

Abstract
ATR-X (alpha thalassemia/mental retardation, X-linked) syndrome is a human congenital disorder that causes severe intellectual disabilities. Mutations in the ATRX gene, which encodes an ATP-dependent chromatin-remodeler, are responsible for the syndrome. Approximately 50% of the patient missense mutations are clustered in a cysteine-rich domain termed ADD (ATRX-DNMT3-DNMT3L, AD-DATRX), indicating its importance. However, the function of ADDATRX has remained elusive. Here we identify ADDATRX as a novel histone H3 binding module, whose binding is promoted by lysine 9 trimethylation (H3K9me3) but inhibited by H3K4me3. The co-crystal structures of ADDATRX bound to H31–15K9me3 peptide reveals an atypical composite H3K9me3-binding pocket, which is distinct from the conventional trimethyllysine-binding aromatic cage. Importantly, H3K9me3-pocket mutants and ATR-X syndrome mutants are defective in both H3K9me3 binding and localization at pericentromeric heterochromatin. Thus, we have discovered a unique histone recognition mechanism underlying the ATR-X etiology.
INTRODUCTION
The basic unit of chromatin is the nucleosome, which is composed of 146 base pairs of DNA wrapped around an octamer of the four histone molecules, H3, H4, H2A, and H2B. Multiple lysine residues on histone H3 are methylated with one, two or three methyl moieties (me1 to me3) by histone methyltransferases (HMTs) and are removed by histone demethylases (HDMs). Methylation events at different lysine residues (e.g. H3K4 and H3K9) are recognized by specific effectors, or “reader”, protein modules, such as PHD (Plant Homeo Domain) fingers for H3K4me31–4 and Chromodomains for H3K9me35,6, which then lead to transcriptional activation or repression, respectively7. Importantly, a number of known or putative HMTs, methyl histone binders, and HDMs have been shown by human genetics studies to be mutated in mental retardation (MR) patients, suggesting important roles for histone methylation dynamics in human brain function8–10. However, how histone methylation contributes to the development of the human nervous system and brain function is not well understood.
Human ATRX encodes an ATP-dependent chromatin remodeling protein11. Male children carrying a mutation in ATRX suffer from a broad range of symptoms, including alpha-thalassemia, psychomotor retardation, facial dysmorphia, and genital abnormalities12. The ATR-X syndrome-associated missense mutations are found predominantly in two discrete regions of ATRX protein, the N-terminal ADD (ADDATRX) and the C-terminal ATP-dependent chromatin-remodeling domain13 (Fig. 1a). Missense mutations in the ATPase domain attenuate the ATPase activity of ATRX14. However, the function of ADDATRX has not been identified. Thus, we sought to uncover the role of ADDATRX to understand the molecular mechanisms underlying ATR-X etiology. Here we report on structural and in vivo functional investigation of ADDATRX as a novel type of histone H3K9me3 “reader” module, whose H3 binding is required for heterochromatin localization, thus shedding new light on the disease mechanisms.
Figure 1. Methylation at K4 and K9 inversely regulates interaction between AD-DATRX and histone H3.

(a) ATRX patient mutations (circles) are predominantly found in either the ADD or the SNF2 (Sucrose Non Fermenting)-type ATP-dependent chromatin remodeling domain. (b) ADDATRX interacts with the unmodified H3 peptide (1–21 aa) (lane 3), and the interaction was enhanced by K9 methylation (lanes 4–6). (c–f) Experimental ITC titration curves are displayed in panel c for unmodified H3 (peptide to buffer control shown in the top), and panel d for H3K9me3 peptide. The fitting curves and the calculated binding affinities are listed in panels e and f. Methylation at K4 (panel e) and K9 (panel f) displayed entirely different effects on the binding affinity. Detailed peptide sequences and complete ITC fitting parameters are summarized in Supplementary Table 1.
RESULTS
Methylation regulates binding of ADDATRX to histone H3
The ADD domains in DNMT3L and DNMT3A, which are components of the de novo DNA methyltransferase complex, have been shown to bind histone H315,16. To determine whether ATRX also binds histones via its ADD domain, ADDATRX (aa 161–292) was fused to GST (glutathione-s-transferase), and incubated with a collection of unmethylated or methylated histone peptides. As shown in Fig. 1b, we detected substantial binding of ADDATRX with the first 21 amino acids of the unmethylated H3 peptide (lane 3), but not with amino acids 2–44 or 69–89 of H3 (lanes 9 and 18) or the H4 tail peptide (lane 22). Consistently, ADDATRX also preferentially pulled down histone H3 when incubated with bulk histones (Supplementary Fig. 1a and 1b). Binding of ADDATRX to the histone H3 peptide was further enhanced by methylation at Lys9, with trimethylation having the most pronounced effect (Fig. 1b, lanes 4–6). These results suggest that ADDATRX binds the H3 N-terminal tail and H3K9 methylation augments this interaction.
The ADDATRX/H3 interactions were quantitatively analyzed by ITC (Isothermal Titration Calorimetry) as well as SPR (Surface Plasmon Resonance) assays. The ADDATRX binds the unmodified H3 tail with a Kd of 3.7μM (ITC, Fig. 1) (3.2 μM by SPR shown in Supplementary Fig. 1c–1e). Methylation at H3K9 improved the binding affinity, with trimethylation exhibiting the most pronounced enhancement (factor of 7.4) (Fig. 1d and f) (enhanced affinity by a factor of 3.8 in the SPR assay shown in Supplementary Fig. 1c–1e). The nearly doubled enthalpy change (−12.2 kcal/mol vs. −6.1 kcal/mol, Supplementary Table 1) associated with binding H3K9me3 versus the unmodified H3 peptide indicates that the recognition of K9me3 group is an enthalpy-driven process. By contrast, methylation at Lys4 dramatically reduced binding in a methyl moiety-dependent manner (Fig. 1e). Furthermore, we calculated a Kd of 4.9 μM when H3 was trimethylated at both Lys4 and Lys9, while retaining the large enthalpy change of about −12.0 kcal/mol (Fig. 1f, Supplementary Fig. 2a and Supplementary Table 1). The H3K4me3-mediated neutralization of binding enhancement by H3K9me3 suggests possible methylation crosstalk between Lys4 and Lys9. The negative regulation of the ADDATRX/H3 interaction by Lys4 methylation is reminiscent of the previous reports on ADDDNMT3L and ADDDNMT3A15,16. An important distinction is that H3K9 trimethylation does not impact histone H3 binding by ADDDNMT3L and ADDDNMT3A15–17. Therefore, the ADDATRX domain represents a new “reader” module that senses methylation states at both H3K4 and H3K9.
H3K9me3 recruits Atrx on pericentromeric heterochromatin
Atrx has been shown to mainly localize to heterochromatic regions, including telomeres and pericentromeric heterochromatin (PCH)18,19. H3K9me3 is a hallmark modification of constitutive heterochromatin, such as PCH20,21. In contrast, H3K4me2 and H3K4me3 are largely absent from PCH22,23. To assess the contribution of H3K9me3 to Atrx PCH localization, we utilized double knockout mouse embryonic fibroblast (DKO MEF) cells, which lack the H3K9me3 histone methyltransferases (HMTases) Suv39h1/h224,25. Both H3K9me3 and the heterochromatin protein, Hp1α (also known as Cbx5), were undetectable at PCH in the DKO MEF cells as previously reported5,26 (Supplementary Fig. 3a and 3b). Immunofluorescence (IF) showed clear PCH foci of Atrx, which overlap with the DAPI-dense signals, in the wildtype (WT) MEF cells (Fig. 2a, panels a–c), but diffused signals in the DKO MEF cells (Fig. 2a, panels d–f). Re-expression of wildtype (WT) Suv39h1 (58.8%), but not the catalytically inactive mutant H324L-Suv39h124 (1.4%), restores PCH localization of Atrx in DKO cells (Fig. 2b and c, right panel). Recovery of H3K9me3 signals was observed only in WT-Suv39h1 positive cells (Fig. 2c, left panel). These results suggest that H3K9me3 itself is essential for tethering Atrx to PCH.
Figure 2. Atrx is recruited to PCH by Suv39h1/2-mediated H3K9 trimethylation.

(a) Atrx fails to locate on PCH in Suv39h1/h2 double knock-out (DKO) cells. (b and c) Suv39h1-WT but not a catalytically inactive mutant Suv39h1-H324L restores the PCH localization of Atrx in DKO cells. Suv39h1-WT (panels a to d) and Suv39h1-H324L (panels e to h) were expressed in DKO cells. (c) Quantification of presence or absence of H3K9me3 signal in the Suv39h1-positive cells (left panel), and punctate PCH localization or diffused distribution of Atrx (right panel). (d) Mecp2 is localized at PCH in both WT- (panels a to c) and DKO- (panels d to f) MEF cells. (e) Schematic diagram of ATRX proteins used for testing their localization. (f) Deletion of PxVxL motif resulted in partial loss of PCH localization. Subnuclear distribution of ATRX proteins represented in (Fig. 4f) was quantified. (g) Targeting of Atrx (panels b and f) to Daxx-positive PML bodies (panels a and e) is not affected in Suv39h DKO cells (panels e to h).
Previous studies implicated the methyl CpG binding protein Mecp2 and the heterochromatin protein Hp1α in the PCH localization of Atrx. Specifically, it has been shown that Mecp2 directly interacts with Atrx, and that loss of Mecp2 disrupts Atrx PCH localization27. Interestingly, while Atrx PCH localization is compromised, Mecp2 remained at PCH in the DKO cells (Fig. 2d), suggesting that Mecp2 is likely to be necessary but not sufficient for Atrx PCH localization. Atrx directly binds Hp1α via the conserved Hp1-interacting motif PxVxL28,29. The deletion mutant ATRX-d/PxVxL, which lacks the Hp1-interacting motif (aa 581–594) (Fig. 2e), showed a decreased PCH localization (Fig. 2f), suggesting that Atrx-Hp1 interaction contributes to the localization. However, PCH localization of the catalytically inactive ATRX mutant K1600R14 was unaffected, indicating that PCH localization is independent of ATRX chromatin remodeling activity (Fig. 2f). RNAi inhibition of Atrx did not affect Hp1α PCH localization (Supplementary Fig. 3c), suggesting that Hp1α protein acts upstream of Atrx. Atrx is also located at promyelocytic leukemia (PML) bodies via physical interaction with Daxx14,30. Atrx remains localized to the Daxx-positive PML bodies in the DKO cells (Fig. 2g), suggesting that recruitment of Atrx to PCH versus PML bodies involves distinctive mechanisms. Taken together, Atrx PCH localization appears to involve multivalent interactions that include H3K9me3 binding and physical interactions with Mecp2 and Hp1 proteins, but H3K9me3 likely plays a dominant role.
Molecular basis for H3K9 trimethylation readout by ADDATRX
To explore the molecular basis for H3 binding by ADDATRX, we solved the crystal structures of ADDATRX in its free state (1.90 Å), in complex with unmodified H31–15 (2.50 Å) and with H31–15K9me3 (0.93 Å) (Table 1). The ADDATRX domain is composed of an N-terminal GATA-like “C2C2” zinc finger and a C-terminal “C4C4” imperfect PHD finger (Fig. 3a). The compact fold of the ADDATRX domain originates from extensive contacts, mostly hydrophobic, between the GATA-like finger and the PHD finger, with buried surface of ~2,000 Å2. In our crystal structure of ADDATRX bound to unmodified H31–15, we could only trace residues 1–7 of the peptide (Supplementary Fig. 4), while residues 1–10 could be traced in the H31–15K9me3 complex (Fig. 3b). Upon complex formation, both bound H3 peptides form an anti-parallel β-sheet with the ADDATRX PHD finger (Fig. 3a). The K9me3 group is inserted into a pocket formed at the junction of the GATA-like finger and the PHD finger (Fig. 3a and b), associated with ~574 Å2 buried surface upon H31–15K9me3 binding. The H3 binding surface is negatively charged, which enables favorable electrostatic recognition of the basic H3 N-terminal tail on complex formation (Fig. 3b). Structural superimposition of free and peptide-bound ADDATRX revealed local conformational adjustments of loops Val178–Tyr187, Arg211–Asp217, Ile256–Gln262, as well as the N- and C-termini (Supplementary Fig. 5a), indicative of a largely pre-formed K9me3 binding pocket (Supplementary Fig. 5b).
Table 1.
Data collection, phasing and refinement statistics
| Free | ADDATRX–H31–15 | ADDATRX–H31–15K9me3 | ||
|---|---|---|---|---|
| Data Collection | Zn-SAD | Native | Native | Native |
| Space group | P32 | P3221 | P65 | C2 |
| Cell dimensions | ||||
| a, b, c (Å) | 74.2, 74.2, 54.1 | 80.6, 80.6, 136.1 | 66.7, 66.7, 131.6 | 83.7, 39.5, 41.1 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | 90, 111.2, 90 |
| Peak | ||||
| Wavelength (Å) | 1.2827 | 0.9795 | 0.9792 | 0.9792 |
| Resolution (Å) | 50–1.9 (1.93–1.90)* | 50–2.5 (2.56–2.50) | 50–1.6 (1.63–1.60) | 50–0.93 (0.95–0.93) |
| Rsym (%) | 6.3 (55.4) | 6.3 (53.2) | 8.3 (56.6) | 6.7 (57.0) |
| I/σI | 43.6 (4.0) | 40.4 (4.0) | 23.3 (2.7) | 35.5 (3.5) |
| Completeness (%) | 99.9 (100.0) | 99.7 (100.0) | 98.5 (97.3) | 93.1 (88.1) |
| Redundancy | 5.6 (5.5) | 12.4 (7.7) | 9.7 (9.3) | 8.5 (8.2) |
| Refinement (F>0) | ||||
| No. reflections (overall) | 26159 | 18264 | 41649 | 76594 |
| No. refelctions (test set) | 1997 | 1473 | 2093 | 3831 |
| Twinning fraction | 0.408 | |||
| Rwork/Rfree (%) | 12.7/15.8 | 19.4/24.1 | 15.7/17.9 | 12.2/13.1 |
| No. non–H atoms | ||||
| Protein | 1935 | 2016 | 1976 | 1085 |
| Ligand/ion | –/6 | 106/6 | 176/6 | 93/3 |
| Water | 334 | 114 | 471 | 240 |
| B-factors (Å2) | ||||
| Protein | 35.5 | 51.1 | 18.6 | 8.1 |
| Ligand/ion | –/37.0 | 60.7 | 21.6/12.1 | 12.9/3.8 |
| Water | 38.0 | 45.5 | 31.5 | 14.0 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.006 | 0.009 | 0.015 | 0.020 |
| Bond angles (°) | 0.92 | 1.13 | 1.37 | 1.41 |
Each data set was collected from a single crystal; values in parentheses are for highest resolution shell.
Figure 3. Molecular basis for H3K9me3 recognition by ADDATRX.

(a) Ribbon representation of ADDATRX domain in complex with H31–15K9me3 peptide. ADDATRX domain is a hybrid of a GATA-like zinc finger (cyan) and a PHD finger (magenta). Three zinc ions are depicted as spheres and the H3 peptide is colored yellow with key residues Lys4 and K9me3 shown in stick representation. (b) Surface electrostatic view of ADDATRX domain in complex with H31–15K9me3 peptide, color-coded with red representing negatively-charged and blue positively-charged potentials. The Fo-Fc omit map was contoured at 3.5 σ level for the bound H3 peptide. (c) Stereo view of the K9me3-binding pocket. The GATA-like segment Tyr203–Ser210 and the PHD finger segment Gln219–-Glu225 are color-coded in cyan and magenta, respectively. Blue dotted lines refer to nonconventional C-H:O hydrogen bonds. (d) A snug fit of the bulky trimethyllysine group inserted into the reader pocket of ADDATRX.
The composite K9me3 binding pocket is formed by segment Tyr203–Ser210 from the GATA-like finger and Gln219–Glu225 from the PHD finger (stereo view in Fig. 3c). The integrity of this pocket is stabilized by three hydrogen-bonding Gln219–Ser210, Tyr203–Glu225, and Tyr204–Arg221 inter-domain pairs (Fig. 3c). One bound water molecule, hydrogen-bonded to residues Met205 and Asp207, fills in the space above the Tyr204 side chain (Fig. 3c). Taken together, the above arrangements generate an exquisitely sculpted pocket with dimensions that are perfectly matched for accommodation of the bulky trimethyllysine group (Fig. 3d). Unlike other classical trimethyllysine readers that usually consist of an aromatic-lined cage for higher methylation state-specific recognition31, the ADDATRX adopts an atypical reader pocket that contains only one aromatic residue and is otherwise rich in polar residues (Fig. 3c and d). The atomic resolution structure of the monoclinic H31–15K9me3 complex enables an accurate assessment of both conventional and non-conventional bonding interactions around the K9me3 group (Supplementary Fig. 6). Specifically, our structural analysis revealed that in addition to a collection of favorable van der Waals contacts associated with a high degree of surface complementarity, the recognition of K9me3 is facilitated by conventional hydrogen bonding between Glu225 and H3K9 backbone; cation-π interaction between the Tyr203 aromatic ring and the cationic trimethyllysine; and most strikingly, a set of carbon-oxygen hydrogen bonds engaging the ε-N-trimethyllysine group and pocket residues Tyr203, Asp207, Gln219 and Ala224 (Fig. 3c and Supplementary Fig. 6). These extensive conventional and nonconventional interactions likely contribute to the nearly doubling of heat generation upon H3K9me3 peptide binding, coupled with the substantially increased affinity compared to an unmethylated H3 peptide (Fig. 1 and Supplementary Table 1).
Effects of H3 sequence context on the ADDATRX/H3 interaction
The recognition of the H3 tail by ADDATRX is dominated by a network of hydrogen bonds and salt bridges (Fig. 4a). In addition to backbone β-sheet formation, sequence specific H3 recognition is notably contributed by 1) anchoring of the free N-terminal amine of H3 by residues Ile256, Asp258, Asn261 and Asp233; 2) unmodified H3K4 recognition by residues Asp212, Asp217 and Glu218; 3) hydrogen bonding between H3T6 and residues Gln219 and Gly227; and 4) H3K9me3 encapsulation within a composite pocket formed at the junction of the GATA-like finger and the PHD finger. The recognition of unmodified H3K4 side chain by direct hydrogen bonding and salt bridge formation with acidic residues, readily explains why the resulting increased hydrophobicity and bulkiness upon Lys4 methylation, would disrupt the above mentioned polar interactions.
Figure 4. Effect of H3 sequence context on ADDATRX binding.

(a) Details of H3-ADDATRX interaction. Histone H3 Ala1–Ser10 containing K9me3 modification is depicted as yellow sticks. ADDATRX is shown as light blue ribbons with key residues that interact with H3 peptide shown as pink sticks. Red dotted lines denote hydrogen bonds. Three zinc ions and water molecules are depicted as large blue and small red spheres, respectively. (b) ITC titration fitting curves and the binding constants of synthetic H3 peptide variants to WT-ADDATRX. R2A, K4A, K9A: Arg2, Lys4, Lys9 to alanine substitution, respectively; AAH3: H3 N-terminal double alanine extension; R2me2s: Arg2 symmetrical dimethylation; R2me2a: Arg2 asymmetrical dimethylation; K9me3: Lys9 trimethylation; K9ac: Lys9 acetylation; n.d., not determined. Detailed peptide sequences and complete ITC fitting parameters are summarized in Supplementary Table 1.
The importance of H3 sequence context was further verified by ITC titrations using various synthetic H3 peptides as summarized in Fig. 4b. Unmodified histone H31–10 (~3.6 μM) and longer H31–15 (~3.7μM) peptides exhibited similar binding affinities, while the binding was totally lost for H31–5 or H35–13K9me3 peptides. These results are consistent with the structural studies, which indicate that H3 aa 1–10 represents the minimal motif for efficient ADDATRX interaction. Notably, dual alanine extension of the H3 N-terminus, R2A, K4A, K9A mutations and K9 acetylation, substantially affect the binding as evidenced by increased disassociation constants ranging from 26.8 to 680 μM. More modest drops in binding affinities by a factor of 2 to 3, compared to the unmodified peptide, were observed upon Arg2 symmetrical or asymmetrical dimethylation, suggesting modulatory methylation crosstalk involving Arg2. Collectively, these results underscore the importance of the free N-terminus, as well as the positively charged side chains of Arg2, Lys4 and Lys9 residues for binding by the ADDATRX domain, and indicate that the binding of H3K9me3 by ADDATRX represents a cooperative process requiring multiple contributing factors for optimal recognition.
Structural comparison with other methylated H3K9 readers
Members of the ‘Royal Family’, such as chromo, MBT and tudor domains, as well as the ankyrin repeat32 have been shown to bind to histone H3K9 methyl marks primarily through an aromatic-lined cage31. However, previous structural analysis showed that H3 sequence motifs essential for this recognition are centralized to the segments adjacent to H3K932–34. For instance, H3 segments Gln5–Ser10, Lys4–Ser10, and Ala7–Gly13 were reported to be responsible for H3 binding by the HP1 chromodomain33 (PDB code: 1KNE), the CHP1 chromodomain34 (3G7L), and the GLP ankyrin repeat32 (3B95), respectively (Supplementary Fig. 7). There is no available information to date on the impact of Lys4 methylation on this recognition5,6,33–35. Though unmodified H3K4 has been reported to contribute to the H3K9me3 peptide binding by the CHP1 chromodomain through interactions with a surface glutamate residue (Supplementary Fig. 7c, left), a role of Lys4 methylation on histone H3 binding was not experimentally tested, and a strong effect on binding affinity by Lys4 methylation was not anticipated given the open binding site and flexibility of H3K434. In contrast, ADDATRX represents a novel type of H3K9me3 reader module whose binding to H3 engages an entire H3 N-terminal tail from Ala1 to Ser10, and is subjected to regulations by methylation at both Lys4 and Lys9. In addition, ADDATRX adopts a polar K9me3 “reader” pocket that is distinct from the prevalent trimethyllysine-binding aromatic cage identified in a series of reader modules including ‘Royal Family’ members and PHD fingers31, thereby shedding light on potential existence of other methyllysine readers with similar recognition strategy.
Integrity of ADD ensures H3 binding and PCH localization of ATRX
To determine the importance of the K9me3 binding pocket, Tyr203, Tyr204, and Asp207 were substituted by alanine, and Gln219 was replaced by proline as it occurs in an individual with ATR-X syndrome13. While D207A showed a slight binding to H3K9me3, Y203A and Y204A showed severely compromised binding to H3K9me3, and Q219P showed no detectable binding (Fig. 5b). Quantitative ITC titration of purified Y203A mutant using H31–15K9me3 peptide revealed a Kd of 4.6 μM and a ΔH of −4.7 kcal/mol, which is comparable to the wild type ADDATRX binding to unmodified H3 peptide, indicative of a specific disruption of K9me3 binding (Supplementary Fig. 3b and Supplementary Table 1). In addition, the above mutants also failed to bind to nucleosomes, which are physiological substrates for Atrx (Fig. 5c), demonstrating the importance of the K9me3 binding pocket. Interestingly, residues accommodating K9me3 in ADDATRX, including Tyr203, Tyr204 and Gln219, are not conserved in the ADD domains of DNMT3A, B, and L, whereas, the residues responsible for K4me0 recognition, such as Asp212, are well conserved (Fig. 5a and Supplementary Fig. 8). Consistently, although K4me0 recognition represents a common function of ADDATRX and ADDDNMT3s, ADDDNMT3s lacks the H3K9me3-binding pocket (Supplementary Fig. 8), thereby defining H3K9me3 recognition as a unique function of the ADDATRX.
Figure 5. Mutations in ADDATRX compromise histone binding and targeting of ATRX onto PCH.

(a) Alignment of the ADD domains. Hs: Homo sapiens (human), Dr: Danio rario (zebrafish), Xt: Xenopus tropicalis (frog). Conserved cysteines are presented as red letters. (b) Mutations in the K9me3-pocket limit the binding of ADDATRX to H3 peptides methylated at Lys9. (c) Mutations in the K9me3-pocket disrupt binding to nucleosomes. GST alone (lane 1) or GST-ADDATRX WT (lane 2) as well as mutants (lanes 3 to 6) were incubated with nucleosomes. (d and e) Four patient mutations resulted in defective H3 peptide binding (d) and nucleosome binding (e). (f) Representative images of subnuclear distribution of recombinant ATRX proteins in NIH3T3 cells. (g and h) Quantification of the distribution patterns is shown for the four K9me3-pocket mutants (g) and additional four syndrome-associated mutants (h).
Next, we determined whether compromised H3 binding is a general consequence of mutations associated with ATR-X syndrome. Four additional human mutations in AD-DATRX, H189N, P190A, R246C and E252L, were analyzed (Fig. 5a). All four mutants exhibited markedly reduced binding to H3 peptides (Fig. 5d) and nucleosomes (Fig. 5e). A previous NMR study showed that folding of ATRX proteins carrying R246C and Q219P mutations remains intact, while another patient mutation, H189D, leads to a disruption of proper folding36. Our results, together with these observations, suggest that loss of H3 binding caused by patient mutations could be due to either local structural changes at the H3 binding interface or a disruption of overall folding of ADDATRX. For the K9me3-pocket mutants, especially Y203A, Y204A, and D207A, their binding with the unmethylated H3 peptide was largely unaffected, suggesting that these mutations did not affect the overall structure of ADDATRX.
To determine the role of ADDATRX in PCH localization, we examined subnuclear distribution of HA-tagged WT and mutant ATRX in NIH3T3 cells by IF. HA-tagged ATRX (HA-ATRX) showed clear PCH foci (“punctate”) in more than 80% of the interphase cells (Fig. 5f, panels a-c). However, HA-ATRX carrying H3K9me3 binding defective or patient point mutations showed either diffused (Fig. 5f, panels d-f) or punctate signals (“abnormal puncta”) (Fig. 5f, panels g–i), which do not overlap with the DAPI-dense signals. The quantification of the IF analysis is shown in Fig. 5g and h. Patient mutations in ADDATRX did not affect PML body localization of ATRX (Supplementary Fig. 9), suggesting that ADDATRX is specifically required for anchoring ATRX onto PCH. Previous studies showed that patient mutations result in a decreased protein level of ATRX36. Collectively, ADDATRX/H3 interaction plays an important role in localizing ATRX to PCH, and patient mutations in ADDATRX disrupt this targeting mechanism and decrease the stability of ATRX protein.
DISCUSSION
In this report, we provided ample evidence demonstrating that ADDATRX is a novel H3K9me3 recognition module, which senses the methylation states at both H3K9 and H3K4. Co-crystal structures at atomic resolution revealed a fascinating composite and atypical H3K9me3 binding pocket formed between the GATA- and the PHD- fingers of ADDATRX. Point mutations in this binding pocket substantially compromised AD-DATRX/H3K9me3 binding in vitro and the PCH localization of ATRX in vivo. Complementing the structural analysis, genetic studies provided further support that H3K9me3 is critical for the PCH localization of Atrx. Importantly, ATRX patient mutations in ADDATRX impaired the PCH localization, further indicating the importance of the ADD domain as a critical determinant of proper ATRX localization in vivo.
PCH is fundamental for sister chromatid cohesion, and proper chromatid cohesion indeed requires the H3K9me3 HMTases37,38. Loss of Atrx has been shown to result in a chromosome segregation defect due to the attenuated cohesion and congression of sister chromatids39, which may lead to increased apoptosis and a reduced number of neurons in the brain39,40. Thus, our findings suggest that Atrx is an important component in the Suv39h/H3K9me3 pathway that is required for faithful chromosome segregation (Fig. 6).
Figure 6. Model of the mechanism underlying ATR-X syndrome caused by the mutations in ADDATRX.
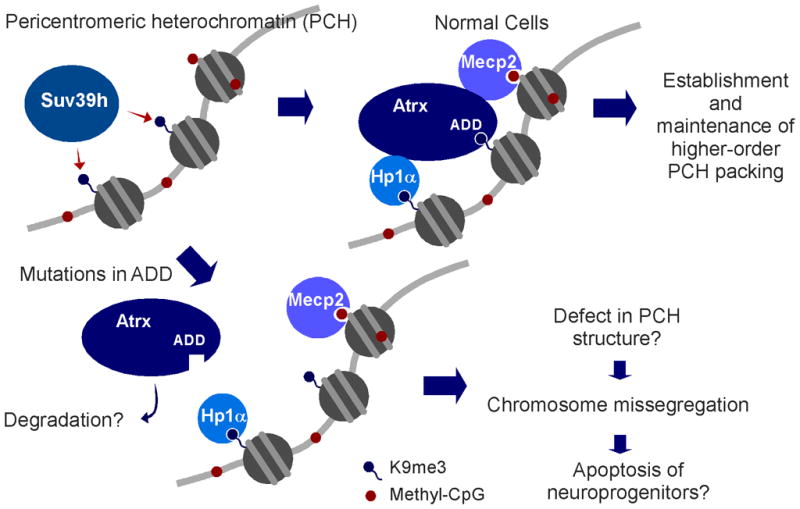
Atrx is recruited on PCH via interaction between ADD and H3K9me3 generated by Suv39h HMTs. Physical associations of Hp1/Atrx and Mecp2/Atrx support the PCH anchoring of Atrx. Patient mutations in ADD disrupt H3K9me3 binding; then Atrx falls off from PCH. Loss of Atrx from PCH may leads to compromised higher-order structure of PCH, followed by chromosome missegregation and apoptosis in neuroprogenitor cells.
While chromosome missegregation would suggest a role for Atrx during M-phase, compromised heterochromatin replication in S-phase could also cause mitotic catastrophe as observed for the loss of other chromatin modifying proteins41,42. Atrx is recruited to the telomere during S-phase and loss of Atrx results in a DNA damage response and de-repression of telomere-associated RNA18,43. Notably, Atrx coordinates deposition of a variant histone with a histone H3.3-specific chaperone Daxx18,44,45. H3.3 deposition at PCH and telomeres44,45 may contribute to heterochromatin formation. Taken together, Atrx may be required for faithful replication of both telomeric and pericentromeric heterochromatin.
Importantly, our structural research has identified an unanticipated mode of histone mark recognition, namely the utilization of integrated modules to generate a composite and atypical binding pocket at the interface between the GATA and PHD fingers of ATRX for Kme3 recognition. The concept of integrated modules acting as a single functional unit was recently highlighted in the structures of DPF3b double PHD finger bound to the H3K14ac mark46 and the PHD-Bromo cassette of TRIM24 bound to the H3K23ac mark47, though in each case the Kac mark was recognized solely by a single module. Taken together, these findings call attention to the role of integrated modules and our newly proposed concept of composite reader pockets, which we anticipate will be increasingly identified amongst various epigenetic regulators associated with histone mark recognition and readout.
In summary, we have uncovered the function of ADDATRX as a recognition motif for the distinctive histone methylation pattern at PCH, representing the first report linking a novel histone methylation sensing mechanism to human brain function. Our study may also open a new avenue for therapeutic intervention of ATR-X syndrome.
ACCESSION CODES
The atomic coordinates and structure factors for the ADDATRX domain in the free and the peptide bound forms have been deposited into Protein Data Bank under accession code 3QLN (free), 3QLC (H31–15 complex), 3QL9 (H31–15K9me3 complex, monoclinic form) and 3QLA (H31–15K9me3 complex, hexagonal form).
METHODS
Expression plasmids
Human ATRX was PCR-amplified from cDNA libraries prepared from HeLa cells and lymphocyte, using Phusion polymerase (Finnenzymes), and cloned into a Gateway entry vector pENTR-D-Topo (Invitrogen). A fully sequenced ATRX-WT cDNA, as well as a series of point mutants, and a deletion mutant d/PxVxL generated by PCR-based mutagenesis, were transferred into Gateway destination vectors pCS2-3HA, which are designed to fuse HA-tags to N-terminus of protein. The ADDATRX domain with its flanking amino acids, positioning 162–293, was cloned into pGEX4T-1 (GE Healthcare) for expression and purification as GST-fusion proteins using E. coli. Point mutations in ADD domain associated with ATR-X syndrome were introduced by PCR-based targeted mutagenesis. Numbering of amino acid residues is based on the Gen-Bank accession numbers NM_000489.3 and NP_000480, for mRNA and protein, respectively.
For crystallographic and ITC binding assays, an ADDATRX construct corresponding to ATRX fragment 167–289 was PCR-amplified from human ETS IMAGE clone: 4695967, and cloned into a modified pGEX6P-1 vector (GE Healthcare) with an N-flanking artificial sequence “GPLGSM” from the vector. The expressed ADD domain based on this human ETS clone contains two amino acid variations, K251R and F284Y, compared to the sequence of NP_000480.
Protein purification and peptide pulldown assays
GST-fused ADD domains were expressed in E. coli strain, purified using glutathione-affinity sepharose (GE Health-care). For the histone peptide pull-down assay, 3 μg of purified proteins, WT as well as the syndrome-associated mutants N179S, H189N, P190A, R246C, and E252L, were incubated with 0.1 μg of biotinylated histone peptide in 100 μl of the binding buffer A (50 mM Tris-HCl pH7.5 containing 150 mM NaCl, and 0.05% (v/v) NP-40) for overnight at 4°C. For the K9me3-binding-defective mutants, Y203A, Y204A, D207A, and Q219P, binding reactions (in parallel with WT) were carried out in modified buffer A containing 500 mM NaCl for 3 hours at 4°C. Protein-peptide complexes were pulled down with streptavidin-conjugated affinity resin (Upstate), washed 5 times using the binding buffer A, and bound proteins are detected as histone binding assay above. Nucleosomes mainly comprised of mono- and di- units were prepared from HeLa cells following the conventional protocol48. Nucleosomes and GST-ADD domains were incubated in binding buffer A, pulled-down by glutathione sepharose resin, washed by binding buffer A and analyzed by Western blot analysis using pan-H3 Abs (Abcam, 1791–100) and anti-GST antibody (Santa Cruz, sc-138).
ADDATRX used for ITC and crystallization was expressed in the E. coli host cell Rossetta2 (Novagen) induced overnight by 0.4 mM isopropyl β–D–thiogalactoside at 20°C in the LB medium supplemented with 0.1 mM ZnCl2. The harvested cells were suspended in 0.4 M KCl, 20 mM Tris pH 7.5. After cell lysis and centrifugation, the GST–fusion protein was purified by GST affinity column, followed by tag cleavage using PreScission protease (GE Healthcare). The ADDATRX was then directly separated from the digestion mixture by size–exclusion chromatography on Superdex™ 75 (GE Healthcare) under the elution buffer: 0.4 M KCl, 20mM Tris, pH 7.5, 5 mM DTT. The resultant peak of AD-DATRX was then concentrated to ~15–40 mg ml−1, split into small aliquots and frozen in liquid nitrogen for future use.
Isothermal titration calorimetry
Calorimetric experiments were conducted at 25.0°C with a MicroCal™ iTC200 instrument (GE Healthcare). The ADDATRX samples were dialyzed against the buffer containing 25 mM HEPES-NaOH, pH 7.5, 100 mM KCl, and 5 mM β-mercaptalethanol. Protein concentration was determined by absorbance spectroscopy at 280 nm with an extinction coefficient of 19940 M−1 cm−1. Peptides were quantified either by the absorbance of an added C-terminal Tyr residue (Tyr ε280=1,280 M−1cm−1), or by weighing in large scale for those without an additional tyrosine residue. Acquired calorimetric titration data were analyzed using software Origin 7.0 (GE Health-care) based on the ‘One Set of Sites’ fitting model.
X-ray crystallographic studies
All crystals were grown by the sitting-drop vapor diffusion method under 4°C. Detailed crystallization conditions are listed in supplementary section. Diffraction data were collected under cryo-conditions using 15–20% (v/v) glycerol as the cryoprotectant. An anomalous data set at zinc peak of the free ADDATRX crystal was collected at the beamline X-29 at the National Synchrotron Light Source (NSLS) of Brookhaven National Laboratory in New York. A native data set of H31–15-ADDATRX complex was collected at the beamline ID24-C at Advanced Photon Source (APS) of Argonne National Laboratory in Illinois. As for the H31–15K9me3 complex crystals, diffraction data were collected at beamline 3W1A of Beijing Synchrotron Radiation Facility (BSRF) at Beijing, China. All diffraction data were processed with the HKL2000 suite49 and data processing statistics are summarized in Table 1.
The structure of free ADDATRX was determined by zinc SAD (Single-wavelength Anomalous Dispersion) method using the PHENIX software suite50. The remaining complex crystal structures were solved by molecular replacement with the program MOLREP51 of the CCP4 program suite52 using the free ADDATRX structure as the search model. Model building and refinement were undertaken with COOT53 and PHENIX50. Detailed structural determination procedures are described in supplementary section (Supplementary Methods) and structural refinement statistics are summarized in Table 1.
Immunofluorescence microscopy
Staining and observation were performed as described previously54. Primary antibodies were used at the following dilutions in the permeabilization buffer: 1:100 dilution was applied for anti-HA (Covance, monoclonal HA.11), anti-DAXX (Santa Cruz, sc-8043), anti-H3-K9me2 (Upstate, 07–441), anti-H3-K9me3 (Upstate, 07–442) Abs, anti-PML (Santa Cruz, sc-5621), monoclonal anti-Suv39h1 (44.1: sc-23961), anti-HP1α (mAbs) (Upstate, 05-689), and 1:500 for anti-ATRX Ab (Santa Cruz, sc-15408). Alexa 594- or Alexa 488-conjugated secondary Abs (Invitrogen) was used at 1:500. Rabbit antiserum for MeCP2 was a generous gift from Dr. Greenberg lab. For the localization of HA-fused ATRX proteins, pCS2-3HA gateway destination vectors carrying HA-ATRX WT and various mutants are transfected in NIH3T3 cells using Fugene HD reagent (Roche Applied Science), and 24 hrs later, subjected to IF using anti-HA mAb as described above. Suv39h1-WT and its catalytically inactive mutant H324L expression plasmids were introduced into Suv39h1/h2 double knockout cells using Fugene HD and recombinant protein expression was detected by IF using anti-Suv39h1 mAb.
Supplementary Material
Acknowledgments
The authors are grateful to the staffs of beamlines X-29 at the Brookhaven National Laboratory at New York, 24IDC at the Advanced Photon Source (APS) at Illinois, and 3W1A at the Beijing Synchrotron Radiation Facility (BSRF) at Beijing, China for their assistance in the synchrotron data collection, with special thanks to Y. Dong of BSRF. We thank A. Vaquero and S. Minucci, for providing the Suv39h1/h2 double knockout cells, and SUV39H1 expression vectors. Anti-MeCP2 antiserum was a generous gift from M. Greenberg. We are grateful to A. Ruthenburg for providing histone H3 peptides, L Liang, Y.Y. Chen and M. Geigges for experimental help. We express great appreciation to C. Richardson, M. Takahashi, and individual members of the Shi, Li and Patel laboratories for helpful discussions. S.I. was a Jane Coffin Childs Memorial Fund Postdoctoral Fellowship Award recipient, and is currently supported by the Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowship for Research Abroad. This work was supported by grants from the National Institutes of Health GM071004 and GM058012 (Y.S.); the Starr Foundation, the Leukemia and Lymphoma Society, the Ab-by Rockefeller Mauze Trust and the Maloris Foundation (D.J. Patel); Canadian Institutes of Health Research (CIHR, D. J. Picketts), the Major State Basic Research Development Program in China (grant # 2011CB965300, H.L.), and Tsinghua University 985 Phase II funds (H.L.).
Footnotes
COMPETING FINANCIAL INTERESTS
Y.S is a co-founder of Constellation Pharmaceuticals. D.J.Patel is on the Epigenetics Advisory Board of Epinova-Glaxo. The remaining authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
S.I., H.L., D.J.Patel and Y.S. participated in the experimental design, contributed to the concept and wrote the paper. S.I. performed pulldown assays and localization analysis. B.X. crystallized the ADDATRX-H3K9me3 complex and participated in calorimetric assays. H.L. performed crystallographic and calorimetric studies. P.W.L., T.R., and J.C.C. performed or helped with pulldown assays. S.G. helped with measuring SPR. C.D.A. supervised pulldown assays. D.J.Picketts provided important reagents and participated in manuscript preparation.
References
- 1.Li H, et al. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–5. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wysocka J, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 3.Shi X, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–9. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pena PV, et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–3. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bannister AJ, et al. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–4. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 6.Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–20. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 7.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 8.van Bokhoven H, Kramer JM. Disruption of the epigenetic code: an emerging mechanism in mental retardation. Neurobiol Dis. 2010;39:3–12. doi: 10.1016/j.nbd.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 9.Iwase S, Shi Y. Histone and DNA Modifications in Mental Retardation. In: Gasser SM, Li E, editors. Epigenetics and Disease: Pharmaceutical Opportunities (Progress in Drug Research) Springer; Basel: 2010. pp. 147–174. [Google Scholar]
- 10.Ropers HH, Hamel BC. X-linked mental retardation. Nat Rev Genet. 2005;6:46–57. doi: 10.1038/nrg1501. [DOI] [PubMed] [Google Scholar]
- 11.Gibbons RJ, Picketts DJ, Villard L, Higgs DR. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome) Cell. 1995;80:837–45. doi: 10.1016/0092-8674(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 12.Gibbons R. Alpha thalassaemia-mental retardation, X linked. Orphanet J Rare Dis. 2006;1:15. doi: 10.1186/1750-1172-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gibbons RJ, et al. Mutations in the chromatin-associated protein ATRX. Hum Mutat. 2008;29:796–802. doi: 10.1002/humu.20734. [DOI] [PubMed] [Google Scholar]
- 14.Tang J, et al. A novel transcription regulatory complex containing death domain-associated protein and the ATR-X syndrome protein. J Biol Chem. 2004;279:20369–77. doi: 10.1074/jbc.M401321200. [DOI] [PubMed] [Google Scholar]
- 15.Ooi SK, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–7. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otani J, et al. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009;10:1235–41. doi: 10.1038/embor.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010;38:4246–53. doi: 10.1093/nar/gkq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldberg AD, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–91. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDowell TL, et al. Localization of a putative transcriptional regulator (ATRX) at pericentromeric heterochromatin and the short arms of acrocentric chromosomes. Proc Natl Acad Sci U S A. 1999;96:13983–8. doi: 10.1073/pnas.96.24.13983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allshire RC, Karpen GH. Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat Rev Genet. 2008;9:923–37. doi: 10.1038/nrg2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pidoux AL, Allshire RC. The role of heterochromatin in centromere function. Philos Trans R Soc Lond B Biol Sci. 2005;360:569–79. doi: 10.1098/rstb.2004.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sullivan BA, Karpen GH. Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat Struct Mol Biol. 2004;11:1076–83. doi: 10.1038/nsmb845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martens JH, et al. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. Embo J. 2005;24:800–12. doi: 10.1038/sj.emboj.7600545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rea S, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–9. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 25.O’Carroll D, et al. Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol Cell Biol. 2000;20:9423–33. doi: 10.1128/mcb.20.24.9423-9433.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rice JC, et al. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell. 2003;12:1591–8. doi: 10.1016/s1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- 27.Nan X, et al. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc Natl Acad Sci U S A. 2007;104:2709–14. doi: 10.1073/pnas.0608056104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lechner MS, Schultz DC, Negorev D, Maul GG, Rauscher FJ., 3rd The mammalian heterochromatin protein 1 binds diverse nuclear proteins through a common motif that targets the chromoshadow domain. Biochem Biophys Res Commun. 2005;331:929–37. doi: 10.1016/j.bbrc.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 29.Berube NG, Smeenk CA, Picketts DJ. Cell cycle-dependent phosphorylation of the ATRX protein correlates with changes in nuclear matrix and chromatin association. Hum Mol Genet. 2000;9:539–47. doi: 10.1093/hmg/9.4.539. [DOI] [PubMed] [Google Scholar]
- 30.Xue Y, et al. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci U S A. 2003;100:10635–40. doi: 10.1073/pnas.1937626100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–40. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collins RE, et al. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat Struct Mol Biol. 2008;15:245–50. doi: 10.1038/nsmb.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacobs SA, et al. Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. Embo J. 2001;20:5232–41. doi: 10.1093/emboj/20.18.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schalch T, et al. High-affinity binding of Chp1 chromodomain to K9 methylated histone H3 is required to establish centromeric heterochromatin. Mol Cell. 2009;34:36–46. doi: 10.1016/j.molcel.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fischle W, et al. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870–81. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Argentaro A, et al. Structural consequences of disease-causing mutations in the ATRX-DNMT3-DNMT3L (ADD) domain of the chromatin-associated protein ATRX. Proc Natl Acad Sci U S A. 2007;104:11939–44. doi: 10.1073/pnas.0704057104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allshire RC, Nimmo ER, Ekwall K, Javerzat JP, Cranston G. Mutations derepressing silent centromeric domains in fission yeast disrupt chromosome segregation. Genes Dev. 1995;9:218–33. doi: 10.1101/gad.9.2.218. [DOI] [PubMed] [Google Scholar]
- 38.Guenatri M, Bailly D, Maison C, Almouzni G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J Cell Biol. 2004;166:493–505. doi: 10.1083/jcb.200403109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ritchie K, et al. Loss of ATRX leads to chromosome cohesion and congression defects. J Cell Biol. 2008;180:315–24. doi: 10.1083/jcb.200706083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berube NG, et al. The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J Clin Invest. 2005;115:258–67. doi: 10.1172/JCI22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen T, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–6. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 42.Ha GH, et al. Mitotic catastrophe is the predominant response to histone acetyl-transferase depletion. Cell Death Differ. 2009;16:483–97. doi: 10.1038/cdd.2008.182. [DOI] [PubMed] [Google Scholar]
- 43.Wong LH, et al. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010;20:351–60. doi: 10.1101/gr.101477.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A. 2010;107:14075–80. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253–65. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng L, et al. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature. 2010;466:258–62. doi: 10.1038/nature09139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsai WW, et al. TRIM24 links recognition of a non-canonical histone signature to breast cancer. Nature. 2010;468:927–32. doi: 10.1038/nature09542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ausubel FM. Current protocols in molecular biology. J. Wiley; New York: 2001. [Google Scholar]
- 49.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 50.Adams PD, et al. PHENIX: a comprehensive Python-based system for macro-molecular structure solution. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 52.Collaborative Computational Project, N. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 53.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 54.Iwase S, et al. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–88. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.