

Abstract
Previous studies have indicated that the δ-carboline (2) ring system derived from the natural product cryptolepine (1) may represent a pharmacophore for anti-infective activity. This paper describes the design and synthesis of a small library of substituted δ-carbolines and the evaluation of the antifungal and antibacterial activities. An evaluation of the anti-bacterial activity of a previously reported library of ring-opened analogs was also conducted to provide an opportunity to test the hypothesis that both group of compounds may have the same biological target. Results indicate that against a selected group of fungal pathogens, substituted δ-carbolinium analogs displayed higher potency and several fold lower cytotoxicity than cryptolepine the parent natural product. Both the δ-carbolinium compounds and their ring-opened analogs, exhibited equally high anti-bacterial activity against the selected pathogens and especially against the gram positive bacteria evaluated.
Keywords: δ-carboline, antifungal, antibacterial, opportunistic, synthesis, quaternary
1. Introduction
We have previously reported that the δ-carboline or indolo[3,2-b]pyridine (2) ring system derived from the natural product cryptolepine (depicted in the salt form, 1), may represent a new pharmacophore for anti-infective activity [1]. By itself however, δ-carboline has no antiinfective properties just like its parent indolo[3,2-b]quinolines typified by quindoline [2]. When quindoline is alkylated on the pyridine nitrogen with appropriate alkyl groups and especially with the ω-cyclohexylpentyl moiety (3), the resulting compounds show anti-infective properties against a variety of opportunistic microorganisms including, C. albicans, C. neoformans, A. fumigatus, and several others. Our more recent work [3] on ring-opened analogs of cryptolepine suggested that alkylating the diarylamine with the ω-cyclohexylpentyl group while maintaining a methyl group on the pyridine nitrogen may produce an optimal combination of potency and toxicity profile. Hence in this paper, we seek to explore the synthesis of a small library of substituted δ-carbolines with the ω-cyclohexylpentyl moiety on the indole nitrogen and a methyl group on the pyridine nitrogen and to evaluate them against opportunistic fungal and bacterial infections associated with immune-compromised conditions. In addition, we report the expansion of the evaluation of a previous library of ring-opened analogs of δ-carbolines (4) against bacterial pathogens including S. aureus and MRSA, which have attracted world-wide attention as major nosocomial infection-causing pathogens and the most frequent cause of skin and soft tissue infections presenting to emergency rooms in the USA [4]. These results will enable a comparison between the anti-infective activities of δ-carbolinium compounds and their ring-opened analogs.

2. Chemistry
Using methods we and others previously reported, [5, 6] we were able to obtain the compounds of interest in good yields. The first step in the synthesis of the δ-carbolinium compounds, 11a-j, involved coupling commercially available substituted phenyl boronic acids or halides 5, with 3-aminopyridine, 6 to give substituted phenylaminopyridines, 7. Ring closure of was accomplished by refluxing intermediate 7 in trifluoroacetic acid, with palladium acetate, to give 8. Compound 9, ω-cyclohexylpentyl iodide was obtained as previously reported [7] and was used to alkylate the secondary diarylamine of 8. This was accomplished by deprotonation with sodium hydride followed by alkylation with 9. The pyridine nitrogen of 10 was methylated with methyl iodide to yield the desired target compounds, 11a-j (Scheme I).
Scheme I.

Reagents and conditions: (i) (Y = B(OH)2), CH2Cl2, Cu(OAc)2, TEA, Molecular sieve, rt, 24 h, (Y = Br, I) 1, 4-Dioxane, PhONa, xantphos, Pd2(dba)3, 80°C, 24h; (ii) Pd(OAc)2, CF3COOH, 80°C, 6 hr; (iii) DME, NaH, 0°C, RT, 12 h; (iv) CH3I, Toluene, 110°C, 12-24 h.
Results and Discussion
N1-alkylated δ-carboline was previously identified as an anti-infective pharmacophore [1]. To begin the current study, we synthesized and evaluated the unsubstituted N5-ω-cyclohexylpentyl N1-methyl δ-carbolinium compound (11a). The results confirmed our hypothesis that ring D in cryptolepine is not required for activity as 11a displayed high activity against both fungi and bacteria (Table 1 and 3). Compounds with anti-infective activities between 1 - 5 μg/mL are considered potent, moderately potent from 6-10 μg/mL, weakly active from 10 – 20 μg/mL and inactive above 20 μg/mL. Compounds with activities in the sub-microgram per mL range and toxicities against Vero cells >10 μg/mL are the target for our design studies. Compound 11a showed considerable potency against all the three fungi we have targeted, i.e., C. albicans, C. neoformans and A. fumigatus all potentially deadly opportunistic fungi. However, the cytotoxicity of 11a is less than 10 μg/mL and hence does not meet our desired target profile. Observations from our previous works suggested that changes in the steric, hydrophobic and electronic space surrounding the tetracyclic ring of cryptolepine results in changes in both activity and cytotoxicity [8]. It was therefore of interest to exploit the steric, hydrophobic and electronic space around the δ-carbolinium moiety in an attempt to improve the overall potency and cytotoxicity profile of this scaffold.
Table 1.
Antifungal activity data of δ-carbolinium analogs,11a-j.
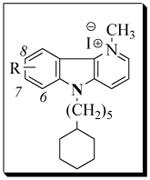 |
|||||
|---|---|---|---|---|---|
| Compoundsa | IC50/MIC/MFC/ (μg/mL) | TC50 (μg/mL) | |||
| Entry | R | Ca | Cn | Afu | Vero Cells |
| 11a | H | 3.0/5.0/>20 | 0.45/0.63/5.0 | 0.45/0.63/5.0 | 7.5 |
| 11b | 6-Cl | 2.5/5.0/5.0 | 0.3/0.6/0.6 | 16/20/20 | >10 |
| 11c | 7-Cl | 2.9/5.0/10 | 0.3/0.6/0.6 | 16/20/>20 | >10 |
| 11d | 8-Cl | 2.0/5.0/5.0 | 0.4/0.6/0.6 | 16/20/20 | >10 |
| 11e | 8-CF3 | 5.8/10/20 | 0.4/0.6/1.3 | 18/>20/>20 | >10 |
| 11f | 8-F | 6.5/20/20 | 0.6/1.3/1.3 | 12/20/>20 | >10 |
| 11g | 8-CH3 | 2.9/5.0/20 | 1.3/2.5/2.5 | 17/20/>20 | >10 |
| 11h | 8- SCH3 | 2.1/5.0/5.0 | 0.3/0.6/0.6 | 13/20/20 | >10 |
| 11i | 8- OCH3 | 7.4/20/>20 | 0.70/1.3/1.3 | 16/>20/>20 | >10 |
| 11j | 8-CN | 19/20/>20 | 2.8/5.0/5.0 | NA | >10 |
| Cryptolepine b | 250 (MIC) | 12.5 (MIC) | NT | 3.2 | |
| Amphotericin B | 0.2/0.6/1.3 | 0.3/0.63 /0.63 | 0.7 /1.3/5.0 | 7.6 | |
Abbreviations: Ca = Candida albicans, Cn = Cryptococcus neoformans, Afu = Aspergillus fumigatus. NT = Not tested. NA = not active at 20 μg/mL. IC50 = The concentration that affords 50% inhibition of growth, MIC = Minimum inhibitory concentration and is the lowest test concentration that allows no detectable growth; MFC = Minimum fungicidal concentration and is the lowest test concentration that kills the organism; TC50 = The concentration that is toxic to 50% of cells. ;
All compounds were subjected to CHN analysis and each passed within 0.4% of the theoretical value.
Data previously reported [2].
Table 3.
Antibacterial activity of δ-carbolinium analogs, 11a-i
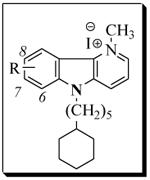 |
|||||
|---|---|---|---|---|---|
| Compounds | IC50/MIC/MBC/ (μg/ml) | ||||
| Entry | R | Sa | MRSA | E. coli | M. int. |
| 11a | H | NT | 1.5/2.5/5.0 | 5.0/>20/>20 | 0.55/1.3/2.5 |
| 11b | 6-Cl | 0.2/0.3/10 | 1.2/2.5/5.0 | 5.7/>20/>20 | 0.5/0.6/5.0 |
| 11c | 7-Cl | 0.4/0.6/5.0 | 1.2/2.5/2.5 | 5.9/>20/>20 | 0.7/1.3/5.0 |
| 11d | 8-Cl | 0.5/1.3/10 | 1.3/2.5/10 | 5.9/>20/>20 | 0.7/1.3/2.5 |
| 11e | 8-CF3 | 0.4/0.6/10 | 1.2/2.5/10 | 7.6/>20/>20 | 0.8/1.3/2.5 |
| 11f | 8-F | 0.7/1.3/20 | 2.8/5.0/20 | 13/>20/>20 | 1.2/2.5/5.0 |
| 11g | 8-CH3 | 0.4/0.6/10 | 1.6/5.0/10 | 5.6/>20/>20 | 1.5/2.5/5.0 |
| 11h | 8-SCH3 | 0.4/0.6/5.0 | 1.1/2.5/10 | 5.6/>20/>20 | 0.7/1.3/2.5 |
| 11i | 8-OCH3 | 0.7/1.3/20 | 3.3/5.0/20 | 12/>20/>20 | 1.6/2.5/2.5 |
| 11j | 8-CN | 2.6/5.0/20 | 5.8/10/20 | NA | 2.1/2.5/5.0 |
| Cryptolepine a | 10 (IC50) | 20 (IC50) | NT | 15 (IC50) | |
| Ciprofloxacin | 0.07/0.3/0.5 | 0.07/0.3/0.5 | 0.005/0.3/0.6 | 0.3/0.5/NA | |
Abbreviations: Sa = Staphylococcus aureus, MRSA = Methicillin-Resistant Staphylococcus aureus, M. Int. = Mycobacterium intracellulare. NT = Not tested. NA = not active at 20 μg/mL. IC50 = The concentration that affords 50% inhibition of growth, MIC = Minimum inhibitory concentration and is the lowest test concentration that allows no detectable growth; MFC = Minimum bactericidal concentration and is the lowest test concentration that kills the microorganism; TC50 = The concentration that is toxic to 50% of cells.
Data previously reported [10].
We first selected the western hemisphere of the structure to explore, using the electron withdrawing and hydrophobic chloro substituents, 11b-d. The synthesis of the designed targets with substitution at the 6-, 7- and 8-positions went smoothly without a hitch. However, all attempts to synthesize the 9-chloro analog were unsuccessful presumably because there was steric hindrance to closing the ring with a meta-substituent on the precursor structure. Evaluation of 11b-d against the three fungi showed similar potencies across the board with the highest potencies against C. neoformans (IC50< 0.4 μg/mL). Cytotoxicity against Vero cells did improve (>10 μg/mL) as a result of the substitutions as expected. To further explore other substitutions in this hemisphere, we selected position 8 using a Craig plot guided selection of substituents [9]. The stronger electron withdrawing and hydrophobic trifluoromethyl substituent (11e) did not fare any better and has diminished potency against C. albicans while it was essentially inactive against A. fumigatus. Similar observations were made for the smaller fluoro substituent (11f). The electron donating and hydrophobic methyl group (11g) was selected from the next quadrant of the Craig plot.
Evaluation showed no significant changes in potency compared to the chloro substituent although activity against C. neoformans was relatively subdued. Similar observations were made when a thiomethyl substituent (11h) was evaluated except that the loss of activity against C. neoformans observed for the methyl group was restored. The electron donating and minimally hydrophilic methoxy compound (11i) represented substituents in the third quadrant in the Craig plot. Activity was generally lower and even more subdued with the more hydrophilic and electron withdrawing cyano group (11j). Thus, it will appear that hydrophilicity decreases activity while lipophilicity increases activity on ring A of the δ-carbolinium scaffold. Changes in the electronic effect of the substituents do not appear to have any significant contribution to activity based on these observations. All the compounds, 11b-j meet our desired cytotoxicity target of >10 μg/mL against Vero cells.
Table 2 depicts the antifungal inhibitory activity results of previously reported ring-opened analogs of δ-carbolinium compounds, 12 a-d, g,h,k,m,p & s). These and the corresponding δ-carbolinium compounds provide the opportunity for comparing each substituted analog. The δ-carbolinium compounds showed consistently higher potency against C. neoformans than the corresponding ring-opened analogs. However, the observed trends remain very similar.
Table 2.
Antifungal activity data of 3-phenylaminopyridinium analogs, 12a-s
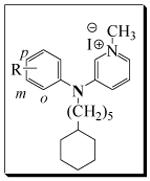 |
|||||
|---|---|---|---|---|---|
| Compounda | IC50 /MIC/MFC (μg/mL) | TC50 (μg/mL) |
|||
| Entry | R | Ca | Cn | Afu | Vero Cells |
| 12a | H | 8.3/20/>20 | 1.5/2.5/2.5 | 20/>20/>20 | >10 |
| 12b | o-Cl | 2.8/5.0/5.0 | 2.0/2.5/2.5 | 18/>20/>20 | NT |
| 12c | m-Cl | 3.0/5.0/10 | 0.72/1.3/1.3 | 12/20/20 | >10 |
| 12d | p-Cl | 1.5/3.1/>20 | 1.0/1.6/NT | 1.0/1.6/>20 | >10 |
| 12g | p-CF3 | 1.0/2.5/10 | 1.5/2.5/5.0 | 1.5/2.5/5.0 | >10 |
| 12h | p-F | 9.6/20/>20 | 1.4/2.5/2.5 | NA | >10 |
| 12k | p-CH3 | 1.5/2.5/10 | 1.5/2.5/5.0 | 1.5/5.0/5.0 | >10 |
| 12m | p-SCH3 | 2.0/5.0/10 | 1.2/1.3/2.5 | 6.4/10/10 | >10 |
| 12p | p-OCH3 | 5.1/10/20 | 1.4/2.5/2.5 | NA | >10 |
| 12s | p-CN | 19/20/>20 | 2.6/5.0/5.0 | NA | >10 |
| Cryptolepine b | >250 (MIC) | 12.5 (MIC) | NT | 3.2 | |
| Amphotericin B | 0.2/0.6/1.3 | 0.3/0.63 /0.63 | 0.7 /1.3/5.0 | 7.6 | |
Abbreviations: Ca = Candida albicans, Cn = Cryptococcus neoformans, Afu = Aspergillus fumigatus. NT = Not tested. NA = not active at 20 μg/mL; IC50 = The concentration that affords 50% inhibition of growth, MIC = Minimum inhibitory concentration and is the lowest test concentration that allows no detectable growth; MFC = Minimum fungicidal concentration and is the lowest test concentration that kills the organism; TC50 = The concentration that is toxic to 50% of cells.
The syntheses and characterization of all compounds in this Table were previously reported [3].
Data previously reported [2]
On the other hand, ring-opened analogs were more potent against A. fumigatus as compared to their δ-carbolinium counterparts and there is a preference for p-substituents compared to the o- and m-substituents. The only exception is the unsubstituted analogs (11a and 12a) where the reverse is the case. We have currently no explanation of this observation. Interestingly, activities are generally similar against C. albicans for both the closed and open ring systems.
The potency of the δ-carbolinium and their ring-opened analogs against the various fungi examined above, and the low cytotoxicity they displayed inspired our evaluation of these compounds against a select number of bacterial pathogens. Both the δ-carbolinium compounds and their ring-opened analogs, 12a-s [3] were evaluated against four bacterial pathogens and the results are shown in Tables 3 and 4. Staphylococcus aureus, methicillin-resistant Staphylococcus aureus (MRSA), M. intracellulare and E. coli were selected for evaluation. Staphylococcus aureus, and especially MRSA, have become some of the deadliest and most common nosocomial infection causing pathogens [4, 11]. Resistance development by S. aureus to current drugs has made vancomycin and a few of the new antibacterial agents the last lines of defense against MRSA [12, 13]. And yet over the last 2 decades, the development of new antibacterial drugs has slowed down significantly [14]. M. intracellulare [15] is an opportunistic pathogen associated with AIDS and E. coli and is included to represent a gram-negative bacterium in the biological evaluation.
Table 4.
Antibacterial activity of 3-phenylaminopyridinium Analogs, 12a-s
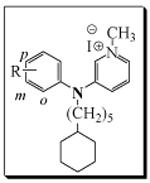 |
|||||
|---|---|---|---|---|---|
| Compound | IC50 /MIC/MBC (μg/mL) | ||||
| Entry | R | Sa | MRSA | E. Coli | M. int. |
| 12a | H | 0.7/1.3/10 | 3.4/10/20 | 7.6>20/>20 | 2.1/25/2.5 |
| 12b | o-Cl | 0.3/0.6/10 | 1.1/2.5/10 | 7.6/>20/>20 | 2.1/2.5/5.0 |
| 12c | m-Cl | 0.34/0.63/5.0 | 1.3/2.5/5.0 | 6.5/>20/>20 | 2.1/2.5/5.0 |
| 12d | p-Cl | 1.5/3.1/NT | NT | NT | 1.0/1.6/NT |
| 12e | o-CF3 | 0.3/0.3/10 | 0.6/1.3/5.0 | 9.1/>20/>20 | 1.2/3.5/2.5 |
| 12f | m-CF3 | NT | 1.5/2.5/5.0 | 4.5/>20/>20 | 2.0/2.5/2.5 |
| 12g | p-CF3 | NT | 0.9/2.5/2.5 | 3.5/20/20 | 1.0/1.3/1.3 |
| 12h | p-F | 0.7/1.3/10 | 3.0/5.0/20 | 18/>20/>20 | 3.2/5.0/10.0 |
| 12i | o-CH3 | 0.3/0.6/10 | 1.0/2.5/10 | 9.4/>20/>20 | 1.6/2.5/2.5 |
| 12j | m-CH3 | 0.7/1.3/5.0 | 1.2/2.5/5.0 | 7.7/>20/>20 | 1.1/1.3/5.0 |
| 12k | p-CH3 | NT | 1.0/2.5/2.5 | 4.5/20/>20 | 1.5/2.5/2.5 |
| 12l | p-C2H5 | 0.2/3.0/5.0 | 0.7/1.3/2.5 | 4.0/20/20 | 0.6/1.3/5.0 |
| 12m | p-SCH3 | 0.3/0.6/5.0 | 1.0/2.5/2.5 | 5.2/20/20 | 1.0/1.3/5.0 |
| 12n | o-OCH3 | 0.4/0.6/20 | 1.2/5.0/20 | 14/>20/>20 | 1.9/2.5/2.5 |
| 12o | m-OCH3 | 0.8/1.3/20.0 | 4.8/10/20 | NA | 3.3/5.0/10 |
| 12p | p-OCH3 | 0.3/0.6/10.0 | 1.9/5.0/10 | 5.4/>20/>20 | 1.0/1.3/5.0 |
| 12q | o-CN | 1.3/2.5/10.0 | 6.4/20/20 | NA | 3.5/5.0/20 |
| 12r | m-CN | 0.8/1.3/20 | 3.9/10/10 | NA | 2.0/2.5/10 |
| 12s | p-CN | 2.5/5.0/20 | 6.2/10/20 | NA | 2.8/5.0/5.0 |
| Cryptolepine a | 10 ( IC50) | 20 ( IC50) | NT | 15 ( IC50) | |
| Ciprofloxacin | 0.07/0.3/0.5 | 0.07/0.3/0.5 | 0.005/0.3/0.6 | 0.3/0.5/>20 | |
Abbreviations: Sa = Staphylococcus aureus, MRSA = Methicillin-resistant Staphylococcus aureus, M. Int. = Mycobacterium intracellulare. NT = Not tested. NA = not active at 20 μg/mL· IC50 = The concentration that affords 50% inhibition of growth, MIC = Minimum inhibitory concentration and is the lowest test concentration that allows no detectable growth; MFC = Minimum bactericidal concentration and is the lowest test concentration that kills the microorganism; TC50 = The concentration that is toxic to 50% of cells. The syntheses and characterization of all compounds in this Table were previously reported [3].
Data previously reported [10].
All the δ-carbolinium compounds, 11a-i have shown sub-microgram potencies (IC50) against S. aureus except for the hydrophilic cyano substituent (11j). Similarly, the compounds were potent against MRSA (1.2 ≤ IC50 ≤ 5.8 μg/mL) and here too the more hydrophilic substituents showed subdued activity. We can only speculate that perhaps the increased hydrophobicity enables the quaternary compounds to cross microbial membranes much more efficiently than their hydrophilic counterparts. While the more hydrophobic compounds fared better in this assay, there was no clear indication of the contributions made by differences in electronic parameters. The δ-carbolinium compounds were equally potent against M. intracellulare, showing IC50 values in the range of 0.5 - 2.1 μg/mL, with the majority in the sub-microgram/mL range. Interestingly, all the δ-carbolinium compounds displayed potent bactericidal properties and especially against M. intracellulare.
4. Conclusion
Using the method previously reported by our labs, we were able to synthesize a number of substituted δ-carbolinium analogs 11a-j in relatively good yields. Biological evaluations of the compounds indicated that substituted δ-carbolinium analogs possess high anti-fungal activity. On average, these analogs are over 100-fold (Cn) and over 150-fold (Ca) more potent than cryptolepine, the original natural product. The in vitro cytotoxicity assay against mammalian kidney fibroblast (Vero) cells indicates that removal of ring D from the cryptolepine scaffold and introduction of an ω-cyclohexylpentyl group on the indole nitrogen resulted in a decrease in cytotoxicity. Anti-bacterial evaluation showed all substituted δ-carbolinium analogs to be more potent than cryptolepine with especially potent activity against M. intracellulare (IC50 values in the range of 0.5 - 2.1 μg/ml). While very potent activities against the gram-positive bacteria were observed, the compounds showed weak or no activity against the gram-negative bacterium, E. coli. In general, the anti-bacterial activities of the ring-opened analogs 12a-s were remarkably similar to their δ-carbolinium counterparts.
5. Experimental
5.1 Reagents and general procedures
Reagents and solvents were obtained from commercial suppliers (Sigma-Aldrich, Alfa Aesar and VWR) and except for tetrahydrofuran (THF) which was dried over sodium metal, all other reagents and solvents were used without further purification. Flash chromatography was performed with Selecto Scientific silica gel (63-200 mesh). Analytical TLC was performed on pre-coated Whatman Ltd aluminium plates (Silica gel 60 Å). NMR spectra were obtained on Varian 300-MHZ NMR spectrometer. Melting points were determined on a Gallenkamp (UK) apparatus. CHN analyses, performed with a Perkin Elmer 2400 CHN elemental analyzer, were carried out by Atlantic Microlab, Inc., Norcross, GA and are within 0.4% of theoretical values unless otherwise noted.
5.2 General Procedure for the synthesis of N-phenylpyridin-3-amine derivatives (7)
Method A was used for halogen and trifluomethyl-substituted analogs, 7a-f and method B was utilized in the case of 7g-j.
5.2.1. Method A
Y = B(OH)2: To a solution of amino pyridine (1 g, 10.63 mmol ) in CH2Cl2 (30 mL) was added portion wise phenyl boronic acid (1.63g), Et3N (1.5g, 10.4 mmol), Cu(OAc)2 (1.90g, 10.4 mmol) and molecular sieves (2g) under stirring. The reaction mixture was stirred at rt for 12-24 h and monitored by TLC. Once reaction is completed, the mixture was quenched by the drop-wise addition of aq NH3 (15 ml). The resulting mixture was extracted with CH2Cl2 (3×20 mL) washed with brine then with water, and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue purified by column chromatography using a mixture of EtOAc and hexanes as eluent.
5.2.2. Method B
Y = Br or I: A mixture of phenyl halide (200 mg), sodium phenolate (191 mg, 1.65 mmol), Pd2(dba)3 (12.6 mg, 0.014 mmol), xantphos (19,1 mg, 0.033 mmol) and heteroarylamine (114 mg, 1.21 mmol) in 1,4-dioxane (6.5 mL), was refluxed at 80 °C for 6 h. After cooling to rt, the mixture was diluted with EtOAc (100 mL), washed with 1N aqueous NaOH (3 × 70 mL) and brine (100 mL), dried over anhydrous Na2SO4, filtered, and the solvent removed under vacuum. The resulting residue was purified over silica gel using hexanes/EtOAc as eluent to obtain the product as a yellow solid.
5.2.3. N-Phenylpyridin-3-amine (7a)
Yellow solid (108 mg, 60 % yield), mp: 390-391°C; 1H NMR (300 MHz, CDCl3): δ8.38 (d, 1H, J = 2.4Hz), 8.16 (d, 1H, J = 4.5Hz), 7.42-7.38 (m, 4H), 7.02-6.97 (m, 3H), 5.71 (s, 1H).
5.2.4. N-(2-Chlorophenyl)pyridin-3-amine (7b)
Yellow solid (97mg, 45 % yield), mp:432-433°C; 1H NMR (300 MHz, CDCl3): δ8.43 (d, 1H, J = 2.1Hz), 8.28 (d,1H, J = 4.2Hz), 7.62-7.03 (m, 6H), 6.01 (s, 1H).
5.2.5. N-(3-Chlorophenyl)pyridin-3-amine (7c)
Yellow solid (104 mg, 48 % yield), mp: 431-432°C; 1H NMR (300 MHz, CDCl3): δ8.41 (s, 1H), 8.23 (d, 1H, J = 2.7Hz), 7.41-7.43 (m, 3H), 7.26-6.92 (m, 3H), 5.98 (s, 1H).
5.2.6. N-(4-Chlorophenyl)pyridin-3-amine (7d)
Yellow solid (119 mg, 55 % yield), mp: 432-433°C; 1H NMR (300 MHz, CDCl3): δ8.39 (d, 1H, J = 2.7Hz), 8.23 (d, 1H, J = 4.8Hz), 7.39 (d, 1H, J = 8.1Hz), 7.26-7.21 (m, 3H), 7.02-6.99 (m, 2H), 5.83 (s, 1H).
5.2.7. N-(4-(Trifluoromethyl)phenyl)pyridin-3-amine (7e)
Yellow solid (128 mg, 51 % yield), mp: 418-419°C; 1H NMR (300 MHz, CDCl3): δ8.47 (s, 1H), 8.30 (s, 1H), 7.50 (t, 2H, J = 8.4Hz), 7.28 - 7.05 (m, 4H), 5.98 (s, 1H).
5.2.8. N-(4-Fluorophenyl)pyridin-3-amine (7f)
Yellow solid (115 mg, 58 % yield), mp: 403-404°C; 1H NMR (300 MHz, CDCl3): δ8.33 (s, 1H), 8.14 (d, 1H, J = 3.9Hz), 8.03 (d, 2H, J = 5.4Hz), 7.32-6.99 (m,4H), 5.73 (s, 1H).
5.2.9. N-(p-Tolyl)pyridin-3-amine (7g)
Yellow solid (123 mg, 63 % yield), mp: 414-415°C; 1H NMR (300 MHz, CDCl3): δ8.30 (d,1H, J = 2.4 Hz), 8.12 (d, 1H, J = 3Hz), 7.26-6.89 (m, 6H), 5.47 (s, 1H), 2.26 (s, 3H).
5.2.10. N-(4-(Methylthio)phenyl)pyridin-3-amine (7h)
Yellow solid (149 mg, 65 % yield), mp: 448-449°C; 1H NMR (300 MHz, CDCl3): δ8.36 (s, 1H), 8.17 (s, 1H), 7.72-7.02 (m, 6H), 5.68 (s, 1H), 2.47 (s, 3H).
5.2.11. N-(4-Methoxyphenyl)pyridin-3-amine (7i)
Yellow solid (140 mg, 66 % yield), mp: 436-437°C; 1H NMR (300 MHz, CDCl3): δ8.32 (s,1H), 8.04 (s, 1H), 7.27-7.09 (m, 4H), 6.91 (t, 2H, J = 4.3Hz), 5.93 (s, 1H), 3.81 (s, 3H).
5.2.12. 4-(Pyridin-3-ylamino)benzonitrile (7j)
Yellow solid (84 mg, 41 % yield), mp: 479-480°C; 1H NMR (300 MHz, CDCl3) δ8.49 (d, 2H, J = 3Hz), 8.12 (d, 1H, J = 1.8Hz), 7.81 (t, 2H, J = 5.1Hz), 6.74-6.69 (m, 3H), 5.3 (s, 1H).
5.3. General Procedure for the synthesis of 5H-pyrido[3, 2-b]indole derivatives (8)
A mixture of 7 (400 mg), CF3COOH (8 mL), Pd(OAc)2 (300 mg, 1.34 mmol) was refluxed for 6h at 72 °C, and allowed to cool to rt. Thereafter, it was poured into ice cold H2O (15 mL), neutralized with aqueous NH3, extracted with EtOAc (3 × 50 mL), washed with brine and dried over anhydrous sodium sulfate. The solvent was removed under reduce pressure and the crude product was purified by column chromatography (hexane:EtOAc 2:3).
5.3.1. 5H-Pyrido[3,2-b]indole (8a)
Yellow solid (150 mg, 38 % yield), mp: 206-207°C; 1H NMR (300 MHz, DMSO-d6):δ 11.2 (s, 1H), 8.39 (d, 2H, J = 7.3Hz), 8.15( dd, 2H, J = 5.4, 1.6Hz), 7.97-7.88 (m, 3H).
5.3.2. 6-Chloro-5H-pyrido[3,2-b]indole (8b)
Yellow solid (119 mg, 30 % yield), mp: 208-209°C; 1H NMR (300 MHz, DMSO-d6):δ11.1 (s, 1H), 8.38 (d, 2H, J = 3Hz), 8.10-8.01(m, 2H), 7.86 (dd, 2H, J = 4.6, 1.2Hz).
5.3.3. 7-Chloro-5H-pyrido[3,2-b]indole (8c)
Yellow solid (154 mg, 39 % yield), mp: 208-209°C; 1H NMR (300 MHz, DMSO-d6):δ11.4 (s, 1H), 8.45 (dd, 2H, J = 6.5, 3.1Hz), 8.2 (d, 2H, J = 5.6 Hz), 7.9 (t, 2H, J = 2.6Hz).
5.3.4. 8-Chloro-5H-pyrido[3,2-b]indole (8d)
Yellow solid (162 mg,41 % yield), mp: 208-209°C; 1H NMR (300 MHz, DMSO-d6):δ 11.2 (s,1H), 8.97 (d, 1H, J = 1.5 Hz), 8.93 (d, 2H, J = 5.7Hz), 8.62 (d, 2H, J = 2.1Hz), 8.12 (d, 1H, J = 6Hz).
5.3.5. 8-(Trifluoromethyl)-5H-pyrido[3,2-b]indole (8e)
Yellow solid (131 mg, 33 % yield), mp: 203-204°C; 1H NMR (300 MHz, CDCl3):δ11.3 (s, 1H), 8.61 (d, 2H, J = 4.5Hz), 8.25 (d, 2H, J = 1.3 Hz), 7.95 (dd, 2H, J = 5.2, 2.1Hz).
5.3.6 . 8-Fluoro-5H-pyrido[3,2-b]indole (8f)
Yellow solid (170 mg, 43 % yield), mp: 207-209°C; 1H NMR (300 MHz, DMSO-d6):δ11.0 (s, 1H), 8.43 (d, 2H, J = 2.1Hz), 8.01 (t, 2H, J = 1.8 Hz), 7.98 (d, 2H, J = 6.3 Hz).
5.3.7. 8-Methyl-5H-pyrido[3,2-b]indole (8g)
Yellow solid (150 mg, 38 % yield), mp: 212-213°C; 1H NMR (300 MHz, DMSO-d6):δ 11.4 (s, 1H), 8.40 (dd, 2H, J = 4.2, 1.5Hz), 8.21 (s, 1H), 8.0 (t, 2H, J = 5.0Hz), 7.95 (d, 1H, J = 1.4 Hz), 3.41 (s, 3H).
5.3.8. 8-(Methylthio)-5H-pyrido[3,2-b]indole (8h)
Yellow solid (40 % yield), mp: 201-202°C; 1H NMR (300 MHz, DMSO-d6):δ11.5 (s, 1H), 8.62 (d, 2H, J = 7.2Hz), 8.41-8.0 (m, 3H), 7.88 (d, 1H, J = 1.4Hz), 4.1 (s,3H).
5.3.9. 8-Methoxy-5H-pyrido[3,2-b]indole (8i)
Yellow solid (159 mg, 36 % yield), mp: 189-190°C; 1H NMR (300 MHz, DMSO-d6):δ11.3 (s,1H), 8.40 (dd, 2H, J = 5.1, 2.0Hz), 8.1 (d, 1H, J = 5.3 Hz), 7.86 (t, 2H, J = 2.8 Hz), 7.84 (d, 1H, J = 4.1Hz), 3.4 (s, 3H).
5.3.10. 5H-Pyrido[3,2-b]indole-8-carbonitrile (8j)
Yellow solid (138 mg, 35 % yield), mp: 221-222°C; 1H NMR (300 MHz, DMSO-d6):δ11.03 (s, 1H), 8.6 (d, 2H, J = 3.4Hz), 8.3 (d, 2H, J = 5.1Hz), 8.11-7.93 (m, 2H).
5.4. Synthesis of 5-Iodopentylcyclohexane (9)
A mixture of (5-bromo-pentyl)-cyclohexane [2] (2 g, 8.6 mmol) in acetone (20 mL) and NaI (2.57g, 17.15 mmol) was heated at 60 °C for 12 h and then allowed to cool to rt. The solvent was removed under vacuum, the residue was diluted with H2O (50 mL) and extracted with EtOAc (3 × 30 mL). The pooled organic layer was washed with brine (30 mL), dried over anhydrous Na2SO4, solvent was removed under vacuum and the crude product purified on column chromatography using hexanes as eluent. The pure product 9, was an oily liquid (2.17g, 90 % yield). 1H NMR (300 MHz, CDCl3): δ 0.8 (t, 2H, J = 10.2 Hz), 1.00-1.38 (m, 9H), 1.52-168 (m, 6H), 1.70-1.80 (m, 2H), 3.10 (t, 2H, J = 6.9 Hz).
5.5. General Procedure for the synthesis of derivatives of 5-(5-Cyclohexylpentyl)-5H-pyrido[3,2-b]indole, 10
To a solution of 8 (100 mg) in dried 1, 2-dimethoxyethane (5 ml), NaH (20 mg, 0.83 mmol) and then 9 (636 mg, 2.27 mmol) were added. The reaction was stirred at rt for 12 h, solvent was removed under reduced pressure and the residue partitioned between H2O (10 ml) and EtOAc (2 × 30 ml). The pooled organic phase was washed with brine, dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified by column chromatography (hexane: EtOAc 4:1).
5.5.1. 5-(5-Cyclohexylpentyl)-5H-pyrido[3,2-b]indole (10a)
Yellow oily liquid (76.2 mg, 40 % yield), 1H NMR (300 MHz, CDCl3):δ 8.55 (dd, 1H, J = 1.2, 1.2Hz), 8.40 (dd, 1H, 6.0, 1.8Hz), 7.69-7.44 (m, 3H), 7.37-7.29 (m, 2H), 4.30 (t, 2H, J = 4.8Hz), 2.04-1.63 (m,8H), 1.34-1.13 (m, 9H), 0.88(t, 2H, J = 8Hz).
5.5.2. 6-Chloro-5-(5-cyclohexylpentyl)-5H-pyrido[3,2-b]indole (10b)
Yellow oily liquid (96.3 mg, 55 % yield), 1H NMR (300 MHz, CDCl3):δ 9.01(d, 2H, J = 6.6 Hz), 8.85 (t, 2H, J = 6.2Hz), 8.02 (d, 1H, J = 8.1Hz) 7.85 (d, 2H, J = 2.5 Hz), 4.1(t, 2H, J = 5.1Hz), 2.1-1.78 (m, 10H), 1.68-1.44 (m, 7H), 0.98 (t, 2H, J = 1.8Hz).
5.5.3. 7-Chloro-5-(5-cyclohexylpentyl)-5H-pyrido[3,2-b]indole (10c)
Yellow oily liquid (80.6 mg, 46 % yield), 1H NMR (300 MHz, CDCl3):δ 8.96 ( d, 2H, J = 4.8Hz), 8.6 (t, 2H, J = 5.8 Hz), 8.21 (d, 1H, J = 2.1Hz), 7.81 (d, 2H, J = 3.4Hz), 4.3 (t, 2H, J = 4.8Hz), 2.03-1.78 (m, 6H), 1.54-1.16 (m, 8H), 1.02-0.89(m, 5H).
5.5.4. 8-Chloro-5-(5-cyclohexylpentyl)-5H-pyrido[3,2-b]indole (10d)
Yellow oily liquid (91.1 mg, 52% yield), 1H NMR (300 MHz, CDCl3):δ 8.99 (d, 1H, J = 3.5 Hz), 8.97 (d, 2H, J = 4.3Hz), 8.62 (d, 2H, J = 2.1Hz), 7.84 (t, 2H, J = 4.5Hz), 4.36 (t, 2H, J = 1.6Hz), 1.85-1.65 (m, 10H), 1.29-1.16 (9m, 7H), 0.86 (t, 2H, J = 2.8Hz).
5.5.5. 5-(5-Cyclohexylpentyl)-8-(trifluoromethyl)-5H-pyrido[3,2-b]indole (10e)
Yellow oily liquid (65.5 mg, 42 % yield), 1H NMR (300 MHz, CDCl3):δ 8.71 (s, 1H), 8.61 (d, 1H, J = 3.5Hz), 7.81-7.79 (m, 3H), 7.6 (d, 2H, J = 4.3Hz), 4.36-4.17 (m, 2H), 1.92 (t, 2H, J = 7.8Hz), 1.88-1.16 (m, 12H), 1.58-0.89 (m,7H).
5.5.6. 5-(5-Cyclohexylpentyl)-8-fluoro-5H-pyrido[3,2-b]indole (10f)
Yellow oily liquid (71 mg, 39% yield), 1H NMR (300 MHz, CDCl3):δ 8.81(d, 2H, J = 3.6Hz), 8.78 (d, 1H, J = 4.2Hz), 8.2 (d, 2H, J = 1.8Hz), 7.86 (t, 2H, J = 5.6Hz), 3.8 (t, 2H, J = 2.7Hz), 1.98-1.76 (m, 10H), 1.52-1.18 (m, 6H), 0.98 (t, 3H, J = 4.8Hz).
5.5.7. 5-(5-Cyclohexylpentyl)-8-methyl-5H-pyrido[3,2-b]indole (10g)
Yellow oily liquid (103 mg, 56% yield), 1H NMR (300 MHz, CDCl3):δ 8.63 (d, 1H, J = 4.7Hz), 8.45 (d, 2H, J = 1.5 Hz), 8.01 (dd, 2H, J = 6.3, 2.1Hz), 7.74 (d, 2H, J = 2.6Hz), 4.01-3.80 (m, 2H), 1.98 (t, 3H, J = 6.8 Hz), 1.78 – 1.43 (m,10H), 1.21 0.89 (m, 9H).
5.5.8. 5-(5-Cyclohexylpentyl)-8-(methylthio)-5H-pyrido[3,2-b]indole (10h)
Yellow oily liquid (87 mg, 51 % yield), 1H NMR (300 MHz, CDCl3):δ 8.87 (d, 2H, J = 7.8Hz), 8.41 (dd, 2H, J = 5.4, 1.6Hz), 7.98 (d, 1H, J = 7.3 Hz), 7.46 (t, 2H, J = 4.1Hz), 3.85 (d, 2H, J = 1.2Hz), 2.7 (s, 3H), 1.96-1.73 (m,12H), 1.15-0.84 (m, 7H).
5.5.9. 5-(5-Cyclohexylpentyl)-8-methoxy-5H-pyrido[3,2-b]indole (10i)
Yellow oily liquid (94 mg, 53 % yield), 1H NMR (300 MHz, CDCl3):δ 8.99 (d, 2H, J = 4.6 Hz), 8.81(t, 2H, J = 7.8Hz), 7.97-7.54 (m, 3H), 4.1 (t, 2H, J = 6.1Hz), 2,31(s, 3H), 1.84-1.76 (m, 10H), 1.31-1.12 (m,7H), 0.86 (t, 2H, J = 1.4Hz).
5.5.10. 5-(5-Cyclohexylpentyl)-5H-pyrido[3,2-b]indole-8-carbonitrile (10j)
Yellow oily liquid (68 mg, 38 % yield), 1H NMR (300 MHz, CDCl3):δ 8.76 (d, 2H, J = 6.8Hz), 8.67 (t, 2H, J = 2.1Hz), 7.81-7.43 (m, 3H), 3.61 (t, 2H, J = 4.5Hz), 2.15-1.84 (m,10H), 1.57-1.16 (m, 6H), 0.87 (t, 3H, J = 7.2Hz).
5.6. General procedure for the synthesis of derivatives of 5-(5-cyclohexylpentyl)-1-methyl-5H-pyrido[3,2-b]indol-1-ium iodide, 11
To a solution of 10 (100 mg) in toluene (3 ml), methyl iodide (0.3 g, 2.1 mmol) was added in a pressure tube and sealed. The reaction mixture was stirred at 110 °C for 24 h, and then allowed to cool to rt, diluted with Et2O (15 mL), to form a precipitate which was filtered and washed with Et2O (3 × 20 ml). The crude product was recrystallized from MeOH.
5.6.1. 5-(5-cyclohexylpentyl)-1-methyl-5H-pyrido[3,2-b]indol-1-ium iodide (11a)
Yellow solid (28.5 mg, 32.9%), mp: 228-230 °C; (DMSO-d6): δ 8.96 (d, 1H, J = 8.7 Hz), 8.90 (d, 1H, J = 6.0 Hz), 8.56 (d, 1H, J = 8.4 Hz), 8.20-8.00 (m, 2H), 7.90 (t, 1H, J = 7.5 Hz), 7.54 (t, 1H, J = 7.2 Hz), 4.80 (s, 3H), 4.66 (t, 2H, J = 6.9 Hz), 1.78(t, 2H, J = 6.6 Hz), 1.64-1.52 (m, 5H), 1.30-1.20 (m, 4H), 1.14-1.00 (m, 6H), 0.80-0.74 (t, 2H, J = 10.2 Hz). Anal. Calcd for C23H31IN2: C, 59.74; H, 6.76; N, 6.06. Found: C, 59.48; H, 6.71; N,5.95
5.6.2. 6-Chloro-5-(5-cyclohexylpentyl)-1-methyl-5H-pyrido[3,2-b]indol-1-ium iodide (11b)
Yellow solid (35 % yield), mp: 246-248 °C; 1H NMR (300 MHz, CDCl3): δ 9.01 (d, 2H, J = 6 Hz), 8.63 (d, 1H, J = 1.8 Hz), 8.14 - 8.08 (m, 2H), 7.95 - 7.91 (m, 1H), 4.82 (s, 3H), 1.76 - 1.58 (m, 6H), 1.21 - 1.00 (m, 12 H), 0.78 (t, 3H). Anal. Calcd for C23H30ClIN2: C, 55.69; H, 6.09; N, 5.64. Found: C, 55.69; H, 6.09; N, 5.61.
5.6.3. 7-Chloro-5-(5-cyclohexylpentyl)-1-methyl-5H-pyrido[3,2-b]indol-1-ium iodide (11c)
Yellow solid (55 % yield), mp: 234-235 °C; 1H NMR (300 MHz, CDCl3): δ 9.62 (d, 1H, J = 5.7 Hz), 8.77 (d, 1H, J = 9Hz), 8.34 (d, 2H, J = 1.5 Hz), 8.18 – 8.13 (m, 2H), 5.04 (s, 3H), 4.58 (t, 2H, J = 7.2 Hz), 1.90 (d, 2H, J = 7.5Hz), 1.65 – 1.56 (m, 12H), 1.33 – 1.12 (m, 5H). Anal. Calcd for C23H30ClIN2: C, 55.60; H, 6.09; N, 5.64. Found: C, 55.35; H, 6.09; N, 5.66.
5.6.4. 8-Chloro-5-(5-cyclohexylpentyl)-1-methyl-5H-pyrido[3,2-b]indol-1-ium iodide (11d)
Yellow solid (57 % yield), mp: 244-245 °C; 1H NMR (300 MHz, CDCl3): δ 8.54 – 8.52 (m, 2H), 8.34 (s, 1H), 7.65 – 7.62 (m, 2H), 7.47 (d, 2H, J = 1.2 Hz), 4.23 (s, 3H), 1.64 – 1.31 (m, 14H), 1.28 – 1.13 (m, 6H). Anal. Calcd for C24H32ClIN2: C, 55.60; H, 6.09; N, 5.64. Found: C, 55.39; H, 6.02; N, 5.65.
5.6.5. 5-(5-Cyclohexylpentyl)-1-methyl-8-(trifluoromethyl)-5H-pyrido[3,2-b]indol-1-ium iodide (11e)
Yellow solid (55 % yield), mp: 239-240 °C; 1H NMR (300 MHz, CDCl3): δ 9.64 (d, 1H, J = 6.0 Hz), 8.88 (d, 1H, J = 8.4 Hz), 8.60 (s, 1H), 8.27 – 7.83 (m, 3H), 5.08 (s, 3H), 4.65 (t, 2H, J = 7.5 Hz), 2.04 – 1.92 (m, 2H), 1.65 – 1.55 (m, 13H), 1.36 -1.15 (m, 4H). Anal. Calcd for C24H32F3IN2.0.05H2O: C, 54.35; H, 5.70; N, 5.28. Found: C, 54.25; H, 5.69; N, 5.27.
5.6.6. 5-(5-Cyclohexylpentyl)-8-fluoro-1-methyl-5H-pyrido[3,2-b]indol-1-ium iodide (11f)
Yellow solid (60 % yield), mp: 244-245 °C; 1H NMR (300 MHz, CDCl3): δ 9.62 (d, 1H, J = 5.7 Hz), 8.74 (d, 1H, J = 8.4 Hz), 8.17 - 8.04 (m, 2H), 7.73 - 7.64 (m, 2H), 5.05 (s, 3H), 1.92 (t, 2H), 1.65 1.57 (m, 13H), 1.35 - 1.14 (m, 9H). Anal. Calcd for C23H30FIN2.0.05H2O: C, 57.50; H, 6.29; N, 5.83. Found: C, 57.40; H, 6.28; N, 5.82.
5.6.7. 5-(5-Cyclohexylpentyl)-1,8-dimethyl-5H-pyrido[3,2-b]indol-1-ium iodide (11g)
Yellow solid (52 % yield), mp: 249-250°C; 1H NMR (300 MHz, CDCl3): δ 9.64 (d, 1H, J = 6.6 Hz), 8.81 (d, 1H, J = 8.7 Hz), 8.60 (s, 3H), 8.25 – 7.83 (m, 3H), 5.09 (s, 3H), 4.63 (t, 2H, J = 7.2 Hz), 1.95 (t, 2H), 1.65 – 1.53 (m, 11H), 1.46 1.34 (m, 9H). Anal. Calcd for C24H33ClIN2.0.05H2O: C, 60.50; H, 6.98; N, 5.88. Found: C, 60.39; H, 6.97; N, 5.87.
5.6.8. 5-(5-Cyclohexylpentyl)-1-methyl-8-(methylthio)-5H-pyrido[3,2-b]indol-1-ium iodide (11h)
Yellow solid (49 % yield), mp: 239-240 °C; 1H NMR (300 MHz, CDCl3): δ 9.66 (d, 1H, J = 6Hz), 8.66 (d, 1H, J = 8.4 Hz), 8.15 – 8.02 (m, 2H), 7.72 – 7.61 (m, 2H), 5.05 (s, 3H), 4.56 (t, 2H, J = 7.2 Hz), 1.93 (t, 2H, J = 7.2 Hz), 1.65 – 1.53 (m, 14H), 1.34 – 1.14 (m, 4H), 0.85 (t, 2H). Anal. Calcd for C24H33IN2S.0.05H2O: C, 56.69; H, 6.54; N, 5.28. Found: C, 56.69; H, 6.53; N, 5.50.
5.6.9. 5-(5-Cyclohexylpentyl)-8-methoxy-1-methyl-5H-pyrido[3,2-b]indol-1-ium iodide (11i)
Yellow solid (46 % yield), mp: 244-245 °C; 1H NMR (300 MHz, CDCl3): δ 9.67 (d, 1H, J = 5.7 Hz), 8.70 (d, 1H, J = 9 Hz), 8.34 (d, 1H, J = 1.8 Hz), 8.17 – 7.66 (m, 4H), 5.05 (s, 3H), 4.56 (t, 2H, J = 7.5 Hz), 1.92 (t, 2H, J = 6.6 Hz), 1.65 – 1.46 (m,4H), 1.35 1.13 (m, 13H), 0.83 (t, 2H). Anal. Calcd for C23H32IN2O.0.05H2O: C, 58.54; H, 6.75; N, 5.69. Found: C, 58.43; H, 6.74; N, 5.68.
5.6.10. 8-Cyano-5-(5-cyclohexylpentyl)-1-methyl-5H-pyrido[3,2-b]indol-1-ium iodide (11j)
Yellow solid (41 % yield), mp: 124-125 °C; 1H NMR (300 MHz, CDCl3): δ 9.60 (d, 1H, J = 5.7 Hz), 8.99 (d, 1H, J = 8.4), 8.41 (d, 4H, J = 5.7 Hz), 4.68 (s, 3H), 3.96 (t, 2H, J = 7.5 Hz), 2.04 – 1.63 (m, 10H), 1.28 – 1.13 (m, 6H), 0.92 – 0.77 (m, 3H). Anal. Calcd for C24H30IN3.0.05H2O: C, 59.14; H, 6.20; N, 8.62. Found: C, 59.03; H, 6.19; N, 8.60.
5.7. Procedure for the synthesis of compounds 12a-s
These were previously reported [3].
5.8. Antifungal and Antibacterial testing
Compounds were evaluated in vitro against a panel of microorganisms, including C. albicans ATCC 90028 (Ca), C. krusei ATCC 6258 (Ck), C. neoformans ATCC 90113 (Cn), Staphylococcus aureus ATCC 29213 (Sa), methicillin-resistant S. aureus ATCC 33591(MRSA), A. fumigatus ATCC 204305 (Af), and M. intracellulare ATCC 23068 (Mi) as previously reported [16] All organisms were obtained from the American Type Culture Collection (Manassas, Va.). Susceptibility testing was performed using a modified version of the NCCLS methods [17 - 19] for all organisms except for M. intracellulare, for which the modified Alamar blue procedure described by Franzblau et al.[20] was followed. Briefly, samples (dissolved in DMSO) were serially diluted by using 0.9% saline and transferred in duplicate to 96-well microplates. Microbial inocula were prepared after comparison of the absorbance (at 630 nm) of cell suspensions to the 0.5 McFarland standard and dilution of the suspensions in broth (Sabouraud dextrose and cation-adjusted Mueller–Hinton broth [Difco] for the fungi and bacteria, respectively, and 5% Alamar blue [BioSource International] in Middlebrook 7H9 broth with oleic acid–albumin–dextrose–catalase enrichment for M. intracellulare) to afford recommended inoculum sizes. Microbial inocula were added to the samples to achieve a final volume of 200 μL and final sample concentrations starting with 20 μg/mL. Growth, solvent, and medium controls were included on each test plate. The plates were read at either 530 nm or excitation and emission wavelengths of 544 and 590 nm (Alamar Blue method) prior to and after incubation. Percent growth was calculated and plotted with the concentration tested to afford the concentration that inhibits 50% of growth (IC50).
5.9. Cytotoxicity assay
In vitro cytotoxicity was determined against mammalian kidney fibroblast (VERO) cells. The assay was performed in 96-well tissue culture-treated microplates and compounds were tested up to a highest concentration of 10 μg/mL as described earlier [21]. In brief, cells (25,000 cells/well) were seeded to the wells of the plate and incubated for 24 h. Samples were added and plates were again incubated for 48 h. The number of viable cells was determined by the neutral red assay as previously described [21]. IC50 values were determined from dose curves of growth inhibition versus concentration. Doxorubicin was used as a positive control, while DMSO was used as the negative (vehicle) control.
Research Highlights.
N-Substituted δ-carbolines have been identified as novel anti-opportunistic infection agents Several synthetic analogs show potency over 100-fold that of the original natural product.
Toxicity has also been attenuated several fold over the parent compounds the introduction of omega-cyclohexylpentyl moiety
Overall, the optimized compounds are fungicidal and bactericidal in contrast to the parent compounds which are only fungistatic and bacteriostatic.
Acknowledgments
We are grateful for financial support from the National Institutes of Health, National Institute of Allergy and Infectious Diseases, through AREA grant number R15 Al37976-01, Research Centers at Minority Institutions (RCMI) grant number G12 RR 03020, and a Title III grant to Florida A&M University. This research was also supported in part by the Pharmaceutical Research Center NIH/NCRR 1 C06-RR12512-01 Grant. We appreciate the generous contributions of Dr. Wang Zhang, Senior Technician responsible for instrumentation for his contribution to the generation of NMR spectra of compounds in this paper, Mrs. Barbara Bricker who proof-read the manuscript and Ms. Marsha Wright for biological evaluation. Antifungal and antibacterial testing were supported by the NIH, NIAID, Division of AIDS, Grant No. AI 27094 and the USDA Agricultural Research Service Specific Cooperative Agreement No. 58-6408-2-0009, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Mardenborough LG, Fan P, Ablordeppey SY, Nimrod A, Clark AM. Identification of a novel antifungal agent based on a pharmacophoric group in cryptolepine. Med. Chem. Res. 1999;9:118–132. [Google Scholar]
- [2].Ablordeppey SY, Fan P, Clark AM, Nimrod A. Probing the N-5 region of the indolo-quinoline alkaloid, cryptolepine for anticryptococcal activity. Bioorg. Med. Chem. 1999;7:343–349. doi: 10.1016/s0968-0896(98)00244-2. [DOI] [PubMed] [Google Scholar]
- [3].Mazu TK, Etukala JR, Eyunni SK, Zhu XY, Jacob MR, Khan SI, Walker LA, Ablordeppey SY. Identification of 3-phenylaminoquinolinium and 3-phenylaminopyridinium salts as new agents against AIDS-related opportunistic pathogens. Bioorg. Med. Chem. doi: 10.1016/j.bmc.2010.10.065. Epub Nov. 5, 2010. DOI information: 10.1016/j.bmc.2010.10.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4] a).Boucher HW, Corey GR. Epidemiology of methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 2008;46(Suppl 5):S344–349. doi: 10.1086/533590. [DOI] [PubMed] [Google Scholar]; b) Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK, for the ABCs Invasive methicillin-resistant Staphylococcus aureus infections in the United States. J. Amer. Med. Assoc. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- [5].Fan P, Ablordeppey SY. An alternative synthesis of 10H-indolo[3,2-b]quinoline and its selective N-alkylation. J. Heterocycl. Chem. 1997;34:1789–1794. [Google Scholar]
- [6].Kumar EV, Etukala JR, Ablordeppey SY. Indolo[3,2-b]quinolines: synthesis, biological evaluation and structure activity-relationships. Mini Rev Med Chem. 2008;8:538–554. doi: 10.2174/138955708784534418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Boateng CA, Eyunni SVK, Zhu XY, Etukala JR, Bricker BA, Ashfaq MK, Jacob MR, Khan SI, Walker LA, Ablordeppey SY. Benzothieno[3,2-b] quinolinium and 3-(phenylthio)-quinolinium compounds: Synthesis and evaluation against opportunistic fungal pathogens. Bioorg. Med. Chem. doi: 10.1016/j.bmc.2010.11.008. Epub. Nov 10, 2010, doi:10.1016/j.bmc.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8] a).Ablordeppey SY, Fan P, Li S, Clark AM, Hufford CD. Substituted indoloquinolines as new antifungal agents. Bioorg. Med. Chem. 2002;10:1337–1346. doi: 10.1016/s0968-0896(01)00401-1. [DOI] [PubMed] [Google Scholar]; b) Zhu XY, Mardenborough LG, Li S, Khan A, Zhang W, Fan P, Jacob MR, Khan S, Walker LA, Ablordeppey SY. Synthesis and evaluation of isosteres of N-methyl indolo[3,2-b]-quinoline (cryptolepine) as new antiinfective agents. Bioorg. Med. Chem. 2007;15:686–695. doi: 10.1016/j.bmc.2006.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Van de Waterbeemd H, Rose S. In: The Practice of Medicinal Chemistry. 2nd Edition Wermuth CG, editor. Elsevier Academic Press; 2004. p. 351. [Google Scholar]
- [10].Mardenborough L, Zhu XY, Fan P, Jacob MR, Khan SI, Walker LA, Ablordeppey SY. ldentification of bis-quindolines as new antiinfective agents. Bioorg. Med. Chem. 2005;13:3951–3963. doi: 10.1016/j.bmc.2005.04.008. [DOI] [PubMed] [Google Scholar]
- [11].Navarro MB, Huttner B, Harbarth BS. Methicillin-resistant Staphylococcus aureus control in the 21st century: beyond the acute care hospital. Curr. Opin. Infect. Dis. 2008;21:372–379. doi: 10.1097/QCO.0b013e3283013add. [DOI] [PubMed] [Google Scholar]; b) File TM., Jr. Impact of community-acquired methicillin-resistant Staphylococcus aureus in the hospital setting. Cleve Clin. J. Med. 2007;74(Suppl 4):S6–11. doi: 10.3949/ccjm.74.suppl_4.s6. [DOI] [PubMed] [Google Scholar]
- [12].Anstead GM, Quinones-Nazario G, Lewis JS. Treatment of infections caused by resistant Staphylococcus aureus. Methods Mol. Biol. 2007;391:227–258. doi: 10.1007/978-1-59745-468-1_17. [DOI] [PubMed] [Google Scholar]
- [13].Stryjewski ME, Corey GR. New treatments for methicillin-resistant Staphylococcus aureus. Curr. Opin. Crit. Care. 2009;15:403–412. doi: 10.1097/MCC.0b013e32832f0a74. [DOI] [PubMed] [Google Scholar]
- [14] a).Cattoir V, Daurel C. Update on antimicrobial chemotherapy. Med Mal Infect. 2010;40:135–154. doi: 10.1016/j.medmal.2009.10.009. [DOI] [PubMed] [Google Scholar]; b) Coates A, Hu Y, Bax R, Page C. The future challenges facing the development of new antimicrobial drugs. Nat. Rev. Drug Discov. 2002;1:895. doi: 10.1038/nrd940. [DOI] [PubMed] [Google Scholar]
- [15] a).Inderlied CB, Kemper CA, Bermudez LE. The Mycobacterium avium complex. Clin Microbiol Rev. 1993;6:266–310. doi: 10.1128/cmr.6.3.266. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) MacDonell KB, Glassroth J. Mycobacterium avium complex and other nontuberculous mycobacteria in patients with HIV infection. Semin. Respir. Infect. 1989;4:123–32. [PubMed] [Google Scholar]
- [16] a).Samoylenko V, Jacob MR, Khan SI, Zhao J, Tekwani BL, Midiwo JO, Walker LA, Muhammad I. Antimicrobial, antiparasitic and cytotoxic spermine alkaloids from Albizia schimperiana. Nat. Prod. Commun. 2009;4:791–796. [PMC free article] [PubMed] [Google Scholar]; b) Guoyi M, Khan S, Jacob MR, Tekwani BL, Li Z, Pasco DS, Walker LA, Khan IA. Antimicrobial and antileishmanial activities of hypocrellins A and B, Antimicrob. Agents Chemother. 2004;48:4450–4452. doi: 10.1128/AAC.48.11.4450-4452.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].National Committee for Clinical Laboratory Standards . M7-A5. National Committee for Clinical Laboratory Standards; Wayne, PA: 2000. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. [Google Scholar]
- [28].National Committee for Clinical Laboratory Standards . Approved standard M27-A. National Committee for Clinical Laboratory Standards; Wayne, PA: 1997. Reference method for broth dilution antifungal susceptibility testing of yeasts. [Google Scholar]
- [19].National Committee for Clinical Laboratory Standards . Tentative standard 3962 M24-T2. 2nd ed. National Committee for Clinical Laboratory Standards; Wayne, PA: 2000. Susceptibility testing of mycobacteria, nocardia, and other aerobic actinomycetes. [PubMed] [Google Scholar]
- [20].Franzblau SG, Witzig RS, McLaughlin JC, Torres P, Madico G, Hernandez A, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, Gilman RH. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 1998;36:362–366. doi: 10.1128/jcm.36.2.362-366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mustafa J, Khan SI, Ma G, Walker LA, Khan IA. Synthesis and in vitro cytotoxic activity of N-, F-, and S-ether derivatives of podophyllotoxin fatty acid adducts. Lipids. 2006;40:375–382. doi: 10.1007/s11745-006-1397-x. [DOI] [PubMed] [Google Scholar]