the conjugates is narrow.
Figure 4.

Graph showing the molar mass (g/mol) vs. time (min): Red, G3-OH, Mw/Mn = 1.027; pink, G3-MTX3.4, Mw/Mn = 1.038; blue, G3-MTX5.2, Mw/Mn = 1.102; green, G3-MTX9.9, Mw/Mn = 1.179.
2.6 Molecular Dynamics (MD) Simulations
G3-OH 3 has a saccharide surface. It has been expected that the uncharged saccharide surface at neutral pH could cause significant structural change. Thus, we have carried out MD simulations at neutral pH to investigate the structural properties of G3-OH 3 and G3-MTX 4 with four MTX on the surface and compared their structural behaviors with that of G3-NH2 1. The structure of G3-NH2 1 is expanded due to repulsive interactions between positive charges on protonated amines on the surface of the dendrimer at neutral pH. However, since the surface of G3-OH 3 is uncharged at neutral pH, G3-OH 3 is more compact than G3-NH2 1 (Figure 5). The radius of gyration estimated by MD simulations demonstrated that G3-OH 3 and G3-MTX 4 showed a smaller radius of gyration (13.1 and 14.0 Å, respectively) than G3-NH2 1 (18.8 Å), even if the saccharide linkers have been added on the surface of the G3-NH2 1 in Table 2. The conjugation of four MTX to G3-OH has not significantly affected the size and structure of G3-MTX.
Figure 5.

Equilibrated molecular dynamics structures at pH 7: (a) G3-NH2 1; (b) G3-OH 3; (c) G3-MTX4 4. Drawing methods: van der Waals for (a) and (b) and licorice (for the dendrimer) and van der Waals (for four MTXs) for (c). Colors: white, hydrogen; light blue, carbon; blue, nitrogen; red, oxygen. The figures were prepared using the VMD program.36
Table 2.
Number of atoms and radius of gyration of the PAMAM dendrimers estimated from 1-ns MD simulations at pH 7
| PAMAM Dendrimers | Number of Atoms | Radius of Gyration (Å) |
|---|---|---|
| G3-NH2 1 | 1124 | 18.8 ± 0.2 19.7 ± 0.1a |
| G3-OH 3 | 1924 | 13.1 ± 0.1 |
| G3-MTX 4 with four MTX | 2128 | 14.0 ± 0.2 |
2.7 Surface Plasmon Resonance (SPR) Sensorgrams
We used SPR to study the interaction of G3-MTX 4 with the folate-binding protein (FBP). The results of the binding of G3-MTX with surface-bound FBP are shown in Figure 6. According to the kinetic analysis results, the dissociation constant (KD) for the binding between the G3-MTX nanoconjugate and the FBP reached to 4×10−8 (Table 3), showing 3 orders of magnitude enhancement in binding avidity compared to the free folic acid (2.29 ×10−5) performed under the same conditions. This boosted KD suggests that G3-MTX can achieve multivalent interaction with FBP.
Figure 6.

Surface Plasmon Resonance (SPR) sensorgrams of G3-MTX 4 at different concentrations.
Table 3.
Quantified Binding Constants of the G3-MTX and free FA with FBP Measured by SPR
| kon | koff | KD | |
|---|---|---|---|
| G3-MTX | 2.48E+04 | 9.76E-04 | 3.94E-08 |
| FA | 2.10+03 | 4.8E-02 | 2.29E-05 |
2.8 In Vitro Studies
In order to examine the binding characteristics of the synthesized G3-MTX 4 in the absence of the natural binding moiety FA, we synthesized G3-FI-MTX 6 and determined its binding in KB cells. As shown in Figure 7, the G3-FI-MTX 6 showed a dose-dependent binding, whereas the control conjugate G3-FI 5 failed to show significant binding. The binding of G3-FI-MTX 6 was reversed in the presence of excess free FA, suggesting FR-mediated uptake. To further verify receptor-mediated uptake of the G3-FI-MTX 6 into the KB cells, we examined the energy and pH dependence of the uptake. As given in Figure 8, the uptake of G3-FI-MTX 6 was completely abolished at 4°C, while there was binding at 37°C. These data clearly demonstrated that the conjugate is taken up into the cells through the FR, and not through the Reduced Folate Carrier (RFC) as the conjugate is too large to use this transporter.29 It was also found that there was more binding at pH 7 than pH 3. The internalization of the G3-FI-MTX 6 was quantified by acid treatment of the cells to remove the surface bound conjugate.30 During a 1-hour incubation at 37°C, ~30% of the total cellular fluorescence was removed by acid washing, indicating 70% internalization of the conjugate (Figure 8). The internalization was further verified by confocal microscopy (Figure 9). Specific uptake of the G3-FI-MTX 6, but not the G3-FI 5 was observed during 2- and 6-hour incubations at 300 nM and 1 μM concentrations.
Figure 7.

Binding comparison of G3-FI-MTX 6 (■), G3-FI 5 (◆) and FA + G3-FI-MTX 6 (□) on KB cells. The cells were maintained in FA-free medium and incubated with different concentrations of the indicated conjugates for 2 hours. The cells were also pre-incubated (30 min) with 50 μM free FA prior to addition of the G3-FI-MTX 6. The cells were then rinsed and resuspended in PBS buffer, and the mean FL1 fluorescence of 10,000 cells was quantified in a flow cytometer.
Figure 8.

Effect of temperature and pH on the binding of G3-FI-MTX 6 and G3-FI 5 on KB cells. The cells were maintained in FA-free medium and incubated with 1 μM each of the indicated conjugates for 1 hour at 4°C or at 37°C. The cells were rinsed with PBS and then exposed to normal saline adjusted to pH 3 or 7 for 30 seconds. The cells were rinsed and resuspended in PBS buffer, and the mean fluorescence was quantified as described in Methods.
Figure 9.
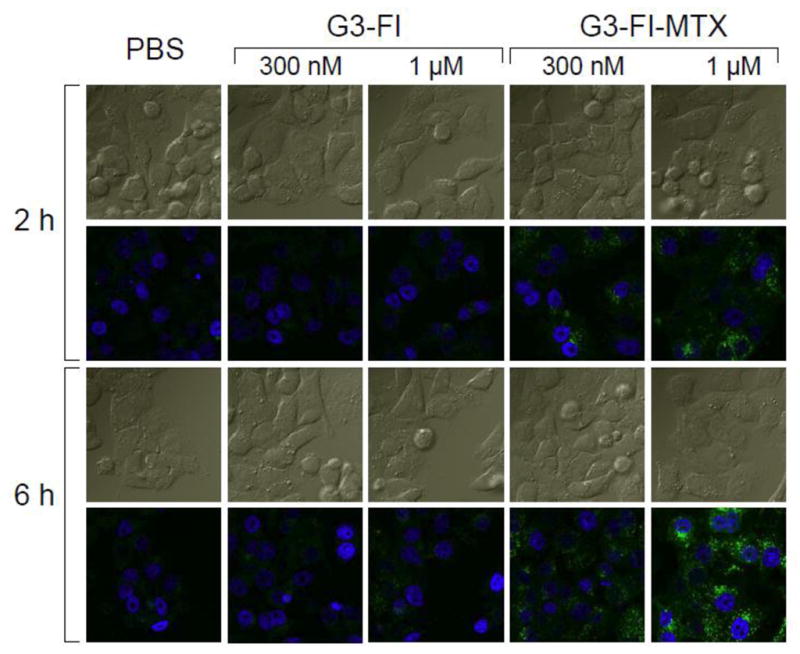
Confocal microscopic analysis of the uptake of G3-FI 5 and G3-FI-MTX 6 into KB cells. Cells grown on cover slips were incubated with 300 nM or 1 μM of the conjugates (green) for 2 hours and 6 hours, fixed with paraformaldehyde, mounted using solution containing the nuclei stain DAPI (blue), and the fluorescence was measured as given in the Experimental Section. The upper rows for each time point represent the DIC light image, and the bottom rows represent the corresponding fluorescent images.
As the fluorescently labeled G3-conjugates given above showed FR-mediated internalization into the KB cells, we subsequently synthesized several conjugates in the absence of the fluorescent agent for further in vitro cytotoxicity and efficacy studies. The cytotoxicity of G3-OH 3, free MTX, and two independently synthesized batches of G3-MTX 4 determined by XTT assay is shown in Figure 10. The cytotoxic potential of the newly synthesized conjugate has been compared to a FA-targeted ‘G5-FA-MTX’ conjugate, synthesized by our previously conventional synthetic approach in which the FA was conjugated through amide-linkage and the MTX through ester linkage via a glycidol moiety, that has been shown to be tumoricidal in vivo.11 The control dendrimer G3-OH 3 failed to show any significant cytotoxicity, whereas the G3-MTX 4 and free MTX evoked a dose-dependent cytotoxicity. At lower concentration, free MTX showed higher cytotoxicity than G3-MTX 4, but at high concentration they have similar cytotoxicity. The higher cytotoxicity of free MTX in the in vitro studies is due to its entry into cells through the RFC over the 2-day incubation period. The FR specificity cell entry of the conjugate suggests an absence of non-specific toxicity, showing different mechanism as compared to free MTX. Although it is possible that the MTX is released from the conjugate following endocytosis and its transit through the acidic endosome/lysosome compartments, we do not propose this as the mechanism of inhibition of dihydrofolate reductase (DHFR) because our previous studies have shown that non-hydrolyzable amide linked G5-MTX conjugates can also inhibit the DHFR similar to free MTX.31 It was also observed that the G3-MTX 4 showed comparable cytotoxicity to the G5-FA-MTX. These results indicate that the multivalent binding and internalization of the newly synthesized G3-MTX conjugate through the FR is sufficient to induce cytotoxicity of FR-expressing tumor cells. Therefore, the multivalent G3-MTX conjugate may have potential as a potent chemotherapeutic for FR-specific targeting of cancer.
Figure 10.

Cytotoxicity comparison of G3-OH (◇), G3-MTX3.4 (■), G3-MTX5.2 (△), and G5-FA5-MTX7 (●), and MTX (○). KB cells plated in 96-well plates were incubated with different concentrations of the indicated dendrimers and conjugates for 2 days, and the cytotoxicity was determined by XTT assay.
3. Conclusion
A low generation saccharide-functionalized polyvalent G3-MTX conjugate has 3 orders of magnitude enhancement in binding avidity to FBP as compared to the free FA. The described method allows a rapid synthetic strategy to produce a simple nanotherapeutic as compared to the more complex ‘G5-FA-MTX’ conjugate, for targeting FR-expressing cells without the need of conjugating also the natural ligand FA. This novel G3-MTX conjugate is shown to internalize through the FR to induce cytotoxicity in the FR-expressing KB cells, similar to the previously explored G5-FA-MTX. Further research on optimizing the structure of the conjugates and in examining the in vitro and in vivo biological activities could lead to their application in targeting FR-expressing tumor treatment.
4. Experimental
4.1 Materials
Amine-terminated G3 PAMAM dendrimer (G3-NH2 1) was synthesized and characterized at the Michigan Nanotechnology Institute for Medicine and Biological Science at the University of Michigan. All other chemicals were purchased from Sigma-Aldrich and used as received, unless otherwise specified. Water used in all experiments was purified by a Milli-Q Plus 185 water purification system (Millipore, Bedford, MA). The Spectra Por dialysis membranes (MWCO 3500) and phosphate buffer saline were acquired from Fisher.
4.2 Nuclear Magnetic Resonance Spectrometry (NMR)
1H NMR spectra were recorded on a 400 MHz Varian Inova 400 nuclear magnetic spectrometer in dimethyl sulfoxide (DMSO-d6).
4.3 Matrix-Assisted Laser Desorption Ionization-Time of Flight (MALDI-TOF) Mass Spectrometry
MALDI-TOF mass spectra were recorded on a Waters TofSpec-2E spectrometer (Beverly, MA, USA), running in linear mode with the high mass PAD detector, and 2,5-dihydroxybenzoic acid (DHB) in acetonitrile/water (50:50, v/v) was used as the matrix. The instrument was calibrated with bovine serum albumin (BSA, Mw = 66.43 × 103) in sinapic acid. The data were acquired and processed by MassLynx 3.5 software.
4.4 High-Performance Liquid Chromatography (HPLC)
A reverse-phase HPLC instrument consisting of a System GOLD 126 solvent module, a Model 507 auto-sampler equipped with a 100 μL loop and a Model 166 UV detector (Beckman Coulter, Fullerton, CA, USA), and a Phenomenex (Torrance, CA, USA) Jupiter C5 silica-based HPLC column (250 × 4.6 mm, 300 Å) were used for analysis of the products in this work. The mobile phase for elution of PAMAM dendrimer products was a linear gradient beginning with 100:0 water/acetonitrile (ACN) (both containing 0.14 wt% TFA) at a flow rate of 1 mL/min., reaching 20:80 (or 50:50) within 35 min. Trifluoroacetic acid (TFA) (0.14 wt% in both water and ACN) was used as a counterion to make the dendrimer-conjugate surface hydrophobic. All samples were dissolved in the aqueous mobile phase (water containing 0.14% trifluoroacetic acid (TFA)) at a flow rate of 1 mL/min, reaching 20:80 (or 50:50) within 35 min. The injection volume in each case was 35 μL with a sample concentration of 1 mg/mL, and the detection wavelength was 210 or 285 nm. The analysis was performed using Beckman’s System GOLD Nouveau software.
4.5 Gel Permeation Chromatography (GPC)
GPC was used to evaluate the molecular weights and molecular weight distribution of the G3-PAMAM dendrimers and conjugates. GPC experiments were performed on an Alliance Waters 2690 separation module (waters Corp., Milford, MA) equipped with a Waters 2487 dual wavelength UV absorbance detector (Waters Corp.), a Wyatt Dawn HELEOS light scattering detector (Wyatt Technology Corp., Santa Barbara, CA), an Optilab rEX differential refractometer (Wyatt Technology Corp.), and TosoHaas TSK-Gel Guard PHW 06762 (75 × 7.5 mm, 12 μm), G 2000 PW 05761 (300 × 7.5 mm, 10 μm), G 3000 PW 05762 (300 × 7.5 mm, 10 μm), and G 4000 PW (300 × 7.5 mm, 17 μm) columns. Column temperature was maintained at 25 ± 0.2°C with a Waters temperature control module. Citric acid buffer (0.1 M) with 0.025% sodium azide in water, pH 2.74, was used as a mobile phase at a flow rate of 1 mL/min. The sample concentration was 2 mg/mL, with an injection volume of 100 μL. The number average of molecular weight, Mn, weight average molecular weight Mw and polydispersity index (PDI) were calculated using ASTRA 5.34.14 software (Wyatt Technology Corp.).
4.6 Synthesis of D-Glucoheptononyl-terminated G3 PAMAM Dendrimer (G3-OH) 3
G3-NH2 1 (691 mg, 0.10 mmol) was dissolved in 10 mL of DMSO in a 25 mL flask. To the solution was added D-glucoheptono-1,4-lactone (799 mg, 3.84 mmol). The mixture was stirred at room temperature under nitrogen overnight and then 40 mL of cold water was added. The product was purified by dialysis against isotonic phosphate-buffered saline (PBS) buffer (3 × 4L) and then water (3 × 4L) with a cellulose dialysis membrane (MWCO = 3500) over 48 hours. The product was dried by lyophilization to yield 3 as a white solid (1.30 g, 96%).
4.7 Synthesis of Conjugate of MTX and D-Glucoheptononyl-terminated G3 PAMAM Dendrimer (G3-MTX) 4
A solution of G3-OH 3 (100 mg, 7.37 μmol) and MTX (33.5 mg, 73.7 μmol) in DMSO (15 mL) was stirred at room temperature under nitrogen. To this solution were added 2-chloro-1-methylpyridinium iodide (23.3 mg, 88.4 μmol) and 4-(dimethylamino)pyridine (21.6 mg, 177 μmol). The mixture was further stirred overnight and then diluted with cold water (55 mL). The product was purified by dialysis against PBS buffer (3 × 4L) and then water (3 × 4L) with a cellulose dialysis membrane (MWCO = 3500) over 48 hours. The final product was dried by lyophilization to yield 4 as a yellow solid (110 mg, 82%).
4.8 Synthesis of Conjugate of FI and D-Glucoheptononyl-terminated G3 PAMAM Dendrimer (G3-FI) 5
A solution of G3-OH 3 (100 mg, 7.37 μmol) and FI (14.4 mg, 29.4 μmol) in DMSO (15 mL) was stirred at room temperature under nitrogen. To the solution was added 2-chloro-1-methylpyridinium iodide (11.3 mg, 44.1 μmol) and 4-(dimethylamino)pyridine (10.8 mg, 88.2 μmol). The mixture was further stirred overnight and then diluted with cold water (65 mL). The product was purified by dialysis against PBS buffer (3 × 4L) and then water (3 × 4L) with cellulose dialysis membrane (MWCO = 3500) over 48 hours. The final product was dried by lyophilization to yield 5 as an orange solid (88 mg, 77%).
4.9 Synthesis of Conjugate of FI, MTX, and D-Glucoheptononyl-terminated G3 PAMAM Dendrimer (G3-MTX-FI) 6
A solution of G3-MTX 4 (50 mg, 3.5 μmol) and FI (5.1 mg, 10.5 μmol) in DMSO (5 mL) was stirred at room temperature under nitrogen. To the solution was added 2-chloro-1-methylpyridinium iodide (3.2 mg, 12.6 μmol) and 4-(dimethylamino)pyridine (3.1 mg, 25.2 μmol). The mixture was further stirred overnight and then diluted with cold water (20 mL). The product was purified by dialysis against PBS buffer (3 × 4L) and then water (3 × 4L) with a cellulose dialysis membrane (MWCO = 10000) over 48 hours. The final product was dried by lyophilization to yield 6 as an orange solid (46 mg, 84%).
4.10 Molecular Dynamics (MD) Simulations
All the PAMAM dendrimers were built using the Insight II software package (Accelrys Inc., San Diego, CA). All the primary amines of G3 PAMAM dendrimer were protonated to simulate the dendrimer at pH 7. The simulation of G3 PAMAM dendrimer in implicit solvent was previously published.32 MD simulations on the dendrimers were carried out using the CHARMM program Version 3533 and the CHARMM22 all-atom topology and parameters.34 After steepest descent minimization for 10,000 steps followed by adopted basis Newton-Raphson minimization process for 10,000 steps to obtain lower energy configurations, we further annealed the minimized dendrimer structures at very high temperature and cooled down to room temperature. We carried out equilibration on the systems for 200 ps and generated dynamics rum for 1 ns at 300 K. The total potential energy function (Utotal) for MD simulations is described as
where ε is the minimum energy of the Lennard-Jones potential, σ the distance yielding a minimum Lennard-Jones potential, q the partial charge on the atom, D the dielectric constant, r the distance between i and j, and i, j are nonbonded atom pairs. We used the dielectric constant D = r without a long-range nonbonded cut-off in the simulations.
The sizes of the simulated dendrimers have been estimated from the radius of gyration, Rg (Å), which is defined as
for a dendrimer composed of N atoms where M is the dendrimer’s total mass, mi and xi are the mass and position of the ith atom, and x0 is center-of-mass of the dendrimer.
4.11 Surface Plasmon Resonance (SPR) Measurements
SPR experiments were performed in Biacore® X (GE healthcare, Piscataway, USA) using a method similar to that which has been previously reported.10 Folate-binding protein (FBP, bovine milk) was immobilized on the surface of a CM5 sensor chip via protein conjugation chemistry to a carboxymethylated dextran-coated layer by a standard amide coupling protocol. Briefly, the process of chip preparation included EDC/NHS activation at 0.4M/0.4M for 10 min., followed by 10 min. of protein immobilization and 10 min. of deactivation by 1M ethanolamine. The immobilization process of FBP on channel 2 resulted in a 13,000 response unit (RU) equivalent to 13 ng/mm2. SPR signals for FBP binding were obtained by injection of each ligand dissolved in HBS-EP buffer at a flow rate of 30 μL/min. After each measurement, the surface of the chip was regenerated by injection of 5 μL of 10 mM glycine-HCl (pH 2.5).
The measured SPR sensorgrams were obtained by subtracting SPR signals from channel 2, the FBP immobilized flow channel, from SPR signals collected from channel 1, the reference flow channel. (ΔRU = RU2 − RU1). Kinetic binding parameters, the rate of association (kon), and the rate of dissociation (koff) were extracted by fitting each binding curve separately using BIA evaluation software with the Langmuir kinetic model and analyzed (dR/dt vs. R) following the method described by Glaser.35 Dissociation constants (KD = koff/kon) for each ligand were the mean of KD at three different concentration of measurement in which chi square (χ2) value of each fitting is lower than 3.
4.12 Measurement of Cellular Binding and Cytotoxicity
KB cells, a sub-line of the cervical carcinoma HeLa cells (ATCC, Manassas, VA, USA), were grown as a monolayer cell culture at 37°C and 5% CO2 in FA-deficient RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS). The 10% FBS provided an FA concentration equivalent to that present in the human serum (~20 nM). For assessment of the cellular binding, KB cells plated in 24-well plates were treated with different concentrations of the conjugates for the indicated time periods on FA-free medium in the absence of serum. The cells were incubated at 37°C, rinsed, and the mean FL1 fluorescence of 10,000 cells was determined by flow cytometry, as described previously.17 For the cytotoxicity experiments, the cells were seeded in 96-well microtiter plates (3000 cells/well) in medium containing dialyzed serum. Two days after plating, the cells were treated with the dendrimer conjugates in tissue culture medium under the indicated conditions. A colorimetric ‘XTT’ (sodium 3-[1-(phenylaminocarbonyl)-3,4-tetrazolium]-bis (4-methoxy-6-nitro) benzene sulfonic acid hydrate) assay (Roche Molecular Biochemicals, Indianapolis, IN) was performed following the vendor’s protocol. After incubation with the XTT labeling mixture, the microtiter plates were read on an ELISA reader (Synergy HT, BioTek) at 492 nm with the reference wavelength at 690 nm.11
Supplementary Material
Acknowledgments
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under award 1 R01 CA119409.
Abbreviation
- PAMAM
polyamidoamine
- G3
generation 3
- MTX
methotrexate
- FA
folic acid
- FR
folate receptor
- SPR
Surface plasmon resonance
- FBP
folate-binding protein
- FI
fluorescein-5(6)-carboxamidohexanoic acid
- PDI
polydispersity index
Footnotes
Supplementary data associated with this article include 1H NMR spectra of MTX, G3-NH2, G3-OH, G3-MTX, G3-FI, and G3-FI-MTX in DMSO-d6 and HPLC and UPLC chromatograms of G3-OH under UV 210 nm and G3-MTX under UV 285 nm.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Kojima C, Kono K, Maruyama K, Takagishi T. Bioconjugate Chemistry. 2000;11:910–917. doi: 10.1021/bc0000583. [DOI] [PubMed] [Google Scholar]
- 2.Jansen JFGA, Debrabandervandenberg EMM, Meijer EW. Science. 1994;266:1226–1229. doi: 10.1126/science.266.5188.1226. [DOI] [PubMed] [Google Scholar]
- 3.Liu MJ, Kono K, Frechet JMJ. Journal of Controlled Release. 2000;65:121–131. doi: 10.1016/s0168-3659(99)00245-x. [DOI] [PubMed] [Google Scholar]
- 4.Stiriba SE, Kautz H, Frey H. Journal of the American Chemical Society. 2002;124:9698–9699. doi: 10.1021/ja026835m. [DOI] [PubMed] [Google Scholar]
- 5.Shen Z, Duan HW, Frey H. Adv Mater. 2007;19:349. [Google Scholar]
- 6.Chen Y, Shen Z, Frey H, Perez-Prieto J, Stiriba SE. Chemical Communications. 2005:755–757. doi: 10.1039/b414046j. [DOI] [PubMed] [Google Scholar]
- 7.Adeli M, Haag R. J Polym Sci Pol Chem. 2006;44:5740–5749. [Google Scholar]
- 8.Xu SJ, Kramer M, Haag R. Journal of Drug Targeting. 2006;14:367–374. doi: 10.1080/10611860600834011. [DOI] [PubMed] [Google Scholar]
- 9.Radowski MR, Shukla A, von Berlepsch H, Bottcher C, Pickaert G, Rehage H, Haag R. Angewandte Chemie-International Edition. 2007;46:1265–1269. doi: 10.1002/anie.200603801. [DOI] [PubMed] [Google Scholar]
- 10.Hong S, Leroueil PR, Majoros IJ, Orr BG, Baker JR, Jr, Banaszak Holl MM. Chem Biol. 2007;14:107–115. doi: 10.1016/j.chembiol.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 11.Kukowska-Latallo JF, Candido KA, Cao ZY, Nigavekar SS, Majoros IJ, Thomas TP, Balogh LP, Khan MK, Baker JR. Cancer Research. 2005;65:5317–5324. doi: 10.1158/0008-5472.CAN-04-3921. [DOI] [PubMed] [Google Scholar]
- 12.Yellepeddi VK, Kumar A, Palakurthi S. Expert Opinion on Drug Delivery. 2009;6:835–850. doi: 10.1517/17425240903061251. [DOI] [PubMed] [Google Scholar]
- 13.Kolhe P, Misra E, Kannan RM, Kannan S, Lieh-Lai M. International Journal of Pharmaceutics. 2003;259:143–160. doi: 10.1016/s0378-5173(03)00225-4. [DOI] [PubMed] [Google Scholar]
- 14.Peer D, Karp JM, Hong S, FaroKHzad OC, Margalit R, Langer R. Nature Nanotechnology. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 15.Kolhatkar RB, Kitchens KM, Swaan PW, Ghandehari H. Bioconjugate Chemistry. 2007;18:2054–2060. doi: 10.1021/bc0603889. [DOI] [PubMed] [Google Scholar]
- 16.Waite CL, Sparks SM, Uhrich KE, Roth CM. Bmc Biotechnology. 2009;9 doi: 10.1186/1472-6750-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas TP, Majoros I, Kotlyar A, Mullen D, Holl MMB, Baker JR. Biomacromolecules. 2009;10:3207–3214. doi: 10.1021/bm900683r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majoros IJ, Thomas TP, Mehta CB, Baker JR. Journal of Medicinal Chemistry. 2005;48:5892–5899. doi: 10.1021/jm0401863. [DOI] [PubMed] [Google Scholar]
- 19.Mullen DG, Desai AM, Waddell JN, Cheng XM, Kelly CV, McNerny DQ, Majoros IJ, Baker JR, Sander LM, Orr BG, Holl MMB. Bioconjugate Chem. 2008;19:1748–1752. doi: 10.1021/bc8002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang YH, Thomas TP, Desai A, Zong H, Leroueil PR, Majoros IJ, Baker JR. Bioconjugate Chemistry. 2010;21:489–495. doi: 10.1021/bc9003958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malek A, Czubayk F, Aigner A. Journal of Drug Targeting. 2008;16:124–139. doi: 10.1080/10611860701849058. [DOI] [PubMed] [Google Scholar]
- 22.Ishida T, Wang X, Shimizu T, Nawata K, Kiwada H. J Control Release. 2007;122:349–355. doi: 10.1016/j.jconrel.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 23.Malek A, Merkel O, Fink L, Czubayko F, Kissel T, Aigner A. Toxicology and Applied Pharmacology. 2009;236:97–108. doi: 10.1016/j.taap.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 24.Xu SJ, Luo Y, Graeser R, Warnecke A, Kratz F, Hauff P, Licha K, Haag R. Bioorg Med Chem Lett. 2009;19:1030–1034. doi: 10.1016/j.bmcl.2008.01.043. [DOI] [PubMed] [Google Scholar]
- 25.Miyazaki M, Torigoe K, Esumi K. Langmuir. 2000;16:1522–1528. [Google Scholar]
- 26.Watanabe S, Iwamura M. J Photoch Photobio A. 2003;155:57–62. [Google Scholar]
- 27.Woller EK, Cloninger MJ. Biomacromolecules. 2001;2:1052–1054. doi: 10.1021/bm015560k. [DOI] [PubMed] [Google Scholar]
- 28.Aoi K, Tsutsumiuchi K, Yamamoto A, Okada M. Macromol Rapid Comm. 1998;19:5–9. [Google Scholar]
- 29.Majoros IJ, Williams CR, Becker A, Baker JR., Jr Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1:502–10. doi: 10.1002/wnan.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gabizon A, Horowitz AT, Goren D, Tzemach D, Mandelbaum-Shavit F, Qazen MM, Zalipsky S. Bioconjugate Chemistry. 1999;10:289–298. doi: 10.1021/bc9801124. [DOI] [PubMed] [Google Scholar]
- 31.Majoros IJ, Baker JR., Jr . Dendrimer Based Nanomedicine. Pan Stanford Publishing; 2008. p. 196. [Google Scholar]
- 32.Lee I, Athey BD, Wetzel AW, Meixner W, Baker JR. Macromolecules. 2002;35:4510–4520. [Google Scholar]
- 33.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 34.MacKerell AD, Bashford D, Bellott M, Dunbrack RLJ, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, III, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Karplus M. J Phys Chem B. 1998;102:3386–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 35.Glaser RW. Analytical Biochemistry. 1993;213:152–161. doi: 10.1006/abio.1993.1399. [DOI] [PubMed] [Google Scholar]
- 36.Humphrey W, Dalke A, Schulten K. J Mol Graphics. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 37.Lifson S, Hagler AT, Dauber P. J Am Chem Soc. 1979;101:5111–5121. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.