

Abstract
The diverse RGS protein family is responsible for the precise timing of G-protein signaling. To understand how RGS protein structure encodes their common ability to inactivate G-proteins and their selective G-protein recognition, we integrated structure-based energy calculations with biochemical measurements of RGS protein activity. We revealed that, in addition to previously identified conserved residues, RGS proteins contain another group of variable modulatory residues, which reside at the periphery of the RGS-domain–G-protein interface and fine-tune G-protein recognition. Mutations of modulatory residues in high-activity RGS proteins impaired RGS function, whereas redesign of low-activity RGS proteins in critical modulatory positions yielded complete gain-of-function. Therefore, RGS proteins combine a conserved core interface with peripheral modulatory residues to selectively optimize G-protein recognition and inactivation. Finally, we show that our quantitative framework for analyzing protein-protein interactions can be extended to analyze interaction specificity across other large protein families.
‘Regulator of G-protein Signaling’ (RGS) proteins play a critical role in numerous G-protein-dependent signaling pathways. RGS proteins “turn off” heterotrimeric (αβγ) G-proteins and thereby determine the duration of G-protein-mediated signaling events 1–5. Like many signaling proteins, RGS proteins comprise a large and diverse family. In humans, there are about 20 “canonical” RGS proteins that down-regulate activated G-proteins of the Gi and Gq subfamilies 6,7. In these RGS proteins, the RGS homology domain of ~120 amino acids functions as a GTPase activating protein (GAP) for GTP-bound Gα subunits 3–5. In recent years, RGS proteins have been implicated in a wide range of pathologies, including cancer, hypertension, arrhythmias, drug abuse, and schizophrenia 7–10, making RGS proteins promising drug targets 7,8. Therefore, identifying the determinants of G-protein recognition by RGS proteins is essential for understanding these signaling pathways and eventually for manipulating them with drugs.
While multiple RGS proteins are often expressed in the same cell, several studies have shown that only particular RGS proteins mediate a given biological function 11–17. This raises a significant interest in understanding the interaction specificity of RGS proteins. In many cases this specificity may originate from precise subcellular targeting, contributions from additional non-catalytic domains, adapter proteins, or participation in scaffolded protein complexes 7,9,13,15,18,19. However, there are clear cases where the ability to recognize a given G-protein is defined by the RGS domain itself 7,9,13,15. Nevertheless, the only two well-studied examples of RGS domain specificity are RGS9, whose specific recognition of Gαt requires the adapter protein PDEγ 18,20, and RGS2, which was shown to specifically down-regulate G-proteins of the Gq, but not Gi, subfamilies 21,22 cf. 23. The key determinants of RGS2 specificity were identified 22 by analyzing the multiple sequence alignment of RGS proteins in the context of the RGS4–Gαi1 crystal structure 24. This alignment revealed three crucial positions that are highly conserved in the RGS family, but are different in RGS2. Changing these three RGS2 residues to their counterparts in RGS4 yielded a gain-of-function phenotype that enabled RGS2 to efficiently down-regulate Gαi 22,25. Many additional studies showed that the GAP activity of individual RGS proteins toward a given Gα may vary (reviewed in refs. 6–8,13), but the molecular determinants for this selectivity have not been identified.
Critical insights into understanding the GAP activity of RGS proteins have been obtained using X-ray crystallography. To date, eight different structures of Gα subunits in complex with canonical RGS domains have been solved 24–28. These studies, combined with biochemical examinations, established that RGS domains bind Gα subunits and stabilize their catalytic residues allosterically in a conformation optimal for GTP hydrolysis 6,24,29–31. RGS protein residues in the vicinity of the Gα–RGS-domain interface show substantial diversity, suggesting that they may set interaction specificity. However, low sequence identity among RGS domains (as low as 30%; Supplementary Table 1) makes it difficult to pinpoint RGS domain residues that determine selective interaction with a specific Gα subunit 27,32.
In this study, we integrated functional assays with structure-based computations to determine the structural features within a large array of human RGS proteins that control their ability to inactivate a representative G-protein, Gαo (also known as GNAO1). We combined the experimental benchmark of the ability of ten RGS domains to activate Gαo GTPase with comparative structural analysis, electrostatic calculations of interaction energies using the Finite-Difference Poisson-Boltzmann method (FDPB), and in silico mutagenesis. Using a consensus approach across the eight available RGS-domain–G-protein crystal structures, we developed a structure-to-sequence map predicting which residues within the RGS domains are essential for their GAP function and which residues can modulate specific interactions with the cognate Gα subunit. We validated these predictions by site-specific mutagenesis of critical residues revealed in this map that allowed us to impair the GAP function in high-activity RGS proteins and completely restore this function in low-activity RGS proteins. Finally, we explored the general utility of this approach by applying it to the interaction between the Escherichia coli colicin E7 and its inhibitory immunity proteins, a well-established system for studying protein-protein interaction specificity. Our computational analysis successfully pinpointed not only specificity determinants revealed in previous computational studies of these proteins, but also those previously identified only by in vitro evolution. Therefore, our approach enables extending the analysis of interaction specificity to the level of whole families and complements existing protein design methodologies.
RESULTS
RGS proteins differ in their ability to activate Gαo GTPase
We measured the GAP activity of ten individual human RGS domains using single turnover GTPase assays with the G-protein Gαo (Fig. 1a). Six of these domains (RGS1, RGS4, RGS7, RGS8, RGS10, and RGS16) exhibited the same level of high GAP activity, RGS2 had no measurable activity (as expected from refs. 21,22,25), whereas three RGS domains (RGS14, RGS17, and RGS18) had low but discernable activities. Interestingly, this quantitative comparison showed little correlation between the GAP activities of individual RGS domains and the degree of their sequence identity. Indeed, the sequence identity among the six highly active RGS domains typically ranged between 37% and 60%, with only one pair sharing 73% identity (Supplementary Table 1). This is the same range as the identity among the sequences of no-activity (RGS2) and low-activity (RGS14, RGS17, and RGS18) RGS domains (37–56%), or between the sequences of the no or low-activity and high-activity groups of RGS domains (36–60%). Therefore, sequence identity among RGS domains does not serve as a reliable predictor for RGS protein GAP activity on Gαo.
Figure 1.
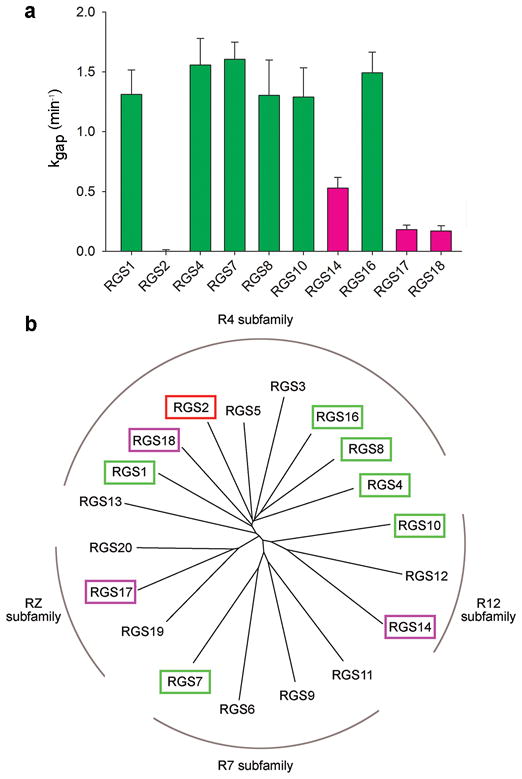
The GAP activities of 10 representative RGS domains do not correlate with their sub-family classification. (a) The kgap constant for each domain was calculated as described in Supplementary Methods from single exponential fits to the time course of GTP hydrolyzed by Gαo (400 nM) with or without added RGS protein (20 nM). The data are shown as mean±s.e.m; n≥4. High-activity, low-activity, and no-activity RGS proteins are colored green, magenta, and red, respectively. (b) Phylogenetic tree of 20 human RGS domains. RGS proteins whose activity was tested in this study are colored as in a.
Consequently, the GAP activity of these ten RGS domains did not correlate with their sequence-alignment-based classification into sub-families (Fig. 1b; note that the same subfamily classification was reached based on the identity of additional non-catalytic domains in the corresponding full-length RGS proteins 6,7,33). Large differences in GAP activity were observed within the same subfamily (e.g. RGS4 vs. RGS18 vs. RGS2), while similar activities were observed in RGS domains representing different subfamilies (e.g. RGS4, RGS7, RGS10). This analysis demonstrates that RGS protein GAP function is determined at a finer resolution (i.e. individual amino acids) than provided by current RGS protein classifications.
Residue-level energy analysis of RGS–G-protein interactions
To map the contributions of individual RGS domain residues to their GAP activity, we characterized the eight available crystal structures of canonical RGS domains bound to Gα subunits, using a comparative structural and energetic analysis (Fig. 2a,b). The number of RGS protein residues in the vicinity of the RGS-domain–Gα interface is large (e.g. the eight crystal structures contain 62–67 RGS domain residues within 10 Å of the Gα subunit) and the sequence diversity among these residues is considerable 27. Therefore, we followed Sheinerman et al. 34 and used the FDPB method coupled with in silico mutagenesis to calculate which RGS protein residues make substantial electrostatic contributions (ΔΔGelec) to the interaction with the cognate Gα partner. In these calculations we considered all residues within 15 Å of the RGS-domain–Gα interface (89–93 residues per RGS domain). We separated the electrostatic contributions of each residue into those coming from the side chain and/or those originating from the main chain (Supplementary Fig. 1; see Methods for details). We also estimated the non-polar energetic contributions of each residue by converting surface area buried in the complex to the equivalent energy contribution 34. Because these energetic contributions were calculated in a static snapshot of a complex, we did not expect the obtained per-residue ΔΔG values to exactly match experimentally determined ΔΔG values (see refs. 34,35 for a detailed discussion). Rather, we aimed to generate a list of residues likely to be important for interactions with a Gα partner. Therefore, we constructed a residue-level sequence “map” that listed all RGS protein residues predicted to contribute substantially (at least 1 kcal mol−1) to the interaction (see Methods). We classified these residues into two major groups: 1) “Significant & Conserved” residues that make the same type of substantial energy contribution in the majority of structures (marked with red asterisks in Fig. 2a). Note that if the energy contribution comes only from the residue backbone, amino acids in Significant & Conserved positions may not be conserved at the sequence level (e.g. position 131). 2) Putative “Modulatory” residues, which make substantial energy contributions only in some of the structures and are not conserved across the structures (marked with purple triangles in Fig. 2a). We identified 12 RGS domain residues as Significant & Conserved and between 6 and 8 residues in each structure as Modulatory.
Figure 2.

Positions of Significant & Conserved and Modulatory residues in multiple RGS proteins. (a) Residue-level sequence “map” summarizing the structure analysis and energy calculations of eight RGS–Gα crystal structures. The PDB IDs of these structures are: 1AGR (RGS4); 2IK8 (RGS16h, human RGS16); 3C7K (RGS16m, mouse RGS16); 2IHB (RGS10); 2GTP (RGS1); 2ODE (RGS8); 1FQJ (RGS9); 2V4Z (RGS2*, gain-of-function RGS2 triple mutant, see Methods). The sequences in the multiple sequence alignment are taken from the crystal structures. RGS protein residues that contribute substantially to the interaction with the Gα subunit are color-coded in the panel according to the type of their energetic contribution (see legend). Putative Significant & Conserved and Modulatory positions are marked above the alignment by red asterisks and purple triangles, respectively. (b) 3D visualization of the different types of energetic contributions by individual RGS protein residues, depicted as spheres and colored as in a. The eight superimposed RGS domain structures are viewed through the semi-transparent surface of Gα. (c) Significant & Conserved and Modulatory residues in the eight superimposed RGS domain structures, shown as spheres and colored red and purple, respectively. The orientation is the same as in b. (d) Same as c, rotated 90° about the Y axis.
Interestingly, Significant & Conserved residues are located mainly in the center of the RGS-domain–Gα interface, while putative Modulatory residues are located mostly at this interface’s periphery (Fig. 2c,d). This arrangement raises the possibility that Significant & Conserved residues are essential for RGS protein GAP activity, while different combinations of Modulatory residues may further tune RGS-domain–Gα interactions, ultimately defining whether a given RGS protein is a good or a poor GAP – the hypothesis tested in this study.
Comparison of predictions with previous mutagenesis studies
To evaluate whether a substantial energetic contribution of an RGS protein residue (Fig. 2a) serves as a reliable predictor of significance for RGS protein GAP function, we first employed published mutagenesis studies. Ref. 36 describes comprehensive mutagenesis of 39 RGS4 residues, analyzed using GTPase assays and/or the inhibition of G-protein signaling in yeast. 23 of these mutants did not affect RGS4 function. Consistent with those experiments, our calculations showed no substantial energetic contribution for 22 of those residues. The only discrepancy was Lys162, predicted to make a conserved non-polar energetic contribution in all RGS-domain structures (Fig. 2a), while the K162A mutation was not found to impair RGS4 activity in ref. 36, though it was tested only in the less-direct yeast assay.
Among the sixteen positions substantially impairing RGS4 activity 36, seven were not located near the RGS-domain–Gα interface and instead are a part of the RGS domain’s hydrophobic core, conserved across all available crystal structures (Supplementary Fig. 2). Presumably, mutating these large hydrophobic residues to alanines impaired RGS4 GAP activity indirectly through improper folding of these mutants. All of the other activity-impairing mutations (three of which were also revealed in refs. 24,29,37,38) corresponded to positions identified as Significant & Conserved in our calculations – thereby substantiating the predictions of our computational analysis. The remaining three Significant & Conserved residues in RGS4 (Ala124, Val127 and Ser131) were not mutated in previous studies. However, the energetic contributions of these residues originate from their backbones rather than their side chains and thus are not amenable to straightforward validation by the mutagenesis of side-chains. Therefore, previous mutagenesis studies are in full agreement with the predictions of our computationally-derived residue-level map.
Design of loss-of-function RGS4 and RGS16 mutants
Next, we tested whether the putative Modulatory residues listed in our map (Fig. 2a) indeed play a role in RGS protein GAP activity. Almost none of these residues were mutated in past studies, probably because the lack of conservation at these positions suggested a lack of functional role. For these mutagenesis experiments, we picked representative Modulatory positions in human RGS4 and RGS16 (Fig. 3a,b). Single alanine substitutions of such residues in RGS4 had either a minor or a moderate effect on GAP activity (Fig. 3c). However, the loss-of-function effect was additive – the GAP activity of a triple mutant (RGS4d) was abolished. Therefore, mutations in a sufficient number of Modulatory residues causes complete loss-of-function, just as the effect of a mutation in the Significant & Conserved residue Asn128 (RGS4e in Fig. 3c), previously shown to be critical for the functions of RGS4 24,29 and RGS16 37,38.
Figure 3.

Mutations in Modulatory positions impair the GAP activities of RGS4 and RGS16 in an additive manner. (a) Sequences of RGS4 mutations in Modulatory positions (RGS4a–d) and a Significant & Conserved position (RGS4e). (b) Sequences of RGS16 mutations in Modulatory positions (RGS16a–d) and a Significant & Conserved position (RGS16e). The annotated sequences of wild-type RGS4 and RGS16 in a and b are from Figure 2a. (c) GAP activities of RGS4 mutants determined by single turnover GTPase assays. 400 nM GTP-loaded Gαo was incubated with or without RGS4 (40 nM) for 1 min. GAP activities are expressed as a percentage of wild-type RGS4 activity. Experiments were conducted in triplicate with error bars representing s.e.m. (d) GAP activities of the RGS16 mutants, determined as in c.
Similarly, mutating individual Modulatory residues in RGS16 had either no effect or a moderate effect on its GAP activity (Fig. 3d). But, like in RGS4, the effect of double or triple mutants was additive and impaired the ability of RGS16 to activate Gαo GTPase to a much higher degree than single mutations. These results affirm the importance of Modulatory residues’ contributions in attaining the maximal GAP activity of RGS proteins and thereby validate our approach for pinpointing critical residues using the structure-to-sequence map.
Design of gain-of-function RGS17 and RGS18 mutants
The ultimate test for the utility of our energy contribution map was to take low GAP-activity RGS proteins and redesign them into mutants having high GAP activity (Figs. 4 and 5). We selected two low-activity RGS proteins representing different subfamilies, RGS17 and RGS18. The high-activity template for redesign was RGS16, as it is best represented in available RGS-domain–Gα crystal structures 27,28. The RGS domain of RGS16 is different from RGS17 and RGS18 in 70 and 56 positions, respectively. To identify which of these residues in RGS17 and RGS18 are responsible for their impaired GAP activity, we focused on the positions defined as either Significant & Conserved or Modulatory, cutting the number of candidate residues down to thirteen in RGS17 and eight in RGS18. To further reduce the number of positions to mutate, we dismissed residues found at the corresponding positions in any of the high-activity RGS proteins (marked in bold black in Figs. 4a and 5a). For example, Arg154 in RGS17 corresponds to a glutamic acid in RGS16; yet in the high-activity RGS1 this position is also an arginine, suggesting that Arg154 in RGS17 is not tied to its low GAP activity.
Figure 4.

Redesign of RGS17 gain-of-function mutants. (a) Sequences of RGS16 (annotated as in Fig. 2a), RGS17 and its mutants. RGS17 residues in Significant & Conserved or Modulatory positions that are different from RSG16 are marked as follows: those predicted to interfere with high GAP activity are orange and those appearing in other high activity RGS proteins are bold black. Residues in RGS17 mutants that were replaced by RGS16 residues are blue. (b) Positions of the four RGS17 sites that were mutated in the redesign experiments. The RGS16 residues that were used as the template for the redesign are visualized on the superimposed structures of Gαi1–RGS16 (PDB ID 2IK8) and Gαo–RGS16 (PDB ID 3CK7), viewed through the semi-transparent surface of the Gα subunit. Corresponding RGS17 residue numbers are in parentheses. (c) GAP activities of the redesigned RGS17 mutants compared to activities of wild-type proteins. kgap values were determined as in Figure 1 and shown as mean±s.e.m.(n≥4).
Figure 5.

Redesign of RGS18 gain-of-function mutants. (a) Sequences of RGS16, RGS18 and its mutants, color-coded as in Figure 4a. (b) Positions of the three RGS18 sites that were mutated in the redesign experiments, visualized as in Figure 4b. Corresponding RGS18 residue numbers are in parentheses. (c) GAP activities of the redesigned RGS18 mutants compared to activities of wild-type proteins. kgap values were determined as in Figure 4c.
We first applied these residue selection criteria to RGS17 and identified four sites that may be responsible for its low GAP activity: positions 143–145, 150, 183–184, and 192 (Fig. 4a,b). Two of these sites are predicted to impair activity because they lack side-chains directly interacting with Gαo in high-activity RGS proteins. Ser150 is found at the RGS17 position occupied by a Significant & Conserved asparagine in all high-activity RGS proteins (Fig. 2). Indeed, the corresponding N128S mutation in RGS4 abolished its GAP function (Fig. 3c and ref. 29). Similarly, Asn192 in RGS17 corresponds to a lysine in all high-activity RGS proteins. The two remaining RGS17 sites (143–145, 183–184), containing mostly Modulatory residues, are likely to impact its GAP activity indirectly by displacing neighboring residues that do interact with Gαo directly. Note that Ser145, despite occupying the position of a Significant & Conserved alanine in high activity RGS proteins, presumably affects the GAP activity of RGS17 indirectly: while the backbone of the corresponding RGS16 alanine interacts favorably with the Gα subunit, the aliphatic side chain points into the RGS domain core. Thus, a serine in this position would likely necessitate a local repacking of the RGS protein, thereby affecting interactions with the Gα subunit indirectly.
We measured the GAP activity of representative RGS17 mutants bearing different combinations of amino acid replacements at these four sites (Fig. 4c). Interestingly, the RGS17-to-RGS16 replacements of both “direct” contributors (S150N or N192K), separately or together, did not increase RGS17 GAP activity at all (Fig. 4c). Even combining the S150N+N192K double mutation with the replacement of the entire 143–145 site containing Ser145 caused only a minor activity increase. However, simultaneous substitution of all four RGS17 sites resulted in the same GAP activity as in RGS16. Therefore, optimizing Modulatory positions in this protein was critical for achieving a complete gain-of-function.
A similar redesign was applied to RGS18, also using the RGS16 template. Unlike RGS17, all Significant & Conserved positions in RGS18 are not different from high-activity RGS proteins. Yet, RGS18 has four Modulatory positions in three distinct sites that could potentially impair its GAP activity: 141, 156+158 and 186 (Fig. 5a,b). In contrast to the minimal effect of partial mutagenesis in RGS17, two out of three single site mutants in RGS18 (H156E+K158R and Q186K) markedly increased its GAP activity (Fig. 5c). Combining H156E+K158R with K141E caused a slight additional improvement and mutating all three sites simultaneously yielded full gain-of-function.
To test whether the increased GAP activity of the redesigned gain-of-function mutants was a result of increased affinity to the Gα subunit, we assessed the binding of the series of redesigned RGS18 mutants (Fig. 5a,c) to Gαo using Surface Plasmon Resonance spectroscopy (SPR) (Table 1 and Supplementary Fig. 3). In correlation with their low GAP activity, the KD values of RGS18 and its K141E mutant for Gαo were each in excess of 3 μM. However, the redesigned mutants that showed higher GAP activity had lower KD values, with the highest-activity mutant (RGS18e) exhibiting the lowest KD of 69 nM. These measurements show a strong correlation between the GAP activity and Gαo binding affinity for each RGS18 mutant. Taken together, our data demonstrate that optimizing Modulatory residues is sufficient for the restoration of maximal GAP activity of RGS18.
Table 1.
Quantification of Gαo binding affinity of RGS18 mutantsa
Gαo is bound to the transition state analog GDP–aluminum fluoride.
KD values were calculated as weighed averages ± s.e.m. from 2–3 independent experiments (see Methods for details)
These KD values are underestimations because dose response saturation was not reached.
Comparison to alternative computational approaches
We compared our computational approach to other methods that predict residues contributing substantially to protein-protein interactions. We applied Rosetta’s computational alanine scanning 39 to the RGS-domain–Gα structures analyzed above. This method identified potential hot spots in each RGS protein corresponding to between five and eight of our Significant & Conserved residues and between zero and two Modulatory residues (Supplementary Table 2). As expected from an alanine-scanning protocol, Rosetta did not identify residues making substantial energy contributions via their backbones, but it also did not identify most Modulatory residues. This suggests that the majority of Modulatory positions in RGS domains do not make sufficient energy contributions to be identified as hot spots by computational alanine scanning. Indeed, we typically had to mutate multiple Modulatory residues to observe large changes in RGS activity (Fig. 3). Another reason why our approach identified a larger number of critical RGS residues may be that long-range electrostatics, which are not explicitly taken into account by Rosetta, play a particularly important role at the RGS-domain–Gα interface. Therefore, the physics-based energy calculations used in this study seem better suited to identify residues in RGS proteins that are engaged in modulatory interactions.
We next used Consurf 40 to test whether a sequence-based approach, which searches for phylogenetic relations between close homologs, can identify RGS residues that contribute to interactions with Gα subunits. Consurf calculated that the majority of Significant & Conserved residues had a conservation score above average, as expected from residues that share a similar functional role among all high activity RGS proteins. Seven additional residues at or near the RGS-domain–Gα interface were also identified as evolutionary conserved, although mutations in most of these residues had no effect on GAP function 36. The vast majority of RGS Modulatory residues had average or below average conservation scores and therefore were not pinpointed by this analysis.
A more complete result was obtained by the Evolutionary Trace method 32. This study identified an evolutionary privileged surface containing 17 RGS domain residues, 10 of which form a cluster of well-conserved contact residues judged not to have a role in determining specificity (we classified eight of them as Significant & Conserved). Five out of the seven remaining residues were defined as a second cluster of “class specific” residues (we classified four of them as Modulatory). In the case of RGS9, this cluster was suggested to form a binding site for the RGS9 adapter protein, PDEγ, a concept experimentally confirmed in a subsequent study 18. However, this study did not address the role of these evolutionary privileged residues in setting RGS–G-protein specificity. Rather it highlighted that certain class specific residues can participate in specific interactions with proteins other than Gα subunits (e.g. RGS9 interaction with PDEγ). This sequence-level superposition of overlapping interaction surfaces may provide an additional challenge for sequence-based methods (e.g. Consurf and Evolutionary Trace), but not structure-based methods, like the approach used in our study.
Computational analysis of colicin E7–Im protein interactions
To explore the general applicability of our approach, we considered the interaction between the DNase colicin E7 (E7) and the inhibitory immunity protein Im7, a system used extensively to study specificity determinants in protein-protein interactions 41,42, interface specificity redesign 43–45 and in vitro evolution studies 46. To map the contributions of individual residues to the interaction, we applied our consensus-based comparative structural and energetic analysis to the five available crystal structures of E7–Im7 complexes (Fig. 6 and Supplementary Fig. 4), which contained no E7 mutations near the Im7 interface and therefore were considered wild type proteins in regards to Im7 binding (see Methods). We also applied our comparative analysis to the two structures of computationally redesigned E7–Im7 43,44 and to the two structures of E7 bound to non-cognate Im9 proteins selected through in vitro evolutionary for high E7 affinity 46.
Figure 6.

Residues contributing substantially to colicin E7–immunity proteins interactions. Energy calculations were performed on the following structures (PDB IDs): Wild-type E7–Im7 complexes (wt1–5: 7CEI, 2JAZ, 2JB0, 2JBG, 1ZNV); Computationally-redesigned E7–Im7 (cr1–2: 1UJZ, 2ERH); E7 bound to Im9 proteins evolved in vitro to bind E7 with high affinity (ie1–2: 3GJN, 3GKL). (a) Residue-level sequence map of the wild-type and engineered immunity proteins. The sequences in the multiple sequence alignment are taken from the crystal structures. Residues that contribute substantially to the interaction are color-coded according to type of energy contribution (see legend). Consensus analysis was applied to the five wild-type proteins and Significant & Conserved and Modulatory positions were determined for all nine structures as in Figure 2. (b) The nine E7–Im structures, superimposed via the Im proteins. (c) Visualization of the energy contribution types for wild-type E7–Im7 residues, depicted as spheres and colored as in a. The E7 and Im7 structures are shown in an “open book” view (rotated 90° relative to b about the X axis in opposite directions). (d) Energy contributions of residues in the computationally-redesigned E7–Im7, shown as in c. (e) Energy contributions of residues in E7 and the in vitro evolved Im9 proteins, shown as in c.
Using the same criteria as for RGS proteins, we identified eight E7 positions and five Im positions as Significant & Conserved and seven E7 positions and twelve Im positions as Modulatory (Fig. 6a and Supplementary Fig. 4). The majority of these positions were previously shown to contribute to colicin–immunity protein binding and specificity 41,47. Interestingly, both the computationally redesigned and the in vitro evolved protein pairs seem to utilize essentially the same complement of energetically important residues as the wild type proteins. A minority of the residues in the computationally redesigned E7–Im7 uses a different energy type of interaction (Fig. 6a and Supplementary Fig. 4, Fig. 6c vs. Fig. 6d); e.g. the E7 K528Q mutation results in a loss of electrostatic side-chain contribution (Supplementary Fig. 4). In contrast, the in vitro evolved Im9 proteins show a drastically different map of energy contributions (Fig. 6a,e).
Importantly, our analysis identified residues in the Im7 α1–α2 loop as substantial contributors to the interactions with E7. This loop, located at the periphery of the E7–Im7 interface (Fig. 6b), was not identified in previous computational analyses and only recently was implicated as playing a role in binding specificity by the in vitro evolution study 46. We observe substantial contributions from these residues in all structures with a consistent theme of main-chain electrostatic contributions. However, the overall pattern of energy contributions from residues in this loop is quite different in the in vitro evolved Im9 proteins, suggesting in vitro evolution revealed an alternative mode of interaction using this Im substructure.
Discussion
Our study presents a novel approach to pinpoint structural determinants that are critical for fine-tuning protein-protein interaction specificity. Following recent successes in redesigning interaction affinity and/or specificity by combining computational analysis with experimental validation (e.g. ref. 48–50), we integrated the experimental benchmark of enzymatic assays with physics-based energy calculations using a consensus approach across multiple crystal structures. Our central result is that RGS proteins contain a previously uninvestigated group of unconserved residues that contribute to selective functional recognition of Gαo. Accordingly, mutations of these Modulatory residues in two high-activity RGS proteins severely impaired their ability to accelerate Gαo GTPase, whereas redesigning low-activity RGS proteins by mutating critical Modulatory residues increased their GAP activity dramatically.
The typical quantitative impact of a single Modulatory residue on RGS GAP activity was found to be smaller than those of Significant & Conserved residues. However, multiple Modulatory residues affect GAP function in an additive manner. In some cases, a single Modulatory residue made a small incremental contribution. In other cases, several Modulatory residues had to be mutated simultaneously to affect GAP activity substantially. The former is best represented by the loss-of-function mutants of RGS4 and 16; the latter is exemplified by the all-or-none gain-of-function effect of the different RGS17 mutants.
Modulatory residues are located mostly at the periphery of the Gα–RGS-domain interface where they contribute to Gα subunit recognition. The center of this interface is occupied by Significant & Conserved residues that are thought to play the primary role in accelerating Gα GTPase by stabilizing Gα in a conformation optimal for GTP hydrolysis 31. This elegant arrangement likely enables RGS proteins to share a common mechanism of GAP function concomitantly with divergent levels of selectivity towards a given Gα subunit. Furthermore, as illustrated in Figure 7, Significant & Conserved and Modulatory residues show different patterns of Gα interactions. In the eight structures we analyzed, the Significant & Conserved residues interact with all three Gα switch regions (Fig. 7a,b), as expected from the pivotal role of the switch regions in GTP hydrolysis 30,31. Modulatory residues interact with switch regions II and III, as well as with multiple residues in the Gα all-helical domain. The latter is particularly intriguing provided the growing interest in the role of the all-helical domain in facilitating Gα interactions with its regulatory partners 27,33. It is also noteworthy that some Modulatory residues may be involved in interaction with proteins other than Gα, as exemplified by RGS9 interactions with PDEγ.
Figure 7.
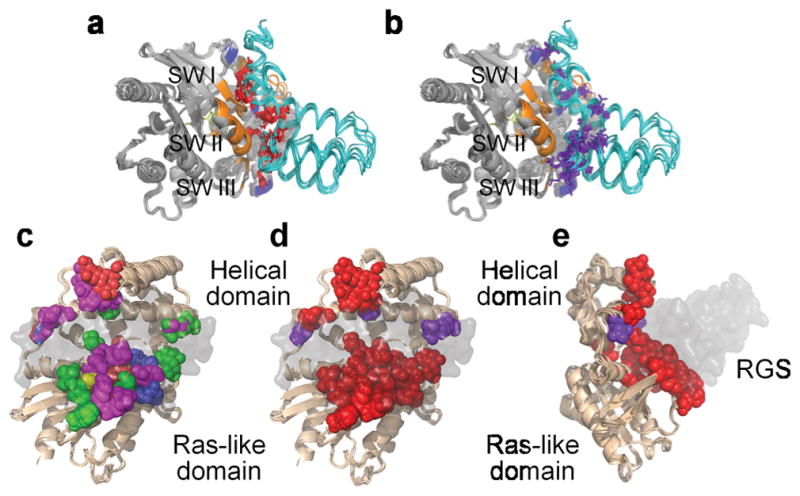
Positions of Significant & Conserved and Modulatory residues in the Gα subunits interacting with RGS domains. (a) Significant & Conserved RGS residues (red) interact with all three Gα switch regions. (b) Modulatory RGS residues (purple) interact with switch II and III and the helical domain of the Gα subunits. In both a and b, Significant & Conserved positions within the Gα subunits are marked in orange and Modulatory positions in blue. (c) The different types of energetic contributions by individual Gα residues, depicted as spheres and colored as in Figure 2. The eight superimposed Gα subunits are viewed through the semi-transparent surface of the RGS domain and are rotated 90° about the Y axis and 30° about the X axis relative to a. (d) Significant & Conserved and Modulatory residues in the Gα structures, shown as spheres and colored red and purple, respectively. (e) Same as d, rotated 90° about the Y axis.
In contrast to the variability of Modulatory residues among RGS proteins, there is a high level of conservation among the Gα residues forming the reciprocal side of this interface (compare Fig. 7d,e to Fig. 2c,d). Almost all of these Gα residues are classified by our energy-based calculations as Significant & Conserved, likely reflecting the fact that Gα subunits analyzed in our calculations are all from the Gi subfamily – Gαi1, Gαi3, Gαo and Gαt. This conservation may explain why some RGS proteins, whose isolated catalytic domains exhibit similarly high GAP activity towards these Gα subunits, rely on additional noncatalytic domains or adapter proteins to discriminate among individual Gi family members 7,13,15,20. Multiple sequence alignment 31 shows that other Gα subfamilies (e.g. Gs, G12/13) are quite different from Gi at the positions interacting with RGS Modulatory residues in Gαi. This hints at how specificity of RGS domain recognition may be achieved across the entire Gα family, which can be investigated in future studies.
From a methodological perspective, our approach to redesigning protein-protein interactions bypasses the well-recognized computational bottleneck of commonly used protein design methods – searching both sequence and 3D-structure space simultaneously to find promising design candidates 45,51. Rather, we used comparative information across the RGS protein family (via our sequence-level map) as a shortcut to identify the RGS domain sites that were most attractive for redesign mutagenesis. Furthermore, our approach does not depend on improving protein-protein interactions by mutating individual residues one at a time and combining mutations showing notable individual experimental effects – the approach used in some of the most successful previous studies (reviewed in ref. 45). Using such a strategy for RGS17 would have failed because individual mutations in this protein did not measurably increase its GAP activity. Our successes in redesigning RGS domain interactions and in predicting the determinants of interactions between colicins and immunity proteins suggest that physics-based energy functions can complement the engineered energy functions commonly used in protein design, both in analyzing design templates and assessing design products.
In conclusion, our work provides a quantitative framework for understanding the determinants of selective RGS protein interactions with a G-protein and enables structure-based redesign of protein-protein interactions at the family level. It can be extended to design a variety of RGS protein and G-protein mutants with distinct activities and selectivities as tools to decipher G-protein signaling networks in living cells. Given the growing number of available structures of representative protein-protein complexes (e.g., ref. 52), this methodology can be easily adapted to study interaction specificity across other large protein families.
Methods
Atomic structural models
The atomic models of the RGS-domains–Gα complexes used in the calculations were taken from the following PDB entries: 1AGR (Gαi1–RGS4); 2IK8 (Gαi1–RGS16); 3C7K (Gαo–RGS16); 2IHB (Gαi3–RGS10); 2GTP (Gαi1–RGS1); 2ODE (Gαi3–RGS8); 1FQJ (Gαi1/t–RGS9); 2V4Z (Gαi3–RGS2-C106S/N184D/E191K triple mutant) 24–28. Colicin–immunity-protein atomic models were taken from the following PDB entries: 7CEI, 2JAZ, 2JB0, 2JBG, 1ZNV (wild-type E7–Im7) 53–55. Although some of these E7 proteins contain point mutations, these mutations are far from the Im7 binding site and therefore these chains were considered wild type); 1UJZ, 2ERH (Computationally-redesigned E7–Im7) 43,44; 3GJN, 3GKL (E7 bound to in vitro evolved Im9) 46. Missing short segments in 2IK8 (Gαi1 residues 112–118), 2IHB (RGS10 residues 103–113), and 2GTP (Gαi1 residues 112–118) were modeled based on the structure of Gαi1–RGS4 (PDB id 1AGR) using the program Nest 56 and partial or missing side chains were modeled using Scap 57. Similarly, a short missing E7 segment in the following structures was modeled based on PDB id 7CEI: 2JBG (residues 547–554), 2JAZ (residues 548–554), 2JB0 (residues 551–552), 1ZNV (residues 547–554), 3GJN (residues 549–554), 3GKL (residues 548–554). Hydrogen atoms were added using CHARMM, and the structures were subjected to conjugate gradient minimization with a harmonic restraint force of 50 kcal mol−1 Å−2 applied to the heavy atoms.
Calculating residue-level electrostatic and non-polar free energies contributions
Electrostatic potentials and free energies were calculated using the DelPhi program. DelPhi yields finite-difference solutions to the Poisson-Boltzmann equation (the FDPB method) for a system where the solvent is described in terms of a bulk dielectric constant and concentrations of mobile ions, while the solutes are described in atomic detail by the coordinates of individual atoms, atomic radii, and partial charges 58. The proteins were mapped onto a fine three-dimensional grid, where each small cube represents a small region of the protein or solvent. Charges and radii were taken from the CHARMM22 parameter set. Regions inside the molecular surfaces of the proteins were assigned a dielectric constant of 2, and those outside a dielectric constant of 80, combined with an ion exclusion layer of 2 Å around the solute. These particular parameters have been optimized for energetic calculations of protein-protein interactions and have been validated extensively for numerous systems (see refs. 34,35 and references therein). The ionic strength was set to 100 mM to approximate the experimental conditions. The numerical calculation of the potential was iterated to convergence, defined as the point at which the potential changes less than 10−5 kT e−1 between successive iterations. A sequence of focusing runs of increasing resolution was employed to calculate the electrostatic potentials (e.g. 0.375, 0.75, 1.5, and 3.0 grid per angstrom). Electrostatic energies were obtained using the calculated potentials, and the net electrostatic energy of a protein-protein interaction was determined as the difference between: (1) the electrostatic free energy of the proteins in complex; and (2) the electrostatic free energies of each of the proteins infinitely far apart (i.e. calculated separately).
Following refs. 34,35, we used the FDPB method, coupled with in silico mutagenesis, to calculate the net electrostatic and polar energetic contributions (ΔΔGelec) of a residue to the interaction with its protein partner resulting from the removal of partial and real charges of each residue. This would correspond to an in silico residue that is identical in shape and dielectric permittivity to the original residue, but is now partially or completely non-polar. For each residue this was repeated twice: once neutralizing backbone and side chain and once neutralizing the side chain only. Thereby, we differentiated between energetic contributions coming from the side chain vs. the main chain (Supplementary Fig. 1). We considered all residues within 15 Å of the RGS-domain–G-protein interface; this distance threshold (~1.5 debye lengths) was a compromise between identifying electrostatic contributions from residues distal to the interface and avoiding excessively long computational times. We checked the consequences of this distance threshold by repeating the calculations for Gαi1–RGS4 without any distance threshold. Indeed, all residues further than 15 Å of the interface contributed < 1 kcal mol−1 to the interaction.
The non-polar energetic contribution (ΔΔGnp) of each residue was calculated as a surface-area proportional term, obtained by multiplying the per-residue surface area buried upon complex formation by a surface tension constant of 0.05 kcal mol−1 Å−2 (Supplementary Fig. 1) 34. Solvent-accessible surface areas were calculated using the surfv program 59.
Test calculations using small translations (0.1–0.2 Å), rotations (5°) of the proteins, or changes in the grid size, estimated the numerical error in ΔΔGelec calculations as <0.5 kcal mol−1. Following the more stringent criteria of ref. 34, we defined residues that made substantial electrostatic contributions to the interactions with their cognate partners as those contributingΔΔGelec ≥1 kcal mol−1 to binding. Similarly, residues contributingΔΔGnp≥1 kcal mol −1 (≥20 Å2 buried upon complex formation) were selected as substantial non-polar energetic contributors. To reduce false positives and negatives, we employed a consensus approach: residues conserved across all structures that have comparable GAP activities (for RGS domains) or affinities (for colicins–immunity proteins) and calculated to have substantial interactions in the majority of structures, were considered to contribute substantially to the interaction in all these structures. Residues conserved across all such structures that were calculated to have substantial interactions in less than two structures were considered false positives. This consensus approach improved the accuracy of our predictions as we encountered several false positives and negatives due to a different side chain rotamer found in only one structure, despite that residue being strictly conserved and in a comparable 3D neighborhood (see Fig. 2 and Supplementary Fig. 1). RGS domain residues thus determined to contribute substantially were mapped onto a sequence map (e.g. Fig. 2a).
Supplementary Material
Acknowledgments
This research was supported by the NIH grants EY012859 (V.Y.A.) and GM082892 (D.P.S.), core grant for vision research to Duke University (EY5722), National Science Foundation through TeraGrid resources (TG-MCB080085T; M.K.), and a long term postdoctoral fellowship from the Human Frontier Science Program (M.K.). We thank the Duke Shared Cluster Resource and the SDSC for computational resources, S.A. Baker (University of Iowa), S. Farsiu, N.P. Skiba, E.S. Lobanova (Duke University) and D. Reichmann (University of Michigan) for helpful suggestions, B. Honig (Columbia University) for insightful guidance (M.K.), and F. Sheinerman, R. Rohs (University of Southern California), S. Fleishman (University of Washington) and E. Alexov (Clemson University) for helpful discussions.
Footnotes
Author contributions
M.K. designed and performed computational analysis and biochemical experiments, analyzed data and prepared the manuscript, A.M.T. performed experiments and prepared the manuscript, D.E.B. performed experiments and prepared the manuscript, D.P.S. supervised project and prepared the manuscript, V.Y.A. supervised project and analysis and prepared the manuscript.
Full methods. Methods for protein expression, purification, GTPase assays and Surface Plasmon Resonance assays can be found in the Supplementary Information.
References
- 1.Siderovski DP, Hessel A, Chung S, Mak TW, Tyers M. A new family of regulators of G-protein-coupled receptors? Curr Biol. 1996;6:211–2. doi: 10.1016/s0960-9822(02)00454-2. [DOI] [PubMed] [Google Scholar]
- 2.Koelle MR, Horvitz HR. EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell. 1996;84:115–25. doi: 10.1016/s0092-8674(00)80998-8. [DOI] [PubMed] [Google Scholar]
- 3.Berman DM, Wikie TM, Gilman AG. GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein α subunits. Cell. 1996;86:445–452. doi: 10.1016/s0092-8674(00)80117-8. [DOI] [PubMed] [Google Scholar]
- 4.Hunt TW, Fields TA, Casey PJ, Peralta EG. RGS10 is a selective activator of G alpha i GTPase activity. Nature. 1996;383:175–7. doi: 10.1038/383175a0. [DOI] [PubMed] [Google Scholar]
- 5.Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha-subunits. Nature. 1996;383:172–5. doi: 10.1038/383172a0. [DOI] [PubMed] [Google Scholar]
- 6.Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem. 2000;69:795–827. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- 7.Hollinger S, Hepler JR. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev. 2002;54:527–59. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- 8.Neubig RR, Siderovski DP. Regulators of G-protein signalling as new central nervous system drug targets. Nat Rev Drug Discov. 2002;1:187–97. doi: 10.1038/nrd747. [DOI] [PubMed] [Google Scholar]
- 9.Neitzel KL, Hepler JR. Cellular mechanisms that determine selective RGS protein regulation of G protein-coupled receptor signaling. Semin Cell Dev Biol. 2006;17:383–9. doi: 10.1016/j.semcdb.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Hurst JH, Hooks SB. Regulator of G-protein signaling (RGS) proteins in cancer biology. Biochem Pharmacol. 2009;78:1289–97. doi: 10.1016/j.bcp.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 11.Wang Q, Liu M, Mullah B, Siderovski DP, Neubig RR. Receptor-selective effects of endogenous RGS3 and RGS5 to regulate mitogen-activated protein kinase activation in rat vascular smooth muscle cells. J Biol Chem. 2002;277:24949–58. doi: 10.1074/jbc.M203802200. [DOI] [PubMed] [Google Scholar]
- 12.Tang KM, et al. Regulator of G-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat Med. 2003;9:1506–12. doi: 10.1038/nm958. [DOI] [PubMed] [Google Scholar]
- 13.Xie GX, Palmer PP. How regulators of G protein signaling achieve selective regulation. J Mol Biol. 2007;366:349–65. doi: 10.1016/j.jmb.2006.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cifelli C, et al. RGS4 regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ Res. 2008;103:527–35. doi: 10.1161/CIRCRESAHA.108.180984. [DOI] [PubMed] [Google Scholar]
- 15.Bansal G, Druey KM, Xie Z. R4 RGS proteins: regulation of G-protein signaling and beyond. Pharmacol Ther. 2007;116:473–95. doi: 10.1016/j.pharmthera.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bansal G, Xie Z, Rao S, Nocka KH, Druey KM. Suppression of immunoglobulin E-mediated allergic responses by regulator of G protein signaling 13. Nat Immunol. 2008;9:73–80. doi: 10.1038/ni1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laroche G, Giguere PM, Roth BL, Trejo J, Siderovski DP. RNA interference screen for RGS protein specificity at muscarinic and protease-activated receptors reveals bidirectional modulation of signaling. Am J Physiol Cell Physiol. 2010;299:C654–64. doi: 10.1152/ajpcell.00441.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sowa ME, et al. Prediction and confirmation of a site critical for effector regulation of RGS domain activity. Nat Struct Biol. 2001;8:234–7. doi: 10.1038/84974. [DOI] [PubMed] [Google Scholar]
- 19.Martemyanov KA, Arshavsky VY. Chapter 7 Biology and Functions of the RGS9 Isoforms. Prog Mol Biol Transl Sci. 2009;86:205–27. doi: 10.1016/S1877-1173(09)86007-9. [DOI] [PubMed] [Google Scholar]
- 20.Skiba NP, Hopp JA, Arshavsky VY. The effector enzyme regulates the duration of G protein signaling in vertebrate photoreceptors by increasing the affinity between transducin and RGS protein. J Biol Chem. 2000;275:32716–20. doi: 10.1074/jbc.C000413200. [DOI] [PubMed] [Google Scholar]
- 21.Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR. RGS2/G0S8 is a selective inhibitor of Gqalpha function. Proc Natl Acad Sci U S A. 1997;94:14389–93. doi: 10.1073/pnas.94.26.14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heximer SP, et al. G protein selectivity is a determinant of RGS2 function. J Biol Chem. 1999;274:34253–9. doi: 10.1074/jbc.274.48.34253. [DOI] [PubMed] [Google Scholar]
- 23.Ingi T, et al. Dynamic regulation of RGS2 suggests a novel mechanism in G-protein signaling and neuronal plasticity. J Neurosci. 1998;18:7178–88. doi: 10.1523/JNEUROSCI.18-18-07178.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tesmer JJ, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4--activated G(i alpha1): stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–61. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 25.Kimple AJ, et al. Structural determinants of G-protein alpha subunit selectivity by regulator of G-protein signaling 2 (RGS2) J Biol Chem. 2009;284:19402–11. doi: 10.1074/jbc.M109.024711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slep KC, et al. Structural determinants for regulation of phosphodiesterase by a G protein at 2.0 A. Nature. 2001;409:1071–7. doi: 10.1038/35059138. [DOI] [PubMed] [Google Scholar]
- 27.Soundararajan M, et al. Structural diversity in the RGS domain and its interaction with heterotrimeric G protein alpha-subunits. Proc Natl Acad Sci U S A. 2008;105:6457–62. doi: 10.1073/pnas.0801508105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slep KC, et al. Molecular architecture of Galphao and the structural basis for RGS16-mediated deactivation. Proc Natl Acad Sci U S A. 2008;105:6243–8. doi: 10.1073/pnas.0801569105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Posner BA, Mukhopadhyay S, Tesmer JJ, Gilman AG, Ross EM. Modulation of the affinity and selectivity of RGS protein interaction with G alpha subunits by a conserved asparagine/serine residue. Biochemistry. 1999;38:7773–9. doi: 10.1021/bi9906367. [DOI] [PubMed] [Google Scholar]
- 30.Kosloff M, Selinger Z. GTPase catalysis by Ras and other G-proteins: insights from Substrate Directed SuperImposition. J Mol Biol. 2003;331:1157–70. doi: 10.1016/s0022-2836(03)00847-7. [DOI] [PubMed] [Google Scholar]
- 31.Sprang SR, Chen Z, Du X. Structural basis of effector regulation and signal termination in heterotrimeric Galpha proteins. Adv Protein Chem. 2007;74:1–65. doi: 10.1016/S0065-3233(07)74001-9. [DOI] [PubMed] [Google Scholar]
- 32.Sowa ME, He W, Wensel TG, Lichtarge O. A regulator of G protein signaling interaction surface linked to effector specificity. Proc Natl Acad Sci U S A. 2000;97:1483–8. doi: 10.1073/pnas.030409597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siderovski DP, Willard FS. The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits. Int J Biol Sci. 2005;1:51–66. doi: 10.7150/ijbs.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheinerman FB, Al-Lazikani B, Honig B. Sequence, structure and energetic determinants of phosphopeptide selectivity of SH2 domains. J Mol Biol. 2003;334:823–41. doi: 10.1016/j.jmb.2003.09.075. [DOI] [PubMed] [Google Scholar]
- 35.Sheinerman FB, Honig B. On the role of electrostatic interactions in the design of protein-protein interfaces. J Mol Biol. 2002;318:161–77. doi: 10.1016/S0022-2836(02)00030-X. [DOI] [PubMed] [Google Scholar]
- 36.Srinivasa SP, Watson N, Overton MC, Blumer KJ. Mechanism of RGS4, a GTPase-activating protein for G protein alpha subunits. J Biol Chem. 1998;273:1529–33. doi: 10.1074/jbc.273.3.1529. [DOI] [PubMed] [Google Scholar]
- 37.Natochin M, McEntaffer RL, Artemyev NO. Mutational analysis of the Asn residue essential for RGS protein binding to G-proteins. J Biol Chem. 1998;273:6731–5. doi: 10.1074/jbc.273.12.6731. [DOI] [PubMed] [Google Scholar]
- 38.Wieland T, Bahtijari N, Zhou XB, Kleuss C, Simon MI. Polarity exchange at the interface of regulators of G protein signaling with G protein alpha-subunits. J Biol Chem. 2000;275:28500–6. doi: 10.1074/jbc.M004187200. [DOI] [PubMed] [Google Scholar]
- 39.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci U S A. 2002;99:14116–21. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–33. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuhlmann UC, Pommer AJ, Moore GR, James R, Kleanthous C. Specificity in protein-protein interactions: the structural basis for dual recognition in endonuclease colicin-immunity protein complexes. J Mol Biol. 2000;301:1163–78. doi: 10.1006/jmbi.2000.3945. [DOI] [PubMed] [Google Scholar]
- 42.Schreiber G, Keating AE. Protein binding specificity versus promiscuity. Curr Opin Struct Biol. 2010 doi: 10.1016/j.sbi.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kortemme T, et al. Computational redesign of protein-protein interaction specificity. Nat Struct Mol Biol. 2004;11:371–9. doi: 10.1038/nsmb749. [DOI] [PubMed] [Google Scholar]
- 44.Joachimiak LA, Kortemme T, Stoddard BL, Baker D. Computational design of a new hydrogen bond network and at least a 300-fold specificity switch at a protein-protein interface. J Mol Biol. 2006;361:195–208. doi: 10.1016/j.jmb.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 45.Mandell DJ, Kortemme T. Computer-aided design of functional protein interactions. Nat Chem Biol. 2009;5:797–807. doi: 10.1038/nchembio.251. [DOI] [PubMed] [Google Scholar]
- 46.Levin KB, et al. Following evolutionary paths to protein-protein interactions with high affinity and selectivity. Nat Struct Mol Biol. 2009;16:1049–55. doi: 10.1038/nsmb.1670. [DOI] [PubMed] [Google Scholar]
- 47.Li W, et al. Highly discriminating protein-protein interaction specificities in the context of a conserved binding energy hotspot. J Mol Biol. 2004;337:743–59. doi: 10.1016/j.jmb.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 48.Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25:1171–6. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Skerker JM, et al. Rewiring the specificity of two-component signal transduction systems. Cell. 2008;133:1043–54. doi: 10.1016/j.cell.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–64. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karanicolas J, Kuhlman B. Computational design of affinity and specificity at protein-protein interfaces. Curr Opin Struct Biol. 2009;19:458–63. doi: 10.1016/j.sbi.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edwards A. Large-scale structural biology of the human proteome. Annu Rev Biochem. 2009;78:541–68. doi: 10.1146/annurev.biochem.78.070907.103305. [DOI] [PubMed] [Google Scholar]
- 53.Doudeva LG, et al. Crystal structural analysis and metal-dependent stability and activity studies of the ColE7 endonuclease domain in complex with DNA/Zn2+ or inhibitor/Ni2+ Protein Sci. 2006;15:269–80. doi: 10.1110/ps.051903406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ko TP, Liao CC, Ku WY, Chak KF, Yuan HS. The crystal structure of the DNase domain of colicin E7 in complex with its inhibitor Im7 protein. Structure. 1999;7:91–102. doi: 10.1016/s0969-2126(99)80012-4. [DOI] [PubMed] [Google Scholar]
- 55.Huang H, Yuan HS. The conserved asparagine in the HNH motif serves an important structural role in metal finger endonucleases. J Mol Biol. 2007;368:812–21. doi: 10.1016/j.jmb.2007.02.044. [DOI] [PubMed] [Google Scholar]
- 56.Petrey D, et al. Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins. 2003;53 (Suppl 6):430–5. doi: 10.1002/prot.10550. [DOI] [PubMed] [Google Scholar]
- 57.Xiang Z, Honig B. Extending the accuracy limits of prediction for side-chain conformations. J Mol Biol. 2001;311:421–30. doi: 10.1006/jmbi.2001.4865. [DOI] [PubMed] [Google Scholar]
- 58.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–9. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 59.Sridharan S, Nicholls A, Honig B. A New Vertex Algorithm to Calculate Solvent Accessible Surface-Areas. Faseb Journal. 1992;6:A174. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.