


Abstract
Rad51 is crucial not only in homologous recombination and recombinational repair but also in normal cellular growth. To address the role of Rad51 in normal cell growth we investigated morphological changes of cells after overexpression of wild-type and a dominant negative form of Rad51 in fission yeast. Rhp51, a Rad51 homolog in Schizosaccharomyces pombe, has a highly conserved ATP-binding motif. Rhp51 K155A, which has a single substitution in this motif, failed to rescue hypersensitivity of a rhp51Δ mutant to methyl methanesulfonate (MMS) and UV, whereas it binds normally to Rhp51 and Rad22, a Rad52 homolog. Two distinct cellular phenotypes were observed when Rhp51 or Rhp51 K155A was overexpressed in normal cells. Overexpression of Rhp51 caused lethality in the absence of DNA-damaging agents, with acquisition of a cell cycle mutant phenotype and accumulation of a 1C DNA population. On the other hand, overexpression of Rhp51 K155A led to a delay in G2 with decondensed nuclei, which resembled the phenotype of rhp51Δ. The latter also exhibited MMS and UV sensitivity, indicating that Rhp51 K155A has a dominant negative effect. These results suggest an association between DNA replication and Rad51 function.
INTRODUCTION
In eukaryotic cells double-strand breaks (DSBs) in chromosomes are induced by harmful exogenous agents such as ionizing radiation and certain chemicals (1,2). DSBs can also occur naturally during meiotic recombination, mating type switching in yeast, V(D)J rearrangement in lymphocytes and DNA replication (3). If DSBs are not properly repaired, even a single unrepaired DSB in a chromosome can lead to cell death. Furthermore, inappropriate repair of DSBs results in formation of tumors. Two major pathways have been identified to repair DSBs; non-homologous end joining (also called illegitimate recombination) and homologous recombination. In Saccharomyces cerevisiae the RAD52 epistasis group of genes, including RAD50–RAD59, MRE11 and XRS2, are involved in homologous recombination and DSB repair (4,5). Mutations in these genes increase sensitivity to ionizing radiation and cause defects in recombination.
The gene product of RAD51 has been regarded as a key player in homologous recombination and recombinational repair. Several features indicate that Rad51 is the structural and functional eukaryotic counterpart of Escherichia coli RecA. Purified Rad51 from budding yeast and human promotes homologous pairing and strand exchange reactions in vitro in an ATP- and single-stranded DNA-binding protein-dependent manner (6,7). Rad51 also forms nucleoprotein filaments in which the DNA is extended and underwound in a manner similar to RecA (8). Unlike RecA, however, Rad51 requires an additional set of proteins, such as Rad52, Rad54 and the Rad55–Rad57 complex, to achieve the maximum level of strand exchange activity (9–15).
Rad51 may also function in cell proliferation. In many species Rad51 expression fluctuates throughout the cell cycle, with a peak occurring during S phase (16–20). Deletion of a Rad51 homolog, rhp51+, in the fission yeast (rhp51Δ) induced delayed growth, heterogeneity in cell size and aberrant nuclear phenotypes, suggesting that Schizosaccharomyces pombe Rad51 may be involved in segregation of chromosomes or maintenance of genomic integrity (21). The deletion phenotype of RAD51 is much more striking in mammals; efforts to generate homozygous rad51 knock-out mice have been unsuccessful due to embryonic lethality, indicating that, unlike S.cerevisiae, Rad51 from higher eukaryotes has an important role that is not yet clearly understood in early development (22,23). This embryonic lethality provides an evident distinction between Rad51 in S.cerevisiae and human with regard to its in vivo function, despite the similarities in biochemical properties. It is of interest, therefore, that tumor suppressor proteins such as p53, BRCA1 and BRCA2, whose homologs are not found in yeast, interact or co-localize in subnuclear structures with Rad51 (18,23–25).
In a previous study we reported that a rhp51Δ mutant of the fission yeast S.pombe showed sensitivity not only to the DNA-damaging agents methyl methanesulfonate (MMS) and UV light, but also to caffeine, which overrides the S/M checkpoint (21). In addition, the mutant displayed a high degree of genomic instability reflected in an accumulation of unusually elongated cells with aberrant nuclei in the absence of DNA-damaging agents. Similar genomic instability was also reported for mutations in rhp54 and rhp55, the fission yeast counterparts of RAD54 and RAD55 (26,27). In contrast, such genomic instability has not been noted for mutations in recombinational repair genes in budding yeast. In an effort to delineate the relevance of Rad51 to chromosome integrity we used a fission yeast system, which is evolutionarily closer to mammalian cells than budding yeast and shows a high degree of genomic instability in this mutant. To address this aim we investigated the cellular and nuclear phenotypes of normal cells overexpressing the wild-type and a dominant negative ATP-binding domain mutant of Rhp51 and compared them with rhp51Δ. We found that overexpression of Rhp51 or the dominant negative ATP-binding domain mutant of Rhp51 induced distinct chromosome instability.
MATERIALS AND METHODS
Strains and media
Schizosaccharomyces pombe haploid strains JY334 (h+ ade6-M216 leu1-32), JY746 (h+ ade6-M216 leu1-32 ura4-D18) and ED668 (h+ ade6-M216 leu1-32 ura4-D18) were used as wild-type cells. The rhp51Δ mutant JAC1/51Δ (h– ade6-704 leu1-32 rhp51::ura4+) was used as a host for complementation by mutant Rhp51. Saccharomyces cerevisiae strain Y190 (MATa ura3-52 his3-200 lys2-801 ade2-101 trp1-901 leu2-3, 112 gal4Δ gal80Δ cyhr2 LYS2::GAL1UAS-HIS3TATA-HIS3 URA3::GAL1UAS-GAL1TATA-lacZ) was used as a host for the yeast two-hybrid assay. Schizosaccharomyces pombe cells were grown and maintained in standard rich medium (YES) or in minimal medium (EMM) supplemented with appropriate nutrients as described in Alfa et al. (28).
Plasmids and site-directed mutagenesis
Site-directed mutagenesis was carried out by the Venkitaraman’s protocol (29). For substitution of Lys155 by Ala in the Walker A motif of the rhp51+ gene the oligomer 5′-CGTACCGGTGCAAGTCAAATC-3′ was hybridized with single-stranded DNA derived from plasmid p51-504, which contains a 0.4 kb EcoRI fragment of the rhp51+ gene cloned in pBluescript SK(+) (Stratagene). The resulting plasmid containing the mutation was sequenced on both strands with overlaps and the mutagenized 0.4 kb EcoRI fragment was replaced by the cognate EcoRI fragment of p51-420 (30) to create the full ORF.
A 2.6 kb BglII–XhoI fragment encompassing the promoter, full ORF and transcription termination sites of the rhp51+ gene was cloned into the BamHI and SalI sites of the multicopy plasmid Splac551 [a derivative of YIplac128 (31) made by inserting the S.pombe ARS1 into a NdeI site] to generate p51.551. To generate plasmid p51.3 the 0.3 kb N-terminal fragment of rhp51+ was generated by PCR using primers 5′-CAAGTTATAATGGCAG-3′ (bold letters represent the first methionine codon) and 5′-GAATATGGTACCCAGTG-3′, cloned into the MscI site of pREP3 and named p51.3N. A 1.8 kb SpeI–XhoI fragment of the rhp51+ gene was then cloned into the SpeI and SalI sites of p51.3N to construct plasmid p51.3, which contains the full ORF of rhp51+. Plasmids p51.41 and p51.81 were constructed by similar procedures with vectors pREP41 and pREP81 (32,33), respectively, except that a new NdeI site was created at the position of the first methionine using primer 5′-CAAGTTCATATGGCAG-3′ (the first methionine codon and the new NdeI site are in bold and underlined, respectively) and the 1.3 kb SpeI–BamHI fragment was inserted into the SpeI and BamHI sites to make the full-length ORF. For generation of plasmid pET51 the rhp51+ ORF was generated by PCR using primers 5′-CAAGTTACCATGGCAGATACAGAGGTGG-3′ (the first methionine codon and a new NcoI site are in bold and underlined, respectively) and 5′-CACTAACTCGAGTTAGACAGGTGCGATAATTTC-3′ (the stop codon and a new XhoI site are in bold and underlined, respectively) and placed in the NcoI and XhoI sites of pET28b (Novagen).
Survival test
Each transformant was grown until mid log phase (OD595 = 0.5–1) in EMM medium and appropriate dilutions containing 500–1000 cells were plated onto solid medium in the presence of MMS or irradiated by UV after plating. The cells were incubated for 4–5 days and the resulting numbers of colonies counted. Experiments were performed at least three times and averaged. For the spotting assay 5 µl of sequential 10-fold dilutions of culture grown in EMM with or without thiamine was spotted onto EMM plates with or without 2 µM thiamine, respectively.
Overexpression of proteins in S.pombe
An exponentially growing culture of cells harboring rhp51+ cloned in pREP vectors in the presence of 2 µM thiamine was washed with sterile water three times. The washed cells were diluted with EMM medium lacking thiamine to a cell density of OD595 ≈ 0.1 and grown at 30°C for 17–25 h. The protein level increased from 14 h after thiamine deprivation and reached a maximum at ∼17 h.
Generation of anti-Rhp51 antibody
Anti-Rhp51 antibody was generated in rabbit using GST–Rhp51 fusion protein as antigen. The antiserum was purified through an antigen-coupled Sepharose column.
Fluorescence microscopy
Indirect immunofluorescence microscopy was performed according to Guthrie et al. (34) using diamidinophenylindole (DAPI) staining, affinity-purified anti-Rhp51 antibody, anti-tubulin TAT1 antibody (35), anti-SPB Sad1 antibody (36), FITC-conjugated donkey anti-rabbit antibody and TRITC-conjugated anti-mouse IgG antibody (Jackson). Fluorescence was viewed with an Olympus IX70-131 microscope with a 100 W light source. Photographs were taken on Kodak Elite Chrome color slide film rated at 100 ASA.
Flow cytometry
For FACS analysis Becton-Dickinson FACStar+ and Cell Quest software (Becton-Dickinson) were used as described (37).
Yeast two-hybrid analysis
The liquid culture β-galactosidase assay for quantitative analysis of protein–protein interaction was performed according to the instructions provided by the manufacturer (Clontech Laboratories, Palo Alto, CA). β-Galactosidase units were calculated as follows:
1 β-galactosidase unit = 1000 × OD420/(t × V × OD600)
where t is the elapsed time (min) of incubation, V is 0.1 ml × concentration factor and OD600 is the A600 of 1 ml of culture
Co-immunoprecipitation
Cells harboring p51.3 or p51.3 K155A with pREP4-Rad22 were induced by thiamine deprivation and the total cell lysates were incubated with affinity-purified anti-Rhp51 or anti-Rad22 antibodies in 0.5 ml of reaction buffer containing 25 mM Tris–HCl pH 7.4, 0.5 mM EDTA, 1% NP-40 and 10% glycerol for 3 h and further incubated for 1 h after addition of 30 µl of 33% protein A–Sepharose. The immune complexes were precipitated, washed with the same buffer six times, resuspended in 1× Laemli buffer and resolved by 8% SDS–PAGE. All procedures were performed at 4°C. Protein complexes were analyzed by immunoblotting.
RESULTS
A single point mutation in the ATP-binding motif of Rhp51 confers an inability for DNA repair
The rhp51+ ORF contains the highly conserved ATP-binding motifs named Walker type A and B in the central region. Among the residues in the Walker A motif Lys155 is known to interact with the β- and γ-phosphates of ADP/ATP (38). Substitution of the conserved lysine residue to alanine has been shown to impair the enzymatic activities of Rad51 and its overexpression has a dominant negative effect on MMS sensitivity in budding yeast (39,40). To examine whether the analogous mutation has a similar dominant negative effect, we substituted Lys155 of Rhp51 by Ala (rhp51 K155A) using site-directed mutagenesis (Fig. 1A). The resulting gene was cloned into the multicopy plasmid Splac551. The wild-type and mutant plasmids were introduced into rhp51Δ cells. These cells were examined for whether they could rescue the DNA repair defect of rhp51Δ cells exposed to sublethal doses of MMS or UV. Unlike the rhp51+ plasmid, the K155A plasmid could not restore resistance to MMS or UV in rhp51Δ cells (Fig. 1B and C). Western blot analysis suggested that protein instability or lack of protein expression was not the reason for its inability in repair activity (data not shown). Thus, a single amino acid substitution in the conserved ATP-binding motif was enough to disable the function of the rhp51+ gene product, indicating that ATP-binding is essential for the repair activity of Rhp51.
Figure 1.

A single point mutation in the conserved ATP-binding motif of Rhp51 impairs its DNA repair activity. (A) Comparison of amino acid sequences of conserved ATP-binding motif A from E.coli RecA, S.cerevisiae Rad51, human Rad51 and S.pombe Rhp51. (B and C) rhp51+ and rhp51 K155A carried by multicopy plasmids were subjected to survival tests to determine their complementation of MMS (B) and UV (C) sensitivity of rhp51Δ cells. Diamond, wild-type cells with vector Splac551; square, rhp51Δ with Splac551; triangle, rhp51Δ with p51.551; circle, rhp51Δ with p51.551 K155A.
A mutation in the ATP-binding motifs of Rhp51 does not affect its interactions with Rad22 or itself
It has been reported that Rad51 binds to Rad52 and self-assembles (41). To examine whether the K155A mutation has an effect on protein–protein interactions of Rhp51 we investigated the interactions of Rhp51 K155A with itself and with Rad22 by the yeast two-hybrid assay. Rad22 is a S.pombe homolog of Rad52, which can bind to DSBs (42). Figure 2A shows that Rhp51 interacts with itself and with Rad22. An interaction between Rhp51 and Rad22 was also observed by co-immunoprecipitation (Fig. 2B) and GST pull-down assay (data not shown), indicating that the two proteins indeed associate directly. Rhp51 K155A also binds normally to itself and wild-type Rhp51, indicating that the K155A mutation does not affect these interactions. Interestingly, the two-hybrid experiment indicated that the association between Rhp51 and Rad22 was greatly enhanced by the K155A mutation. The co-immunoprecipitation experiment also showed that Rhp51 K155A associates normally with Rad22 (Fig. 2B). After co-overexpression of Rhp51-Rad22 and Rhp51, K155A-Rad22 cell lysates were immunoprecipitated with anti-Rhp51 or anti-Rad22 antibodies and then subjected to immunoblotting. As shown in Figure 2B, Rad22 protein was co-precipitated not only with wild-type Rhp51 but also with Rhp51 K155A, indicating that the mutant protein binds Rad22. However, it is not clear whether the K155A mutation strengthens the association with Rad22 as observed in Figure 2A. The amount of Rad22 co-precipitated with Rhp51 K155A slightly increased compared to that of Rad22 precipitated by anti-Rad22 antibody (Fig. 2B, lanes 7 and 8), whereas Rad22 co-precipitated with Rhp51 decreased slightly (Fig. 2B, lanes 3 and 4). In this experiment the expression levels of Rhp51 and Rhp51 K155A were almost the same (Fig. 2C), whereas different levels of expression of Rad22 were observed in the two cell extracts (Fig. 2B, lanes 1 and 5). These data demonstrate that Rhp51 K155A could interact normally with Rhp51 and Rad22, indicating that an inability in protein–protein interactions did not cause the lack of repair activity of the mutant protein.
Figure 2.

Interactions between Rhp51, Rhp51 K155A and Rad22. (A) To measure the interactions yeast two-hybrid liquid β-galactosidase assays were performed and the β-galactosidase activity of each interaction was calculated as described in Materials and Methods. For simplicity the activation domain, DNA-binding domain, empty vector, Rad22, Rhp51 and Rhp51 K155A are designated AD, BD, v, 22, 51 and KA, respectively. (B) The Rhp51–Rad22 and Rhp51 K155A–Rad22 proteins were co-overexpressed in the same cell and immunoprecipitated by anti-Rhp51 and anti-Rad22 antibodies. The co-immunoprecipitated protein complexes were analyzed by immunoblotting with anti-Rad22 antibody. PI, α-Rad22 and α-Rhp51 indicate pre-immune serum, anti-Rad22 antibody and anti-Rhp51 antibody, respectively. (C) Expression levels of Rhp51 and Rhp51 K155A were compared by immunoblotting using anti-Rhp51 antibody.
Overexpression of Rhp51 K155A induces dominant negative effects in response to DNA-damaging agents
Since Rad51 in S.cerevisiae and human interacts with various proteins, the formation of protein complexes is seemingly very important for the action of Rad51. We hypothesized that if Rhp51 K155A, which can interact with Rhp51 and Rad22 but is unable to repair DNA damage, is overexpressed in normal cells it may inhibit the action of endogenous Rhp51 by foraging its protein–protein interactions. As expected, wild-type cells overexpressing Rhp51 K155A lost their DNA repair activity and, consequently, displayed only slightly higher resistance than the rhp51Δ mutant (Fig. 3A and B). The results indicate that the K155A mutation is dominant negative in DNA repair, as was the case with Rad51 in budding yeast.
Figure 3.

Dominant negative effect of overexpression of Rhp51 K155A protein and partial rescue by co-overexpression of Rad22. Overexpression of Rhp51 K155A sensitizes wild-type cells to sub-lethal doses of MMS (A) and UV (B). Diamond, wild-type cells with vector pREP3; square, rhp51Δ with pREP3; triangle, wild-type cells with p51.3 K155A. (C and D) Rad22 was co-overexpressed with Rhp51 K155A and MMS (C) and UV (D) sensitivity were measured by survival tests.
To further investigate whether interference in protein–protein interactions of endogenous Rhp51 protein caused the dominant negative effect we examined the effect of co-overexpression of Rad22. The MMS and UV sensitivity of cells overexpressing Rhp51 K155A was partially rescued by co-overexpression of Rad22 (Fig. 3C and D). Since overexpression of Rad22 itself slightly sensitized wild-type cells to DNA-damaging agents, it is unlikely that excess Rad22 caused the rescue effect in Rhp51 K155A-overexpressing cells. These results suggest that interference in protein–protein interactions of endogenous Rhp51 might be the main reason for the dominant negative effect of Rhp51 K155A in DNA repair.
Overexpression of Rhp51 induces lethality with a cell cycle mutant phenotype
Previously we reported that the Rhp51 null mutant has defects in cell proliferation accompanied by an abnormal nuclear morphology (21). To further characterize the molecular basis of this defect we overexpressed Rhp51 and Rhp51 K155A in wild-type cells under control of an inducible nmt1+ promoter and compared their terminal morphology to rhp51Δ. Overexpression of Rhp51 was carried out by promoters of different strengths and the effect on cell viability in the absence of external damage was measured by a semi-quantitative spotting assay (Fig. 4A). Viability of the cells was proportionally reduced with increasing amounts of Rhp51 and the cells with the highest levels of Rhp51 were totally non-viable. Overexpression from plasmids p51.81, p51.41 and p51.3 enhanced the cellular level of Rhp51 by ∼80, 150 and 700-fold, respectively, over the wild-type (Fig. 4B).
Figure 4.

Overexpression of Rhp51 confers lethality to wild-type cells. (A) Wild-type cells overexpressing Rhp51 under the control of inducible nmt1+ promoters with different strengths were spotted onto plates in the presence (left) or absence (right) of thiamine as serial 10-fold dilutions. (B) Expression of Rhp51 under the control of different strength nmt1+ promoters from 10 µg each cell lysate compared by western blotting. (C) Cut and elongated phenotypes on Rhp51 overexpression plotted as percentages. More than 200 cells in each cell population were counted.
To identify the cause of this lethality we examined the final nuclear morphology by DAPI staining 25 h after thiamine deprivation. As shown in Figure 4C, a large portion of cells containing plasmid p51.3 exhibited a stretched nucleus known as the ‘cut (cell untimely torn) phenotype’. This phenotype was previously reported in mutants of topoisomerse II and various cut genes (43), where coupling between the completion of S phase and the onset of mitosis is disrupted. However, those cells showing Rhp51 overexpression were not as short as known cut cells. About 70% of cells containing p51.3 showed the cut phenotype, while cells harboring plasmids p51.81 and p51.41, expressing much lower levels of Rhp51, rarely showed the cut phenotype (Fig. 4C). Instead, relatively more elongated cells than the wild-type formed a significant portion of cells containing p51.81 and p51.41, suggesting that a cell cycle or DNA damage checkpoint was activated before the cells underwent catastrophic nuclear division.
Overexpression of Rhp51 K155A induces similar cellular and nuclear morphologies in rhp51Δ cells
Unlike overexpression of Rhp51, normal cells overexpressing Rhp51 K155A were viable. However, a reduced growth rate was observed, as seen in rhp51Δ cells (data not shown; 21). Microscopic observation revealed that a considerable number of cells overexpressing Rhp51 K155A were unusually elongated (Table 1). Because such elongated cells were also frequently found in rhp51Δ populations (21), we compared these with rhp51Δ cells by DAPI staining. Fluorescence microscopic observation of nuclear staining revealed that both phenotypes resembled each other, showing elongated cells with decondensed, amorphous and fragmented nuclei (Fig. 5B and D). These results indicate that overexpression of Rhp51 K155A caused dominant negative effects not only on DNA repair but also on normal cell growth.
Table 1. Distribution of lengths of cells overexpressing Rhp51K155A and rhp51Δ.
| Cell length (µm) | Percentage of cells | ||
| |
Wild-type |
rhp51Δ |
Rhp51 K155A |
| 5–10 | 55.8 | 28.4 | 51.7 |
| 10–15 | 39.6 | 44.8 | 31.4 |
| 15–20 | 4.6 | 14.4 | 11.1 |
| 20–25 | 0.0 | 9.3 | 4.8 |
| 25–30 | 0.0 | 2.6 | 1.0 |
| >30 | 0.0 | 0.5 | 0.0 |
| Total no. of cells counted | 217 | 194 | 207 |
Figure 5.
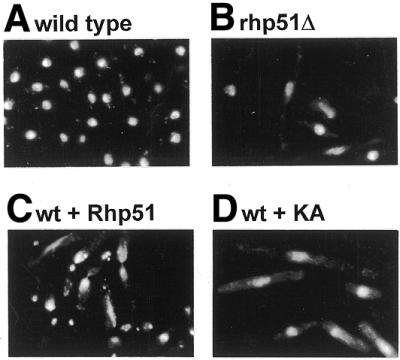
DAPI staining of cells overexpressing Rhp51and Rhp51 K155A. (A) Wild-type, (B) rhp51Δ, (C) wild -type cells overexpressing Rhp51 and (D) wild-type cells overexpressing Rhp51 K155A. Bar 10 µm.
Accumulation of different cell cycle stages on overexpression of Rhp51 and Rhp51 K155A
Detailed cell cycle mutant phenotypes of cells overexpressing Rhp51 were investigated by spindle pole body (SPB) and α-tubulin staining. In normally dividing cells the SPB duplicates and a mitotic spindle is formed between the two new SPBs at late G2 phase. The spindle extends almost immediately and the chromosomes separate at anaphase (Fig. 6A, arrowheads). Therefore, SPB duplication and mitotic spindle formation are landmarks of the onset of mitosis (44). In Rhp51-overexpressing cells showing catastrophic mitosis duplicated SPBs were observed, but no mitotic spindles (Fig. 6C). On the other hand, in cells with a single stretched nucleus a single SPB and interphase spindles were seen (Fig. 6D, arrowheads), indicating that decondensation of the nucleus preceded the appearance of aberrant cytokinesis. Thus this suggests that assembly of the mitotic spindle may have failed due to a lack of chromosome condensation, which provides the spindle-binding sites on chromosomes.
Figure 6.

Indirect immunofluorescence microscopic analysis. Cells overexpressing Rhp51 (C and D) and Rhp51 K155A (E) were collected 25 h after thiamine deprivation and stained with DAPI (left), anti-Sad1 (SPB) (middle) and anti-tubulin (right) antibodies. Wild-type (A) and rhp51Δ (B) cells are also shown. Bar 10 µm.
Staining of cells overexpressing Rhp51 K155A or rhp51Δ cells for SPB and α-tubulin showed that most of the populations of elongated cells contained a single SPB and interphase spindles, indicating that they are largely in interphase (Fig. 5B and E). Anaphase cells formed only a small portion and even cells having two nuclei were largely in interphase, suggested by the lack of a mitotic spindle between the two SPBs (Fig. 5B). These observations indicate that these cells might spend a longer period in G2 phase, where the longitudinal growth of cells takes place, than normal cells.
Next we analyzed the DNA content of these cells by FACS analysis. In normal S.pombe, cells with a 1C DNA content are rarely detected because the short G1 phase has finished by the time they divide (28) (Fig. 7A). In cells overexpressing Rhp51, however, the portion of 1C cells is significantly increased by ∼34.7% 25 h after thiamine deprivation. In addition, populations having >2C DNA contents also increased (Fig. 7C). Owing to the heterogeneity of this population, it is unclear whether appearance of the 1C population is reminiscent of a delay in G1/S progression or the consequence of unequal chromosome segregation. On the other hand, DNA content analysis of rhp51Δ cells and cells overexpressing Rhp51 K155A did not show considerable changes from wild-type cells, indicating that G2 cells dominate in these populations (Fig. 7B and D), as was suggested by the microscopic observations.
Figure 7.

DNA content analysis by FACS of (A) wild-type, (B) rhp51Δ, (C) wild-type cells overexpressing Rhp51 and (D) wild-type cells overexpressing Rhp51 K155A. Cells were collected 15, 20 and 25 h after thiamine deprivation, except for the wild-type and rhp51Δ cells, which were collected at 25 h.
Taken together, overexpression of Rhp51 caused lethality due to aberrant nuclear division accompanied by decondensation of the nucleus and accumulation of cells with a 1C DNA content, suggesting that excess Rhp51 may inhibit DNA replication (see Discussion). On the other hand, all the features of rhp51Δ cells and cells overexpressing Rhp51 K155A, including the elongated cell length, 2C DNA content and lack of mitotic cells, suggest that these cells may be delayed in G2 phase.
DISCUSSION
In the present study we report that distinct genomic instabilities are caused by overexpression of wild-type Rhp51 or the dominant negative K155A mutation of Rhp51. Overexpression of Rhp51 confers lethality to cells, whereas Rhp51 K155A induces cells to exhibit phenotypes similar to the rhp51Δ mutant.
Until now the exact role of ATP hydrolysis by RecA and Rad51 has been controversial (45). In one view ATP hydrolysis appears to be required for disassembly of the RecA monomer from the RecA nucleoprotein filament (46). A mutation in the ATP-binding motif (Rhp51 mutation K155A) completely abolished its DNA repair activity. Rad51 K191A, a similar S.cerevisiae mutant lacking ATP hydrolysis and strand exchange activity, also showed a deficiency in DNA repair (39,47), suggesting that the K155A mutation of Rhp51 is also likely to be enzymatically inactive. On the other hand, the same mutant protein displayed normal self-assembly and binding to Rad22, a Rad52 homolog in fission yeast. Therefore, the DNA damage sensitivity and other phenotypes caused by overexpression of Rhp51 K155A are presumably due to disruption of protein–protein interactions of endogenous Rhp51 by the mutant proteins. Partial rescue by co-overexpression of Rad22 suggests that the interaction with Rad22 may be involved in the dominant negative effect. It is also likely that participation of the mutant protein in formation of the nucleoprotein filament may weaken the strand exchange activity of Rhp51. Similar dominant negative effects were observed in S.cerevisiae after overexpression of a Gal4 DNA-binding domain–Rad51 fusion or Rad51 K191A (40). It is noteworthy, however, that they were not sensitive to UV.
The overexpression phenotypes of the wild-type and a dominant negative mutation of Rhp51 in the absence of DNA-damaging agents provide clues to its role in normal cell growth, especially related to DNA replication. Overexpression of Rhp51 K155A showed characteristics of the rhp51Δ mutant, such as a slow growth rate and cell elongation with a decondensed, fragmented nucleus. In vertebrates Rad51 deficiency results in accumulation of spontaneously occurring DSBs and accumulation of G2/M phase-arrested cells, consequently leading to cell death (48). Studies in bacteria suggested that DSBs actively occur in arrested replication forks and that the repair of such DSBs is a prerequisite for the restart of stalled forks (49,50). If this is also the case in eukaryotes, the accumulation of DSBs in Rad51-deficient cells may be a consequence of stalled replication forks, thereby resulting in a delay in G2 phase. Therefore, the genomic instability caused by a lack of Rhp51 function suggests that recombinational repair mediated by Rhp51 may also be involved in the repair of spontaneous DSBs during DNA replication in fission yeast. Such damage processing or DNA damage itself is a likely candidate as the signal for DNA damage and/or replication checkpoints. The enhanced expression of rhp51+ in S phase further supports its requirement in DNA replication. In that embryonic lethality caused by Rad51 deficiency limits in vivo studies of its function in mammals, overexpression of dominant negative mutations may be an alternative approach to study the nature of the genomic instability.
On the other hand, the phenotypes caused by overexpression of Rhp51 are quite striking. Elevated levels of Rhp51 caused the appearance of cut phenotypes with stretched nuclei and accumulation of cells with a 1C DNA content accompanied by cell death. At the present time the nature of the 1C population is unclear. One example of accumulation of cells with a 1C DNA content is the cut5 mutation (37,43); cut5 is known to participate in DNA replication and damage checkpoints. At the restrictive temperature thermosensitive cut5 mutants transiently accumulate cells with a 1C DNA content, which is a distinct phenomenon among cut mutations.
Since the same level of overexpression of Rhp51 K155A did not kill the cell, it is suggested that Rhp51 requires a functional ATP-binding site in order to bring about cell death followed by aberrant mitosis. Therefore, it is speculated that an active process mediated by Rhp51 would be required to trigger the cut phenotype. Up to now many cut genes have been isolated and characterized (reviewed in 43). It has been shown that the cut phenotype is induced by a failure in chromosome condensation at the onset of mitosis or by a defect in the replication checkpoint. Based on this, we propose two possible roles of Rhp51, which are not mutually exclusive. First, Rhp51 (or homologous recombination) may be involved in chromosome segregation by interacting with the chromosome architecture. The higher order structures of condensed chromosomes, such as sister chromatid cohesion and mitotic chromosome condensation, are regulated by multiprotein complexes known as condensin and cohesin, which are composed of SMC (structural maintenance of chromosome) family proteins and non-SMC components (51). It is known that mutations in SMC or non-SMC components, such as Cut3, Cut14 and Rad18, cause the cut phenotype with a decondensed nucleus (43,52). Hence, the cut phenotype could appear if excess Rhp51 disturbs the stoichiometry of the chromosome architecture, consequently inhibiting chromosome condensation. It is noticeable that two proteins, Rad18 and Rad21, which are epistatic with Rhp51, are involved in SMC complexes as a SMC or non-SMC component, respectively (51,53). Although there is no direct evidence that Rhp51 interacts with Rad18 or Rad21, isolation of brc1, which has BRCT motifs, as a high copy suppressor of the rad18-74 mutation (52) suggests a probable interaction of Rhp51 with SMC components, since the BRCT motif is required for association of BRCA1 and BRCA2 with hRad51 (18).
Secondly, the level of Rhp51 (or recombination) may regulate turning on/off of the DNA damage and/or replication checkpoint. It is probable that excess Rhp51 may compete with the replication machinery for single-stranded DNA substrates during DNA replication and block its normal processing, resulting in replication fork arrest. If the replication checkpoint is not turned on for some reason the cell cycle may proceed without restarting the arrested fork, consequently leading to the cut phenotype. One possible mechanism by which the replication checkpoint could be inhibited by Rhp51 is direct association of Rhp51 with checkpoint components. Cut5 and Crb2 are possible candidates since they contain the BRCT motif (54).
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Jae Bum Kim, Hee Kyung Chung, Kyung Jae Myung, Sang Eun Lee, Neal Sugawara, Andre Walter, James E.Haber and Onyou Hwang for critical reading and comments on this manuscript. We are grateful for the hospitality of Drs K.Gull and I.Hagan for anti-TAT1 and anti-Sad1 antibodies. E.J.P. and S.D.P. are supported by a BK21 Research Fellowship from the Korean Ministry of Education.
References
- 1.Michel B., Ehrlich,S.D. and Uzest,M. (1997) DNA double-strand breaks caused by replication arrest. EMBO J., 16, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- 3.Haber J.E. (1998) Mating-type gene switching in Saccharomyces cerevisiae. Annu. Rev. Genet., 32, 561–599. [DOI] [PubMed] [Google Scholar]
- 4.Shinohara A. and Ogawa,T. (1995) Homologous recombination and the roles of double-strand breaks. Trends Biochem. Sci., 20, 387–391. [DOI] [PubMed] [Google Scholar]
- 5.Baumann P. and West,S.C. (1998) Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci., 23, 247–251. [DOI] [PubMed] [Google Scholar]
- 6.Sung P. (1994) Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science, 265, 1241–1243. [DOI] [PubMed] [Google Scholar]
- 7.Baumann P., Benson,F.E. and West,S.C. (1996) Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell, 87, 757–766. [DOI] [PubMed] [Google Scholar]
- 8.Ogawa T., Yu,X., Shinohara,A. and Egelman,E.H. (1993) Similarity of the yeast RAD51 filament to the bacterial RecA filament. Science, 259, 1896–1899. [DOI] [PubMed] [Google Scholar]
- 9.Sung P. (1997) Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J. Biol. Chem., 272, 28194–28197. [DOI] [PubMed] [Google Scholar]
- 10.Petukhova G., Stratton,S. and Sung,P. (1998) Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature, 393, 91–94. [DOI] [PubMed] [Google Scholar]
- 11.Petukhova G., Van Komen,S., Vergano,S., Klein,H. and Sung,P. (1999) Yeast Rad54 promotes Rad51-dependent homologous DNA pairing via ATP hydrolysis-driven change in DNA double helix conformation. J. Biol. Chem., 274, 29453–29462. [DOI] [PubMed] [Google Scholar]
- 12.Sung P. (1997) Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev., 11, 1111–1121. [DOI] [PubMed] [Google Scholar]
- 13.New J.H., Sugiyama,T., Zaitseva,E. and Kowalczykowski,S.C. (1998) Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature, 391, 407–410. [DOI] [PubMed] [Google Scholar]
- 14.Benson F.E., Baumann,P. and West,S.C. (1998) Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature, 391, 401–404. [DOI] [PubMed] [Google Scholar]
- 15.Shinohara A. and Ogawa,T. (1998) Stimulation by Rad52 of yeast Rad51-mediated recombination. Nature, 391, 404–407. [DOI] [PubMed] [Google Scholar]
- 16.Basile G., Aker,M. and Mortimer,R.K. (1992) Nucleotide sequence and transcriptional regulation of the yeast recombinational repair gene RAD51. Mol. Cell. Biol., 12, 3235–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marsh T.C., Cole,E.S., Stuart,K.R., Campbell,C. and Romero,D.P. (2000) RAD51 is required for propagation of the germinal nucleus in Tetrahymena thermophila. Genetics, 154, 1587–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scully R., Chen,J., Plug,A., Xiao,Y., Weaver,D., Feunteun,J., Ashley,T. and Livingston,D.M. (1997) Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell, 88, 265–275. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto A., Taki,T., Yagi,H., Habu,T., Yoshida,K., Yoshimura,Y., Yamamoto,K., Matsushiro,A., Nishimune,Y. and Morita,T. (1996) Cell cycle-dependent expression of the mouse Rad51 gene in proliferating cells. Mol. Gen. Genet., 251, 1–12. [DOI] [PubMed] [Google Scholar]
- 20.Jang Y.K., Jin,Y.H., Myung,K., Seong,R.H., Hong,S.H. and Park,S.D. (1996) Differential expression of the rhp51+ gene, a recA and RAD51 homolog from the fission yeast Schizosaccharomyces pombe. Gene, 169, 125–130. [DOI] [PubMed] [Google Scholar]
- 21.Jang Y.K., Jin,Y.H., Shim,Y.S., Kim,M.J., Yoo,E.J., Seong,R.H., Hong,S.H. and Park,S.D. (1995) Evidences for possible involvement of Rhp51 protein in mitotic events including chromosome segregation. Biochem. Mol. Biol. Int., 37, 329–337. [PubMed] [Google Scholar]
- 22.Tsuzuki T., Fujii,Y., Sakumi,K., Tominaga,Y., Nakao,K., Sekiguchi,M., Matsushiro,A., Yoshimura,Y. and MoritaT. (1996) Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl Acad. Sci. USA, 93, 6236–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim D.S. and Hasty,P. (1996) A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol. Cell. Biol., 16, 7133–7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sturzbecher H.W., Donzelmann,B., Henning,W., Knippschild,U. and Buchhop,S. (1996) p53 is linked directly to homologous recombination processes via RAD51/RecA protein interaction. EMBO J., 15, 1992–2002. [PMC free article] [PubMed] [Google Scholar]
- 25.Wong A.K.C., Pero,R., Ormonde,P.A., Tavtigian,S.V. and Bartel,P.L. (1997) RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J. Biol. Chem., 272, 31941–31944. [DOI] [PubMed] [Google Scholar]
- 26.Muris D.F., Vreeken,K., Carr,A.M., Murray,J.M., Smit,C., Lohman,P.H. and Pastink,A. (1996) Isolation of the Schizosaccharomyces pombe RAD54 homologue, rhp54+, a gene involved in the repair of radiation damage and replication fidelity. J. Cell Sci., 109, 73–81. [DOI] [PubMed] [Google Scholar]
- 27.Khasanov F.K., Savchenko,G.V., Bashkirova,E.V., Korolev,V.G., Heyer,W.D. and Bashkirov,V.I. (1999) A new recombinational DNA repair gene from Schizosaccharomyces pombe with homology to Escherichia coli RecA. Genetics, 152, 1557–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alfa C.E., Fantes,P., Hyams,J.S., McLeod,M. and Warbrick,E. (1993) Experiments with Fission Yeast: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 29.Venkitaraman A.R. (1989) Use of modified T7 DNA polymerase (Sequenase version 2.0) for oligonucleotide site-directed mutagenesis. Nucleic Acids Res., 17, 3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jang Y.K., Jin,Y.H., Kim,E.M., Fabre,F., Hong,S.H. and Park,S.D. (1994) Cloning and sequence analysis of rhp51+, a Schizosaccharomyces pombe homolog of the Saccharomyces cerevisiae RAD51 gene. Gene, 142, 207–211. [DOI] [PubMed] [Google Scholar]
- 31.Gietz R.D. and Sugino,A. (1988) New yeast–Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene, 74, 527–534. [DOI] [PubMed] [Google Scholar]
- 32.Basi G., Schmid,E. and Maundrell,K. (1993) TATA box mutations in the Schizosaccharomyces pombe nmt1 promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene, 123, 131–136. [DOI] [PubMed] [Google Scholar]
- 33.Maundrell K. (1993) Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene, 123, 127–130. [DOI] [PubMed] [Google Scholar]
- 34.Guthrie C., Fink,G.R. and Colowick,S.P. (1991) Guide to yeast genetics and molecular biology. Methods Enzymol., 194, 821–823. [PubMed] [Google Scholar]
- 35.Woods A., Sherwin,T., Sasse,R., MacRae,T.H., Baines,A.J. and Gull,K. (1989) Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies. J. Cell Sci., 93, 491–500. [DOI] [PubMed] [Google Scholar]
- 36.Hagan I. and Yanagida,M. (1995) The product of the spindle formation gene sad1+ associates with the fission yeast spindle pole body and is essential for viability. J. Cell Biol., 129, 1033–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoo E.J., Jin,Y.H., Jang,Y.K., Bjerling,P., Tabish,M., Hong,S.H., Ekwall,K. and Park,S.D. (2000) Fission yeast hrp1, a chromodomain ATPase, is required for proper chromosome segregation and its overexpression interferes with chromatin condensation. Nucleic Acids Res., 28, 2004–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Story R.M., Bishop,D.K., Kleckner,N. and Steitz,T.A. (1993) Structural relationship of bacterial RecA proteins to recombination proteins from bacteriophage T4 and yeast. Science, 259, 1892–1896. [DOI] [PubMed] [Google Scholar]
- 39.Sung P. and Stratton,S.A. (1996) Yeast Rad51 recombinase mediates polar DNA strand exchange in the absence of ATP hydrolysis. J. Biol. Chem., 271, 27983–27986. [DOI] [PubMed] [Google Scholar]
- 40.Donovan J.W., Milne,G.T. and Weaver,D.T. (1994) Homotypic and heterotypic protein associations control Rad51 function in double-strand break repair. Genes Dev., 8, 2552–2562. [DOI] [PubMed] [Google Scholar]
- 41.Hays S.L., Firmenich,A.A. and Berg,P. (1995) Complex formation in yeast double-strand break repair: participation of Rad51, Rad52, Rad55 and Rad57 proteins. Proc. Natl Acad. Sci. USA, 92, 6925–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim W.J., Lee,S., Park,M.S., Jang,Y.K., Kim,J.B. and Park,S.D. (2000) Rad22 protein, a Rad52 homologue in Schizosaccharomyces pombe, binds to DNA double-strand breaks. J. Biol. Chem., 275, 35607–35611. [DOI] [PubMed] [Google Scholar]
- 43.Yanagida M. (1998) Fission yeast cut mutations revisited: control of anaphase. Trends Cell Biol., 8, 144–149. [DOI] [PubMed] [Google Scholar]
- 44.Nasim A., Young,P. and Johnson,B.F. (1989) Molecular Biology of the Fission Yeast. Academic Press, San Diego, CA.
- 45.Cox M.M. (1994) Why does RecA protein hydrolyse ATP? Trends Biochem. Sci., 19, 217–222. [DOI] [PubMed] [Google Scholar]
- 46.Kowalczykowski S.C. (1991) Biochemistry of genetic recombination: energetics and mechanism of DNA strand exchange. Annu. Rev. Biophys. Biophys. Chem., 20, 539–575. [DOI] [PubMed] [Google Scholar]
- 47.Shinohara A., Ogawa,H. and Ogawa,T. (1992) Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell, 69, 457–470. [DOI] [PubMed] [Google Scholar]
- 48.Sonoda E., Sasaki,M.S., Buerstedde,J.M., Bezzubova,O., Shinohara,A., Ogawa,H., Takata,M., Yamaguchi-Iwai,Y. and Takeda,S. (1998) Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J., 17, 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michel B. (2000) Replication fork arrest and DNA recombination. Trends Biochem. Sci., 25, 173–178. [DOI] [PubMed] [Google Scholar]
- 50.Flores-Rozas H. and Kolodner,R.D. (2000) Links between replication, recombination and genome instability in eukaryotes. Trends Biochem. Sci., 25, 196–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirano T. (1999) SMC-mediated chromosome mechanics: a conserved scheme from bacteria to vertebrates? Genes Dev., 13, 11–19. [DOI] [PubMed] [Google Scholar]
- 52.Verkade H.M., Bugg,S.J., Lindsay,H.D., Carr,A.M. and O’Connell,M.J. (1999) Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol. Biol. Cell, 10, 2905–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fousteri M.I. and Lehmann,A.R. (2000) A novel SMC protein complex in Schizosaccharomyces pombe contains the Rad18 DNA repair protein. EMBO J., 19, 1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saka Y. and Yanagida,M. (1993) Fission yeast cut5+, required for S phase onset and M phase restraint, is identical to the radiation-damage repair gene rad4+. Cell, 74, 383–393. [DOI] [PubMed] [Google Scholar]