

Abstract
Xanthine oxidase (XO) is a critical source of reactive oxygen species (ROS) that contribute to vascular inflammation. Binding of XO to vascular endothelial cell glycosaminoglycans (GAGs) results in significant resistance to inhibition by traditional pyrazolopyrimidine-based inhibitors such as allopurinol. Therefore, we compared the extent of XO inhibition (free and GAG-bound) by allopurinol to febuxostat, a newly approved nonpurine XO-specific inhibitor. In solution, febuxostat was 1000 fold more potent than allopurinol inhibition of XO-dependent uric acid formation (IC50 = 1.8 nM vs. 2.9 μM). Association of XO with heparin-Sepharose 6B (HS6B-XO) had minimal effect on inhibition of uric acid formation by febuxostat (IC50 = 4.4 nM) while further limiting the effect of allopurinol (IC50 = 64 μM). Kinetic analysis of febuxostat inhibition revealed Ki values of 0.96 nM (free) and 0.92 nM (HS6B-XO), confirming equivalent inhibition for both free and GAG-immobilized enzyme. When XO was bound to endothelial cell GAGs, complete enzyme inhibition was observed with 25 nM febuxostat, while no more than 80% inhibition was seen with either allopurinol or oxypurinol, even at concentrations above those tolerated clinically. The superior potency for inhibition of endothelium-associated XO is predictive of a significant role for febuxostat in investigating pathological states where XO-derived ROS are contributive and traditional XO inhibitors are only slightly effective.
Introduction
The molybdoflavin enzyme, xanthine oxidoreductase (XOR) catalyzes the terminal two steps of purine degradation (hypoxanthine → xanthine → uric acid) in humans. XOR is transcribed as a single gene product, xanthine dehydrogenase (XDH) where substrate-derived electrons at the Mo-cofactor of XDH are transferred via two Fe/S centers to a FAD-cofactor where NAD+ is reduced to NADH. During inflammatory conditions, post-translational modification by oxidation of critical cysteine residues or limited proteolysis converts XDH to xanthine oxidase (XO) (1,2). The key difference distinguishing XO from XDH is the structural conformation and electrostatic microenvironment surrounding the FAD-cofactor resulting in XO’s lower affinity for NAD+ and enhanced affinity for O2 (3). Substrate-derived electrons at the Mo-cofactor of XO reduce O2 at the FAD-cofactor both divalently, forming hydrogen peroxide (H2O2) and univalently, generating superoxide (O2 •-). However, conversion to XO is not requisite for ROS production as XDH displays partial oxidase activity under conditions in which NAD+ levels are diminished such as the ischemic/hypoxic microenvironment encountered in vascular inflammation (4). This same inflammatory milieu leads to enhanced XO levels and thus increased XO-derived ROS formation resulting in activation of redox-dependent cell signaling reactions and alterations in vascular function. Evidence of this role for XO is exemplified by numerous studies in which XO inhibition attenuates vascular dysfunction, including congestive heart failure, sickle cell anemia and diabetes (5-8).
The splanchnic system, the site of highest XDH specific activity, readily releases XDH into the circulation in response to ischemic/hypoxic or inflammatory insults (9,10). Once released, XDH is rapidly converted to XO by plasma proteases. Pockets of cationic amino acid motifs present on XO confer a high affinity (Kd = 6 nM) for negatively charged glycosaminoglycans (GAGs) on the luminal face of endothelial cells (11). This XO-GAG association induces substantial sequestration and thus amplification of local endothelial XO concentration, producing a microenvironment primed for enhanced ROS production. Of crucial importance, GAG-association also results in resistance to XOR inhibition by oxypurinol, the active metabolite of allopurinol, increasing the Ki from 230 nM for soluble XO to 405 nM for GAG-bound XOR (12,13). Combined, amplification of endothelial XO-derived ROS formation and GAG immobilization-induced resistance to inhibition results in a setting that exacerbates inflammation with inextinguishable vascular ROS formation.
While inhibition of XO-derived uric acid formation and resultant symptoms of gout has been accomplished successfully for over 50 years by clinical administration of allopurinol, only partial reduction of vascular inflammatory-related symptoms and restoration of function has been observed by allopurinol-based inhibition approaches. This phenomenon may be explained, in part, by examination of allopurinol reaction with the Mo-cofactor of XO. Allopurinol is a classic “suicide inhibitor” as its binding to and reduction of the Mo-cofactor induces self-oxidation to form oxypurinol (the active inhibitory metabolite). Reduction of the Mo-cofactor by allopurinol ultimately leads to electron transfer to the FAD resulting in reduction of O2 (14). It is equally important to note that oxypurinol binding and resultant inhibition requires the Mo-cofactor to be reduced (15). This is accomplished by initial reaction of allopurinol or, in the case of treatment with pure oxypurinol, XO substrates such as xanthine must provide the electrons. In either case, both allopurinol and oxypurinol require enzyme turn-over resulting in ROS formation before inhibition is attained. This undesirable action of allo/oxypurinol combined with the reduced capacity to inhibit endothelial GAG-associated XO may lead to significant misinterpretation of ROS-driven vascular pathology where XO is contributory. These limitations emphasize the need for alternative inhibitors with mechanisms independent of enzyme redox status and unaffected by GAG-immobilization.
Febuxostat, an XO-specific inhibitor recently approved by the FDA for clinical use, is reported to be significantly more potent than allopurinol (16). Kinetic analysis conducted at pH 8.5, near the pH optimum for xanthine/Mo-cofactor reaction, produced a Ki for febuxostat nearly 6000 times lower than allopurinol 0.12 nM vs. 700 nM, respectively (16). Detailed crystallography studies revealed febuxostat reaction with XO is confined to critical amino acid residues in the tunnel leading to the Mo-cofactor where it effectively blocks substrate access to the active site (17). Thus, febuxostat should not be affected by enzyme redox state and interaction with XO should not induce ROS formation. These characteristics of enhanced potency and independence from enzyme turn-over suggest that febuxostat may provide new insights into XO-dependent contributions to inflammatory disease. Therefore, we examined the inhibition properties of febuxostat for soluble XO, GAG-bound XO and endothelial cell-bound XO at physiological pH.
Materials and Methods
Materials
Heparin-Sepharose CL-6B and PD10 G25 Sephadex columns were from GE Healthcare (USA). Xanthine, allopurinol, catalase and native cytochrome c were from Sigma (St. Louis, Mo), while Febuxostat was purchased from Axon Medchem BV (The Netherlands). Xanthine oxidase was purchased from Calbiochem (USA). Medium 199 (M199) and fetal bovine serum (FBS) were from Invitrogen (Carlsbad, CA). Superoxide dismutase (CuZnSOD) was from OXIS International Inc. (Portland, OR). The EPR spin probe 1-hydroxy-4-phosphono-oxy-2,2,6,6-tetramethylpi-peridine (PPH) was from Enzo Life Sciences (Plymouth Meeting, PA).
Enzyme Analysis
Enzymatic activity was determined either spectrophotometrically by the rate of uric acid formation monitored at 292 nm in 50 mM potassium phosphate (KPi), pH 7.4 (ε = 11 mM-1 cm-1) or electrochemically via reverse phase HPLC analysis of uric acid production (ESA CoulArray System, Chelmsford, MA) as previously (12). Activity is expressed as 1 Unit = 1 μmole urate/min. Formation of O2 •- was assessed by the SOD-inhibitable reduction of cytochrome c (550 nm) (18) and EPR spin trapping (see below).
Xanthine Oxidase Binding to Heparin-Sepharose 6B
Xanthine oxidase was bound to Heparin-Sepharose-6B (HS6B) as we previously reported (12). Briefly, HS6B was washed with 5 mM KPi, incubated with XO for 45 min at 25°C and washed free of non-associated enzyme. Activity of the suspension was determined by following the formation of uric acid (λ = 292). Reactions were initiated by addition of xanthine (100 μM) and maintained under continuously stirred conditions at 25°C. Final activity of HS6B-XO used for inhibition studies was identical to studies with XO free in solution.
Cell Culture and Cell-Bound XO Inhibition Studies
Bovine aortic endothelial cells (BAEC) were isolated as we previously described (19). Primary cell culture, routine passage, and experimental manipulations were all conducted in the absence of proteases. Cells were propagated by subculturing in a 1:4 ratio in M199 containing 10% FBS and 10 μM thymidine. For inhibition studies, when cells reached confluence, the medium was replaced with fresh M199(-) (formulated without xanthine and hypoxanthine) containing XO (5 mU/ml) for 20 min at 25°C. We have reported that this process minimizes enzyme internalization (20). The XO-containing media was then removed, cells were washed, fresh M199(-) containing designated concentrations of inhibitor was added and reactions initiated by the addition of 100 μM xanthine. After 1 h, aliquots of the medium were removed and tested for uric acid formation by reverse-phase HPLC electrochemical analysis. Uricase activity of the medium was undetectable as determined by the addition of known concentrations of uric acid with or without cells (37°C) and then monitoring the loss of uric acid over time.
EPR Studies
Confluent bovine aortic endothelial cells (BAEC) were exposed to XO (5 mU/ml) for 20 min at 25°C, harvested by mechanical dissociation, washed thoroughly (3 times with ice-cold PBS, pH 7.4), resuspended as a single-cell suspension (1 × 106 cells /ml) and placed on ice (for less than 30 min) until warmed to 37°C immediately before evaluation in the EPR. Aliquots (50 μL) of the cell suspension were exposed to indicated concentrations of inhibitor and 50 μM of the membrane impermeable EPR spin probe PPH (21) followed by the addition of xanthine (100 μM). The samples were immediately transferred to a 50 μL glass pipette and placed into a temperature and gas-controlled Bruker eScan Table Top EPR spectrometer cavity and analyzed for 10 min at 37°C and 21% O2. Spectra represent 5 signal-averaged scans from t = 9-10 min. The EPR instrument settings were as follows: field sweep 50 G; microwave frequency 9.78 GHz; microwave power 20 mW; modulation amplitude 2 G; conversion time 327 ms; time constant 655 ms; and receiver gain 1×105. To minimize the effects of adventitious metals, all buffers were treated with Chelex resin and contained 25 μM deferoxamine.
RESULTS
Reaction of allopurinol with XO produces O2 •-
Allopurinol reduces the Mo-cofactor of XO by 2 electrons during conversion to oxypurinol. These electrons subsequently reduce the FAD-cofactor where reaction with O2 produces ROS. This is illustrated in Fig. 1 where exposure of purified XO to allopurinol results in significant O2 •- formation while treatment with febuxostat generates no detectable O2 •-, Fig. 1A. Confirmation of O2 •- formation from the XO-allopurinol reaction was accomplished by EPR with PPH, Fig. 1B. The PP• radical intensity increased in a time-dependent manner (A-C), was completely abolished by SOD (D) and was not observed when XO was exposed to 100 nM febuxostat (E).
Fig. 1. Reaction of allopurinol with XO induces ROS formation.
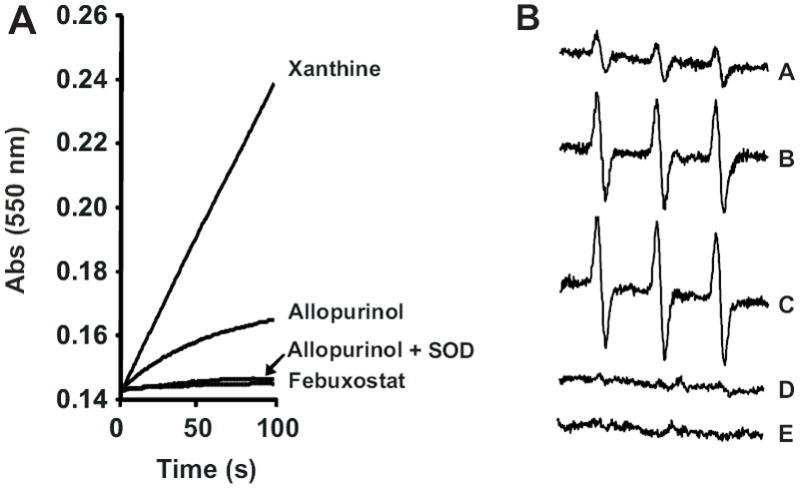
(A) XO (10 mU/ml) was exposed to either xanthine (50 μM), allopurinol (50 μM), Febuxostat (100 nM) or allopurinol (50 μM) + SOD (10 U/ml) and O2 •- formation monitored by the reduction of cytochrome c (λ = 550 nm) over time. (B) Xanthine oxidase (10 mU/ml) was exposed to allopurinol (50 μM) and O2 •- formation monitored by EPR spin trapping with PPH (100 μM). Spectra represent the following conditions: allopurinol 25, 50 and 100 s (A-C, respectively), 100 s + SOD (D) and 100 s + febuxostat (E).
Inhibition of XO in solution
When soluble XO (10 mU/ml in PBS, pH 7.4) was exposed to increasing concentrations of inhibitor while assessing uric acid formation from xanthine (50 μM), febuxostat was > 1000 times more potent than allopurinol (IC50 = 2.9 μM vs. 1.8 nM, respectively), Fig. 2A. Similarly, febuxostat more potently inhibited O2 •- formation than allopurinol (IC50 = 3.9 μM vs. 0.9 nM, respectively). Fig. 2B.
Fig. 2. Febuxostat is more potent than allopurinol at inhibiting XO free in solution.
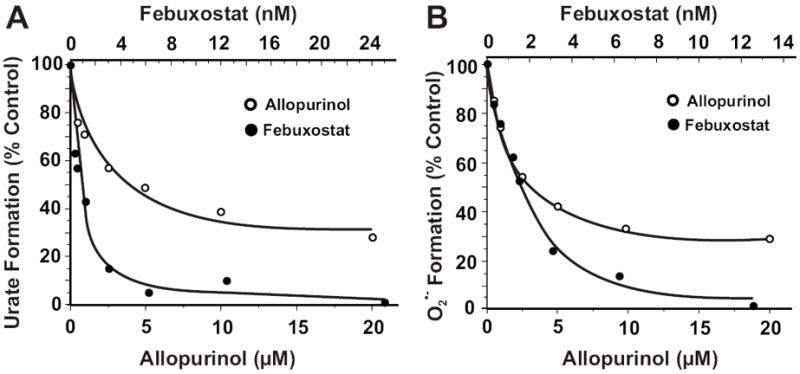
(A) Xanthine oxidase (2 mU/ml, 5 mM KPi, pH 7.4) was exposed to various concentrations of either allopurinol or febuxostat and assessed for formation uric acid (λ = 295 nm) upon the addition of xanthine (50 μM). Shown are the initial reaction rates (V0) plotted as % control (no inhibitor). IC50 values were calculated as the ordinate value of 50% inhibition (allopurinol = 2.9 μM and febuxostat = 1.8 nM). (B) Same as A except O2 •- formation was detected by the reduction of cytochrome c (λ = 550) and IC50 values were 3.9 μM for allopurinol and 0.9 nM for febuxostat.
Inhibition of HS6B-immobilized XO
Immobilization of XO on HS6B significantly increased the IC50 for allopurinol inhibition of uric acid formation (IC50 = 64 μM) while XO binding to HS6B had minimal effect on inhibition of urate formation by febuxostat (IC50 = 4.4 nM), Fig. 3A. The same disparity between the effectiveness of allopurinol and febuxostat was also observed when monitoring O2 •- formation where IC50 = 4.6 nM (febuxostat) and 77 μM (allopurinol), Fig. 3B.
Fig. 3. Febuxostat inhibition of GAG-immobilized XO is superior to allopurinol.
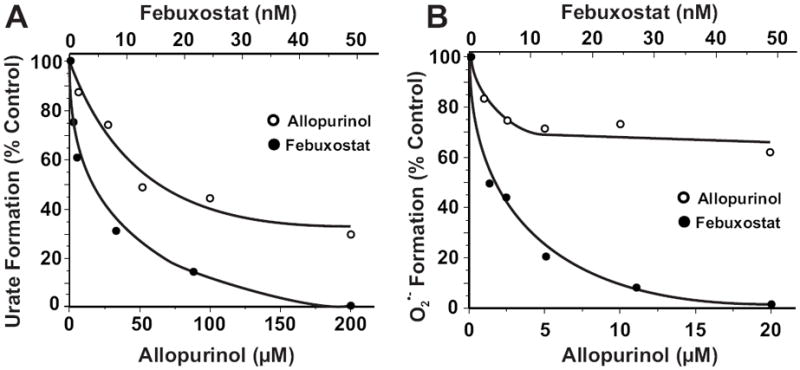
(A) HS6B-XO (2 mU/ml, 5 mM KPi, pH 7.4) was exposed to various concentrations of either allopurinol or febuxostat and assessed for formation of uric acid (λ = 295 nm) upon the addition of xanthine (200 μM). Shown are the initial reaction rates (V0) plotted as % control (no inhibitor). IC50 values were calculated as the ordinate value of 50% inhibition (allopurinol = 64 μM and febuxostat = 4.4 nM). (B) Same as A except O2 •- formation was detected by the reduction of cytochrome c (λ = 550 nm) and IC50 values were 77 μM for allopurinol and 4.6 nM for febuxostat.
Kinetic analysis of febuxostat for free and HS6B XO
Plotting the initial velocity of uric acid formation versus inhibitor concentration using different concentrations of substrate (Dixon Plot) produced inhibition constant (Ki) values for febuxostat, Fig. 4. Inhibition of XO, free in solution, generated a Ki = 0.96 nM for febuxostat, Fig. 4A. Importantly, this inhibition constant was not affected by immobilization of XO where the Ki = 0.92 nM, Fig. 4B. It is important to note that due to the extreme potency of febuxostat, inhibitor concentrations used in this study approached those of the enzyme whereas concentrations of inhibitor significantly higher than the enzyme resulted in extreme difficulty differentiating differences in uric acid formation.
Fig. 4. Kinetic analysis of XO inhibition by febuxostat.
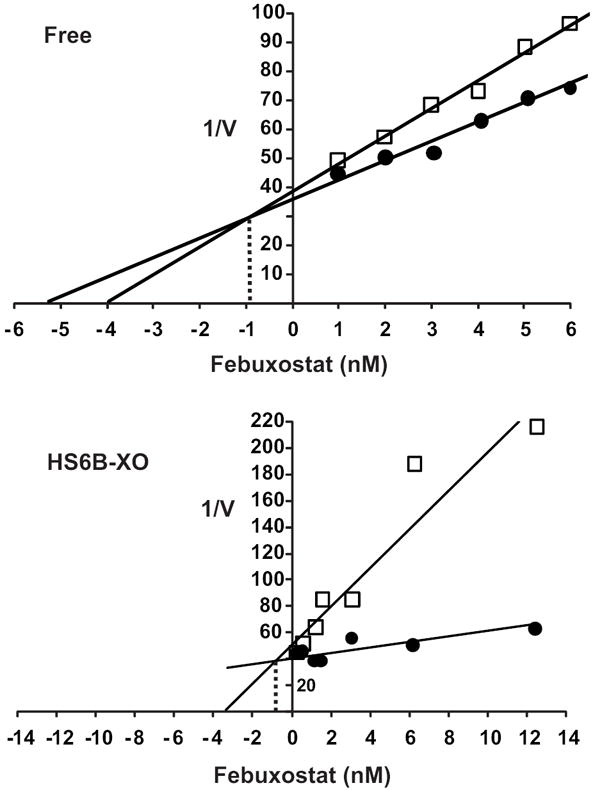
(A) Xanthine oxidase (2 mU/ml, 5 mM KPi, pH 7.4) was exposed to various concentrations of febuxostat and assessed for formation uric acid (λ = 295) using 2 concentrations of xanthine (10 and 100 μM). Shown is a Dixon plot (1/V vs. [inhibitor]) generating an inhibition constant (Ki) = 0.96 nM for febuxostat. (B) HS6B-XO (2 mU/ml, 5 mM KPi, pH 7.4) was used with xanthine (10 and 100 μM) generating an inhibition constant (Ki) = 0.92 nM for febuxostat. The Ki values were calculated as the ordinate value of the intersection of the 2 lines.
Inhibition of endothelial cell-bound XO
Purified XO was bound to BAEC GAGs as we have previously reported (20) and the capacity of febuxostat to inhibit uric acid formation was compared to both allopurinol and oxypurinol, Fig. 5. Complete inhibition was not achieved by either allopurinol or oxypurinol at concentrations above those seen clinically (20-90 μM) (22), Fig. 5A. In contrast, exposure of XO-associated BAEC to 25 nM febuxostat completely inhibited uric acid formation (23), Fig. 5A (inset). Additional analysis were performed where O2 •- formation from endothelial cell-bound XO + xanthine was measured by EPR with the cell impermeable spin probe PPH, Fig. 5B. A robust PP• signal was produced upon the addition of xanthine which was completely inhibited by SOD and was not observed when XO was removed from the surface of the cells by brief treatment with trypsin. Comparison of the two XO inhibitors revealed complete inhibition of endothelial cell-bound XO-derived O2 •- formation by 25 nM febuxostat whereas concentrations of allopurinol up to 200 μM were not effective in extinguishing radical formation.
Fig. 5. Febuxostat is superior to allo/oxypurinol at inhibiting endothelial cell-bound XO.
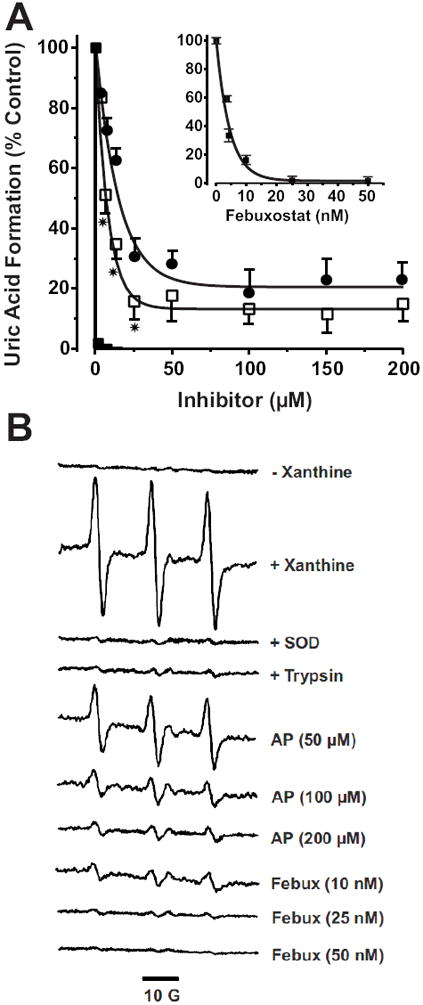
A) BAECs were exposed to purified XO (5 mU/ml) for 20 min 25°C and washed as described in the methods. Inhibitors were added at the indicated concentrations followed by xanthine (100 μM) and uric acid levels assessed after 1 h. Values represent % control (no inhibitor) and are the mean of at least 3 independent determinations (* = p < 0.05). B) Purified XO was bound to BAEC GAGs (as in A), the cells were then harvested by mechanical dissociation and resuspended at 1 × 106 cells/ml. Cell suspensions were exposed to the indicated concentrations of inhibitor followed by xanthine (100 μM) and immediately analyzed by EPR spin trapping of extracellular O2 •- with PPH (50 μM). Spectra represent XO-loaded cells exposed to PPH and the following agents from top to bottom: (- xanthine), (+ xanthine), (+ xanthine and SOD), (100 U/ml), (XO-loaded cells treated with trypsin to remove extracellular XO and then exposed to xanthine), (+ xanthine and 50, 100 or 200 μM allopurinol), (+ xanthine and 10, 25 and 50 nM febuxostat). Each spectrum represents 5 cumulative scans over 1 min from t = 9-10 min at 37°C and 21% O2.
Discussion
Although successful in the prevention of gout associated with elevated plasma uric acid levels, allopurinol (and oxypurinol) treatment for diseases in which XO-derived ROS are thought to contribute has been disappointing. This may, in part, be explained by two factors that strongly influence the extent and nature of XO inhibition by allopurinol. First, inflammatory and/or hypoxic conditions enhance tissue XO expression, and these conditions also promote sequestration and immobilization of XO by endothelial GAGs. This in turn confers resistance of XO to inhibition by allo/oxypurinol (11,12,19). Second, allopurinol reaction with and reduction of the Mo-cofactor results in enzyme turn-over and ultimately the undesirable production of O2 •- (14), Fig. 1. These limitations in the use of allopurinol to modulate XO-derived reactive species in pathogenic events can result in substantially underestimating the impact of XO and affirm the need for new inhibition approaches with mechanisms independent of enzyme redox state as well as superior efficacy towards GAG-bound XO.
Febuxostat is reported to be more potent than allo/oxypurinol and mechanistically independent of reaction with the Mo-cofactor of XO and as such, independent of the redox state of the enzyme (16). However, these kinetic analyses were performed at pH = 8.5 where XO-xanthine interactions are optimal. When inhibition of XO-derived uric acid and O2 •- formation at pH 7.4 was examined febuxostat was found to be greater than 3 orders of magnitude more potent than allopurinol, confirming these previous reports, Fig. 2. More importantly however, the extent of febuxostat inhibition of both uric acid and O2 •- formation remained relatively unaffected by immobilization of XO on HS6B compared to allopurinol, Fig. 3. The IC50 values for febuxostat increased ~2.5-fold for both uric acid and O2 •- while allopurinol IC50 values increased over 22-fold. Complete inhibition of uric acid and O2 •- production from HS6B-XO by febuxostat, however, was attained at or below 50 nM, a concentration well below its reported Cmax of 15 μM (23). In addition, kinetic analysis also revealed nearly equivalent inhibition constants for both soluble and HS6B-XO 0.96 vs. 0.92 nM, respectively. These values are ~8-fold higher than those reported previously and may reflect the pH differences (8.5 vs. 7.4) where slight alterations to the electrostatic microenvironment of the tunnel leading to the active site of XO could modify febuxostat binding. It is also important to note that inhibition of XO with allopurinol is time dependent as time is requisite for Mo-dependent oxidation of allopurinol to oxypurinol and subsequent building of oxypurinol levels necessary to achieve observable inhibition. This factor may skew our results in favor of febuxostat since initial reaction rates were used to compare the two inhibitors. However, these results remain highly suggestive that febuxostat will be more efficacious than allopurinol at inhibiting XO-derived ROS in the vasculature where a significant amount of enzyme will be immobilized on extracellular GAGs.
To bridge the gap between biochemical studies with artificial GAGs and more biologically relevant settings, endothelial cell GAGs were treated with XO and comparative inhibition studies were performed. Both allopurinol and oxypurinol incompletely inhibited uric acid formation from cell-bound XO, even at concentrations above those achievable in the clinic (e.g. 200 μM), Fig. 5A. On the other hand, 30 nM febuxostat abolished uric acid formation from endothelial cell-bound XO. The superior potency of febuxostat was also observed when XO-derived O2 •- production was assessed by EPR, Fig. 5B. Following XO loading of cellular GAGs, the addition of xanthine in the presence of the membrane-impermeable spin probe PPH induced a robust production of PP• radical. This PP• signal was completely abolished by the presence of SOD verifying O2 •- as the seminal reactant. In addition, when the cells were briefly treated with trypsin to remove the XO from the extracellular GAGs and then exposed to xanthine, the radical intensity was reduced to near control levels confirming the detected O2 •- was generated by XO bound to the extracellular surface of the cells. Exposing XO-loaded BAEC to increasing concentrations of allopurinol (50-200 μM) produced a concentration-dependent reduction in PP• signal. However, 200 μM allopurinol, a concentration well above clinically achievable levels (9-90 μM), resulted in only 80% reduction of radical formation (24). The failure of allopurinol to fully inhibit endothelial cell-bound O2 •- production may be due to the combination of immobilization-induced resistance to inhibition and allopurinol-induced O2 •- formation. This is an important point when considering the local vascular inflammatory milieu where substrate (hypoxanthine + xanthine) levels have been reported to reach 50-100 μM and as such would offer robust competition with allopurinol for the Mo-cofactor (25,26). In contrast to allopurinol, febuxostat (25 and 50 nM) was effective at reducing cell-bound O2 •- production by 96 and 100%, respectively. Combined, these data suggest febuxostat is a substantially more effective inhibitor of endothelial cell-associated XO than allopurinol.
Limitations to the extent of inhibition of immobilized XO as well as adventitious ROS formation from allo/oxypurinol clearly identify a critical deficiency for use of these inhibitors to verify contributions of XO to various inflammatory processes. Addressing this crucial issue, we demonstrate the superior potency of febuxostat as an inhibitor of soluble, GAG-bound and endothelial cell-bound XO. Additionally, we determined that febuxostat reaction with XO does not induce ROS formation. Combined, these results establish febuxostat as a critical adjuvant for investigating contributory roles of XO in inflammatory disease.
Acknowledgments
This work was supported by AHA Scientist Development Grant 10SDG3560005 and University of Pittsburgh Vascular Medicine Institute Seed Grant (EEK), Howard Hughes International Research Scholar (RR), NIH HL8115 and HL64937 (BAF).
Abbreviations
- GAGs
glycosaminoglycans
- ROS
reactive oxygen species
- XDH
xanthine dehydrogenase
- XO
xanthine oxidase
- XOR
xanthine oxidoreductase
References
- 1.Amaya Y, Yamazaki K, Sato M, Noda K, Nishino T. Proteolytic conversion of xanthine dehydrogenase from the NAD-dependent type to the O2-dependent type. Amino acid sequence of rat liver xanthine dehydrogenase and identification of the cleavage sites of the enzyme protein during irreversible conversion by trypsin. J Biol Chem. 1990;265:14170–14175. [PubMed] [Google Scholar]
- 2.Waud WR, Rajagopalan KV. The mechanism of conversion of rat liver xanthine dehydrogenase from an NAD+-dependent form (type D) to an O2-dependent form (type O) Arch Biochem Biophys. 1976;172:365–379. doi: 10.1016/0003-9861(76)90088-6. [DOI] [PubMed] [Google Scholar]
- 3.Enroth C, Eger BT, Okamoto K, Nishino T, Nishino T, Pai EF. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: Structure-based mechanism of conversion. Proc Natl Acad Sci USA. 2000;97:10723–10728. doi: 10.1073/pnas.97.20.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris CM, Massey V. The reaction of reduced xanthine dehydrogenase with molecular oxygen. Reaction kinetics and measurment of superoxide radical. J Biol Chem. 1997;272:8370–8379. doi: 10.1074/jbc.272.13.8370. [DOI] [PubMed] [Google Scholar]
- 5.Butler R, Morris AD, Belch JJ, Hill A, Struthers AD. Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension. 2000;35:746–751. doi: 10.1161/01.hyp.35.3.746. [DOI] [PubMed] [Google Scholar]
- 6.Desco MC, Asensi M, Marquez R, Martinez-Valls J, Vento M, Pallardo FV, Sastre J, Vina J. Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol. Diabetes. 2002;51:1118–1124. doi: 10.2337/diabetes.51.4.1118. [DOI] [PubMed] [Google Scholar]
- 7.Farquharson CA, Butler R, Hill A, Belch JJ, Struthers AD. Allopurinol improves endothelial dysfunction in chronic heart failure. Circulation. 2002;106:221–226. doi: 10.1161/01.cir.0000022140.61460.1d. [DOI] [PubMed] [Google Scholar]
- 8.Aslan M, Ryan TM, Adler B, Townes TM, Parks DA, Thompson JA, Tousson A, Gladwin MT, Patel RP, Tarpey MM, Batinic-Haberle I, White CR, Freeman BA. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc Natl Acad Sci USA. 2001;98:15215–15220. doi: 10.1073/pnas.221292098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giler S, Sperling O, Brosh S, Urca I, De Vries A. Serum xanthine oxidase in jaundice. Clin Chim Acta. 1975;63:37–40. doi: 10.1016/0009-8981(75)90375-7. [DOI] [PubMed] [Google Scholar]
- 10.Ramboer C, Piessens F, De Groote J. Serum xanthine oxidase and liver disease. Digestion. 1972;7:183–195. doi: 10.1159/000197273. [DOI] [PubMed] [Google Scholar]
- 11.Houston M, Estevez A, Chumley P, Aslan M, Marklund S, Parks DA, Freeman BA. Binding of xanthine oxidase to vascular endothelium. Kinetic characterization and oxidative impairment of nitric oxide-dependent signaling. J Biol Chem. 1999;274:4985–4994. doi: 10.1074/jbc.274.8.4985. [DOI] [PubMed] [Google Scholar]
- 12.Kelley EE, Trostchansky A, Rubbo H, Freeman BA, Radi R, Tarpey MM. Binding of xanthine oxidase to glycosaminoglycans limits inhibition by oxypurinol. J Biol Chem. 2004;279:37231–37234. doi: 10.1074/jbc.M402077200. [DOI] [PubMed] [Google Scholar]
- 13.Radi R, Rubbo H, Bush K, Freeman BA. Xanthine oxidase binding to glycosaminoglycans: kinetics and superoxide dismutase interactions of immobilized xanthine oxidase-heparin complexes. Arch Biochem Biophys. 1997;339:125–135. doi: 10.1006/abbi.1996.9844. [DOI] [PubMed] [Google Scholar]
- 14.Galbusera C, Orth P, Fedida D, Spector T. Superoxide radical production by allopurinol and xanthine oxidase. Biochem Pharmacol. 2006;71:1747–1752. doi: 10.1016/j.bcp.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Massey V, Komai H, Palmer G, Elion GB. On the mechanism of inactivation of xanthine oxidase by allopurinol and other pyrazolo[3,4-d]pyrimidines. J Biol Chem. 1970;245:2837–2844. [PubMed] [Google Scholar]
- 16.Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem. 2003;278:1848–1855. doi: 10.1074/jbc.M208307200. [DOI] [PubMed] [Google Scholar]
- 17.Okamoto K, Nishino T. Crystal structures of mammalian xanthine oxidoreductase bound with various inhibitors: allopurinol, febuxostat, and FYX-051. J Nippon Med Sch. 2008;75:2–3. doi: 10.1272/jnms.75.2. [DOI] [PubMed] [Google Scholar]
- 18.Fridovich I. Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. J Biol Chem. 1970;245:4053–4057. [PubMed] [Google Scholar]
- 19.Kelley EE, Hock T, Khoo NKH, Richardson GR, Johnson KK, Powell PC, Giles GI, Agarwal A, Lancaster JR, Jr, Tarpey MM. Moderate hypoxia induces xanthine oxidoreductase activity in arterial endothelial cells. Free Radic Biol Med. 2006;40:952–959. doi: 10.1016/j.freeradbiomed.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Kelley EE, Batthyany CI, Hundley NJ, Woodcock SR, Bonacci G, Del Rio JM, Schopfer FJ, Lancaster JR, Jr, Freeman BA, Tarpey MM. Nitro-oleic acid, a novel and irreversible inhibitor of xanthine oxidoreductase. J Biol Chem. 2008;283:36176–36184. doi: 10.1074/jbc.M802402200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dikalov SI, Li W, Mehranpour P, Wang SS, Zafari AM. Production of extracellular superoxide by human lymphoblast cell lines: comparison of electron spin resonance techniques and cytochrome c reduction assay. Biochem Pharmacol. 2007;73:972–980. doi: 10.1016/j.bcp.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hande K, Reed E, Chabner B. Allopurinol kinetics. Clinical Pharmacology & Therapeutics. 1978;23:598–605. doi: 10.1002/cpt1978235598. [DOI] [PubMed] [Google Scholar]
- 23.Becker MA, Schumacher HR, Jr, Wortmann RL, MacDonald PA, Eustace D, Palo WA, Streit J, Joseph-Ridge N. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450–2461. doi: 10.1056/NEJMoa050373. [DOI] [PubMed] [Google Scholar]
- 24.Hande K, Reed E, Chabner B. Allopurinol Kinetics. Clin Pharmacol Ther. 1978;23:598–605. doi: 10.1002/cpt1978235598. [DOI] [PubMed] [Google Scholar]
- 25.Pesonen EJ, Linder N, Raivio KO, Sarnesto A, Lapatto R, Hockerstedt K, Makisalo H, Andersson S. Circulating xanthine oxidase and neutrophil activation during human liver transplantation. Gastroenterology. 1998;114:1009–1015. doi: 10.1016/s0016-5085(98)70321-x. [DOI] [PubMed] [Google Scholar]
- 26.Himmel HM, Sadony V, Ravens U. Quantitation of hypoxanthine in plasma from patients with ischemic heart disease: adaption of a high-performance liquid chromatographic method. J Chromatogr. 1991;568:105–115. doi: 10.1016/0378-4347(91)80344-c. [DOI] [PubMed] [Google Scholar]