

Abstract
Oligopeptidase B is a clan SC, family S9 serine peptidase found in gram positive bacteria, plants and trypanosomatids. Evidence suggests it is a virulence factor and thus therapeutic target in both Trypanosoma cruzi and T. brucei, but little is known about its function in Leishmania. In this study L. major OPB-deficient mutants (Δopb) were created. These grew normally as promastigotes, had a small deficiency in their ability to undergo differentiation to metacyclic promastigotes, were significantly less able to infect and survive within macrophages in vitro, but were virulent to mice. These data suggest that L. major OPB itself is not an important virulence factor, indicating functional differences between trypanosomes and Leishmania in their interaction with the mammalian host. The possibility that an OPB-like enzyme (designated OPB2) in L. major might compensate for the loss of OPB in Δopb was investigated via by mapping its sequence onto the 1.6 Å structure of L. major OPB. This suggested that the residues involved in the S1 and S2 subsites of OPB2 are identical to OPB and hence the substrate specificity would be similar. Consequently there may be redundancy between the two enzymes.
1. Introduction
Leishmania are obligate intracellular protozoa and the causative agent of leishmaniasis, a disease that is endemic in various tropical and subtropical regions [1]. Leishmaniasis is a complex of diseases, with three main clinical forms which depend on the infecting species: visceral leishmaniasis, cutaneous leishmaniasis and mucocutaneous leishmaniasis [2]. Leishmania major causes cutaneous leishmaniasis in humans [1]. It is estimated that there are 1.5 million cases of cutaneous leishmaniasis and 500,000 cases of visceral leishmaniasis annually [3]. Leishmania alternate between a mammalian host and the digestive tract of female phlebotomine sand flies and have several developmental forms. The procyclic promastigote is a flagellated motile form which multiplies in the sand fly gut; the metacyclic promastigote is the non-dividing mammal-infective stage present in the mouthparts of the fly and the amastigote is a non-motile form which lives and multiplies within a parasitophorous vacuole in mammalian macrophages [4].
Oligopeptidase B (OPB) is a serine peptidase of clan SC, family S9 - the prolyl oligopeptidase (POP) family. The POP family are restricted to hydrolysing peptides of up to 30 amino acids in length [5]. Oligopeptidase B is restricted in occurrence, apparently being present only in bacteria, plants and trypanosomatids [6]. OPBs cleave after basic residues, preferring arginine, in a trypsin-like activity [7], though they have a preference for cleaving after di-basic residues [8-10]. Oligopeptidase B was originally identified in Escherichia coli and was included in the POP family due to its homology to porcine prolyl oligopeptidase [11]. OPB has been described in various Leishmania species [12-15]. Detection of OPB activity in the growth medium demonstrated the release of OPB by L. donovani promastigotes [16]. OPB is up-regulated in the amastigote stage of the life-cycle in L. braziliensis and L. donovani compared with promastigotes [17, 18], whereas there are similar levels of expression of OPB in all stages in L. major and L. mexicana [19, 20].
OPB has been reported to be an important virulence factor in some trypanosomatids [6, 21]. In Trypanosoma cruzi, OPB plays a part in the invasion of non-phagocytic cells by trypomastigotes - via triggering an increase in the host cell’s free intracellular calcium [22-24]. T. cruzi OPB null mutant trypomastigotes are 75% less infective to mammalian cells than wild type T. cruzi and exhibited lower parasitaemia in mice [24]. Parasite OPB is released into the bloodstream of animals infected with T. brucei, apparently through lysis of dying parasites [25-27]. The enzyme is thought to reduce the levels of the regulatory peptide hormone atrial natriuretic factor in the plasma of T. brucei-infected dogs and of T. evansi-infected rats [25, 28], potentially leading to the circulatory system lesions which are observed in the trypanosome infections [6]. The trypanocidal drugs pentamidine, diminazene and suramin are all inhibitors of T. brucei OPB [29]. Pentamidine and diminazene contain arginine-like motifs, thus may act through mimicking OPB substrates, whilst suramin has been shown to inhibit a variety of serine peptidases. Two groups of serine peptidase inhibitors, peptidyl chloromethyl ketones and peptidyl phosphate diphenyl esters, have activity against T. brucei OPB and it is thought that the improvement in the survival rate of mice infected with T. brucei resulting from administration of one of these compounds may be mediated through its action on OPB [30].
Six POP family members have been identified in Leishmania [13], including an oligopeptidase B-like protein, or oligopeptidase 2 (OPB2, [31]). L. amazonensis OPB2 was found to have an unusual C-terminal extension and a low identity to OPB, though it was predicted to have a similar structure to OPB. OPB2 was found to be expressed throughout the L. amazonensis lifecycle [31]. Other POP family enzymes have been investigated in trypanosomatids: the prolyl oligopeptidases from T. brucei and T. cruzi. Inhibition of T. cruzi POP prevented entry of trypomastigotes into non-phagocytic host cells [32, 33], whilst T. brucei POP has been shown to be released into the plasma of infected mice and to be able to cleave a number of bioactive peptides that are down-regulated in sleeping sickness patients [34].
To investigate whether OPB is a Leishmania virulence factor, we generated OPB-deficient mutants of L. major and analysed how the gene-deletion affected virulence of the parasite in vitro and in vivo. We also generated OPB over-expressing mutants. Sequence mapping of OPB2, a L. major protein related in primary structure to OPB, onto a high resolution structure of OPB was undertaken to gain insight into the likely substrate specificity of OPB2 and to estimate the likelihood of it compensating for OPB function in the OPB-deficient mutants.
2. Materials and methods
2.1 Parasites
Leishmania major (MHOM/IL/80/Friedlin) promastigotes were grown in modified Eagle’s medium (designated complete HOMEM medium) with 10% (v/v) heat-inactivated fetal calf serum at 25 °C, as described previously [35]. Metacyclic promastigotes were isolated by the peanut agglutinin method [36] from cultures that had been in stationary growth phase for 3-4 days. Briefly, promastigotes were resuspended in PBS, pH7.4 at 2 × 108 cells ml−1. Peanut agglutinin (Vector Laboratories) was added at 50 μg ml−1 and the sample was incubated for 30 min at room temperature. The supernatant contained the non-agglutinated metacyclic promastigotes. In order to calculate the percentage of metacyclic promastigotes in a culture of stationary phase cells, the number of metacyclic promastigotes found after the incubation with peanut agglutinin was compared to the initial concentration of 2 × 108 parasites ml−1. Three replicates were completed per culture. L. major amastigotes were purified from infected BALB/c mice as described [37].
2.2 Antibodies and Western Immunoblotting
Anti-OPB antibodies were raised in a sheep using L. major recombinant OPB[38]. 5 mg of rOPB was linked to an Aminolink column (Pierce) on which the antibodies were affinity-purified according to the manufacturer’s protocol. Parasites were lysed in lysis buffer (50 mM Tris/HCl pH 8.0, 0.25% (v/v) Triton X-100, 20% (v/v) glycerol), with a cocktail of peptidase inhibitors (10 μM E-64, 1 mM 1,10-phenanthroline, 2 μM pepstatin A, 1 mM PMSF, 10 μM antipain, 10 μM leupeptin and 2 mM EDTA). Whole-cell lysates were resolved with a 12% (w/v) SDS-PAGE, transferred to a polyvinylidene fluoride (PVDF) membrane and immunoblotted with the appropriate primary antibodies diluted in PBS with 5% (w/v) skimmed milk and 0.1% (v/v) Tween-20. The primary antibodies used and their respective dilutions were sheep anti-LmjOPB (1:20,000), mouse monoclonal anti-TbEF1α (Upstate, 1:10,000), rabbit anti-HASPB antibody ([39], 1:1000) and mouse anti-GP63 antibody ([40]1:100). Anti-sheep, anti-mouse or anti-rabbit HRP-conjugated secondary antibodies were used at 1:5000 dilution and the SuperSignal West Pico chemoluminescent system (Pierce) was then used to visualise the antigens.
2.3 Generation of Δopb null mutants and cells re-expressing OPB
The 872 bp 5′-flank fragment of OPB (LmjF09.0770) was generated by PCR from L. major genomic DNA with primers OL2395 and OL2415 (Table 1, bold indicates restriction sites added to primers), ligated into the PCR-Script subcloning vector (Stratagene) and digested out using HindIII and SalI. The 539 bp 3′ flanking region of OPB was generated by PCR using the primers OL2356 and OL2459 (Table 1, bold indicates restriction sites added to primers), ligated into the TOPO subcloning vector (Invitrogen) and digested out using XmaI and BglII. The digested-flank fragments were cloned sequentially into digested pGL1062 to give the Hygromycin B-resistance plasmid pGL1693. The cassette used for transfection was released by BglII digestion. The streptothricin acetyltransferase-resistance plasmid, pGL1762, was generated from pGL1693 by replacement of the SpeI/BlpI cassette containing the hygromycin B resistance gene with a SpeI/BlpI cassette containing the streptothricin acetyltransferase resistance gene. A population of parasites resistant to hygromycin was generated after transfection with pGL1693, with the integration of the cassette assessed by PCR. This population was used for a second round of transfection with pGL1762. Two streptothricin acetyltransferase-resistant clones from independent transfection events were selected for analysis, having had the absence of OPB assessed by PCR. These clones were designated Δopb clones 10 and 21. To generate re-expresser plasmid, the OPB locus was digested from pGL1640 (section 2.3) using BglII. This fragment was then cloned into BglII digested and calf alkaline phosphatase (CIP) treated pRIB vector [41], to give plasmid pGL1818. pGL1818 was transfected into Δopb clones 10 and 21 and clones were selected for analysis after selection with puromycin.
Table 1.
Primers used in this study.
| OL2352 | 5′ - A CCC GGG ATG TCG TCG GAC AGC TCC GTC – 3′ |
| OL2353 | 5′ - T AGA TCT TTA CCT GCG AAC CAG CAG GCG – 3′ |
| OL2356 | 5′ - G CCC GGG GAG TCG TGA ACA TTA ACT CC - 3′ |
| OL2395 | 5′ - C AAG CTT CTC CTT CTC GGT GGC ACT TG - 3′ |
| OL2415 | 5′ - CC GTT GGG CAC GCA TGT ACC GTC GAC G - 3′ |
| OL2459 | 5′ - G AGA TCT CGG TAG CGG GAG AGA GAA GG - 3′ |
| OL2550 | 5′ -GCC TGC GAG GGG CGT GGC GCC GGT GGC CTG C– 3′ |
| OL2551 | 5′ - G CAG GCC ACC GGC GCC ACG CCC CTC GCA GGC - 3′ |
2.3 Generation of OPB over-expressing L. major cell lines
OPB (LmjF09.0770) with its start and stop sites intact was obtained by PCR from L. major genomic DNA with primers OL2352 and OL2353 (Table 1). The 2182 bp fragment was ligated into the TOPO sub-cloning vector (Invitrogen), digested with EcoRV and KpnI, blunt ended using Mung bean nuclease and cloned into Eco RV-digested and blunt ended HcN pNUS vector [42], giving plasmid pGL1640. Plasmid expressing LmjOPBS577G was produced by site directed mutagenesis of pGL1640, using the QuikChange mutagenesis kit (Stratagene) with primers, OL2550 and OL2551. These are shown in Table 1, with the codon modified underlined. This produced the active site mutant construct, pGL1714. Populations of parasites were generated after transfection with pGL1640, pGL1714 and pGL1137 (the empty pNUS vector) and selection with Geneticin (G418), with the presence of the plasmids assessed by PCR.
2.4 Analysis of Bz-R-AMC cleavage by L. major promastigotes
6 × 105 L. major stationary phase promastigotes in 100 μl HOMEM medium were used per well in 96-well plates, with control wells of the same volume of HOMEM medium, to account for the inherent cleavage of the substrate. Bz-R-AMC (Bachem) was made up at a stock concentration of 100 mM as per manufacturer’s instructions, then diluted to 10 μM in HOMEM medium and 100 μl was added to both parasite and control wells. Cleavage was determined by the change in fluorescence (λex= 355 nm. λem= 460 nm) at room temperature over 30 mins, in an EnVision 2102 plate reader (Perkin Elmer). The fluorescence readings (minus the fluorescence of the control wells) were converted to a concentration of AMC released using an included AMC standard and then the rate of cleavage of Bz-R-AMC calculated. To determine the effect of peptidase inhibitors, the basic cleavage assay was completed with minor changes. The inhibitor was made up at 20 x the desired concentration in medium. 10 μl of inhibitor was added to the desired wells of a 96-well plate, with 10 μl of HOMEM medium for uninhibited wells. 6 × 105 L. major stationary phase promastigotes in 90 μl HOMEM medium were added to each well, with control wells of the same volume of HOMEM medium. 100 μl of 10 μM Bz-R-AMC was also added to the parasite and control wells. The following inhibitors and concentrations were used: 20 μM E-64d; 10 μM N-methyl-piperazine-Phe-homoPhe-vinylsulfone-phenyl (K11777; [43]); 1 μM 1,10 phenanthroline; 250 μM EDTA; 2 μM pepstatin A; 1 mM PMSF; 100 μM AEBSF; 500 nM antipain and 12.5 μM leupeptin. For determination of IC50 values for inhibition of Bz-R-AMC cleavage, a 5-fold serial dilution of antipain, from 500 nM to 0.16 nM; a 5-fold serial dilution of leupeptin, from 12.5 μM to 4 nM and a 2-fold serial dilution of AEBSF, from 100 μM to 1.56 μM, were used. Fluorescence was read as above, and then the concentration of AMC released was calculated to find the rate of Bz-R-AMC cleavage. The cleavage rates were converted to relative activities compared to uninhibited cells and from these IC50 values were determined, using GraFit 5 data analysis software (Erithacus Software).
2.5 Lysis and fractionation of cells for western immunoblot
Cells were lysed by sonication of cells suspended in PBS, using a Soniprep 150. The following peptidase inhibitors were in the lysates: 10 μM E-64d, 400 μM 1,10-phenanthroline, 2 μM pepstatin A, and 2 mM EDTA. To separate lysates into membrane and soluble fractions, the lysate was centrifuged at 37500 g for 1 h at 4°C. The supernatant (soluble fraction) was separated from the pellet (membrane fraction), and the pellet was washed with PBS, re-centrifuged and then resuspended in the same volume as before in PBS. The soluble and membrane fractions were then used for western immunoblotting as in section 2.2.
2.6 Macrophage infections
Peritoneal macrophages from CD1 mice were adhered overnight in RPMI 1640 medium (PAA) with 10% (v/v) HIFCS at 37 °C in 5% CO2/95% air onto Nunc 16-well Lab-tek™ tissue culture slides (Fisher Scientific) and then infected using metacyclic promastigotes at a ratio of 6 promastigotes per macrophage. After incubation overnight at 37 °C in 5% CO2/95% air, non-phagocytosed promastigotes were removed by three gentle washes with RPMI 1640 and the cultures were then incubated at 37°C in 5% CO2/95% air. The uptake and subsequent intracellular survival was determined by counting the number of infected macrophages in stained slides at one, four and six days following infection. Slides were fixed in 100% methanol and stained with 10% (v/v) Giemsa’s stain to identify the parasites. The percentage of infected macrophages and number of amastigotes were determined by examination of 100 macrophages per replicate.
2.7 Mice infections
Groups of 6 female BALB/c mice were inoculated in the footpad with 1 × 105 metacyclic promastigotes resuspended in 20 μl of PBS, pH 7.4. The thickness of the footpad was measured weekly until the footpad was 5 mm thick, when the mice were culled.
2.8 Statistical Analyses
Values were expressed as means ± standard error of the mean (SEM). Levels of significance were calculated by unpaired t tests. Differences were considered significant at a p value <0.05.
3. Results
3.1 Generation of L. major Δopb null and OPB over-expression mutants
Recombinant OPB was used to raise anti-OPB antibodies in a sheep, which were affinity purified and used for western immunoblotting. This showed that the 83 kDa OPB enzyme was present at comparable levels in the three main life cycle stages of L. major: procyclic promastigotes, purified metacyclic promastigotes and lesion-derived amastigotes (Fig. 1). In order to investigate the function of OPB in L. major, OPB null mutant (Δopb) cell lines were generated (Fig. 2A). Clones 10 and 21 were obtained and lines re-expressing OPB from the ribosomal locus generated. No difference in growth rate was found upon deletion or re-expression of OPB compared to wild type parasites (data not shown). The deletion of OPB and subsequent re-expression was confirmed by Southern blot using OPB, HYG and SAT probes (Fig. 2B) and western blot (Fig. 2C) analyses. Hybridization of XhoI-digested genomic DNA with an OPB probe revealed a 4.7 kb DNA fragment corresponding to the locus of the wild type parasite, which was absent from the Δopb clones and re-expressor lines; the latter, however, contained a 10 kb positive fragment (Fig. 2B). Hybridisation with a probe for HYG revealed a 3.4 kb fragment in the heterozygote, Δopb clones and re-expresser lines, which was absent from the wild type parasites. Hybridisation with a probe for SAT revealed a 2.9 kb fragment in the Δopb clones and re-expresser lines, which was absent from wild type and heterozygote parasites. OPB was detected by western immunoblot with anti-OPB antibody in wild type L. major (Fig. 2C, lane 1) and, at a reduced level, in the heterozygote (lane 2) and also at high abundance in both the re-expresser lines (lanes 5 and 6). The two OPB null mutant clones lacked the protein (lanes 3 and 4). In contrast, a comparable level of expression of the control protein, EF-1α, was found in all lines. These data confirm that OPB was deleted.
Fig. 1.
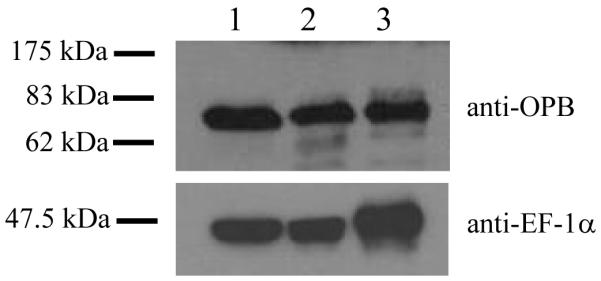
Expression of OPB in L. major
Western immunoblot showing the expression of OPB in the three life cycle stages of L. major.
Lane 1 = procyclic promastigotes, 2 = metacyclic promastigotes, 3 = amastigotes. Purified anti-OPB antibody was used, with anti-T. brucei elongation factor 1α (EF-1α) antibody as a loading control. A lysate of 5 × 106 cells was run per lane.
Fig. 2.

Deletion of L. major OPB.
A: Schematic representation of the OPB locus and the plasmid constructs used for gene replacement. The OPB and antibiotic resistance genes are shown as arrows, flanking DNA sequences are shown as boxes. XhoI fragments are shown. 5′-DHFR and 3′-DHFR, dihydrofolate reductase flanking regions; HYG, hygromycin resistance gene and SAT, streptothricin acetyltransferase resistance gene.
B: Southern blot analysis of genomic DNA digested with XhoI, separated on a 0.7% agarose gel, blotted onto a nylon membrane and hybridized using DNA probes corresponding to a 300 bp fragment of OPB, and the HYG and SAT genes. 1; L. major WT, 2; heterozygote, 3; Δopb clone 10, 4; Δopb clone 21, 5; Δopb clone 10 re-expressing OPB and 6; Δopb clone 21 re-expressing OPB. Molecular size markers are shown on the left.
C: Demonstration of OPB protein levels in L. major WT promastigotes and Δopb by western immunoblot. Lanes as in B. Purified anti-OPB antibody was used and EF-1α antibody as a loading control. A lysate of 5 × 106 cells was run in each lane.
D: Demonstration of over-expression of OPB in L. major cells by western immunoblot. Lane 1; L. major WT, 2; WT[OPB] population, 3; L. major WT, 4; WT[pNUS] population and 5; WT[OPBS577G] population. Purified anti-OPB antibody was used, with anti-EF-1α antibody as a loading control. A lysate of 5 × 106 cells was run in each lane.
Cells over-expressing wild-type OPB (WT[OPB]) or a mutant OPB in which the active site serine 577 was mutated to a glycine (WT[OPBS577G]) were also generated. An increased level of OPB was detected by western immunoblot with an anti-OPB antibody in the WT[OPB] (lane 2) and WT[OPBS577G] cell lysates (lane 5) compared to the L. major WT lysate (lanes 1 and 3) or empty vector control (lane 4, Fig. 2D).
3.2 Characterisation of mutant cell lines
Phenotype analyses were performed with both Δopb clones, but as they behaved similarly data are shown only for clone 10 and the re-expresser generated from it. Metacyclogenesis is central to the ability of Leishmania to successfully infect macrophages [44]. The ability of the two clones to differentiate to metacyclic promastigotes was investigated by western immunoblotting for the surface protein HASPB (Fig. 3A), a known marker for metacyclic promastigotes [39], and comparison of peanut lectin agglutination of cells, which agglutinates the non-metacyclic cells [36]. Comparative analyses showed that the Δopb clone contained less HASPB protein than wild type parasites, whereas the re-expressor line had slightly more (Fig. 3A). The peanut lectin agglutination analyses also showed that there were significantly fewer metacyclic promastigotes in the Δopb culture, with stationary phase wild type promastigote population comprising 35% (± 1.3) metacyclic promastigotes whereas there were only 21% (± 2.4) in the Δopb clone 10 population (p value <0.01). The equivalent population of Δopb re-expressing OPB comprised 27% (± 2.4) metacyclic promastigotes (not significantly different to wild type). These data together suggest a small reduction in metacyclogenesis in Δopb.
Fig. 3.

Analysis of infectivity and metacyclogenesis of Δopb clones and re-expressing cell lines.
A: Western immunoblot to compare HASPB levels. Lane 1; L. major WT cells, 2; Δopb clone 10, 3; Δopb clone 10 re-expressing OPB. Anti-HASPB antibody was used, with anti-EF-1α antibody as a loading control. A lysate of 5 × 106 cells was run per lane.
B: Percentage of infected peritoneal exudate macrophages (PEM). PEM were infected at a ratio of 6 metacyclic promastigotes to 1 macrophage and were incubated for between 24 hours and 6 days, with non-phagocytosed Leishmania washed off after 24 hours. Each value is the mean of 6-8 replicates over two experiments ± SEM. *= p <0.001, in comparison to wild type.
C: Number of amastigotes per 100 PEM. PEM were infected at a ratio of 6 metacyclic promastigotes to 1 macrophage and were incubated for between 24 hours and 6 days, with non-phagocytosed Leishmania washed off after 24 hours. Each value is the mean of 6-8 replicates over two experiments ± SEM.
D: Growth of footpad lesions in BALB/c mice. Footpads infected with Δopb clone 10 were compared to footpads infected with WT L. major. 1 × 105 metacyclic promastigotes were injected per footpad. Each value is the mean of 6 mice ± SEM.
The ability of Δopb metacyclic promastigotes to infect, survive and multiply within peritoneal exudate macrophages (PEM) was assessed by sampling one, four and six days post-infection, with the numbers of infected macrophages (Fig. 3B) and the numbers of amastigotes per 100 macrophages being determined (Fig. 3C). After one day, 82% of macrophages were infected with wild type promastigotes and 66% with Δopb re-expressing OPB, whilst only 47% of macrophages were infected with Δopb. The numbers of macrophages infected with wild type and Δopb re-expressing OPB promastigotes increased slightly by the sixth day, to 88% and 90% respectively. However, only 35% of PEM were still infected with Δopb amastigotes (p < 0.001, compared with the data for wild type parasites). The numbers of amastigotes per 100 PEM increased over the time of the experiment for both PEM infected with wild type and Δopb re-expressing OPB promastigotes, whilst there was no overall change for PEM infected with Δopb. The ability of Δopb to infect BALB/c mice was assessed in comparison with wild type parasites (Fig. 3D). Footpad measurements indicated that Δopb parasites were not significantly different from wild type parasites in their ability to establish an infection in mice.
The ability of living L. major promastigotes to cleave Bz-R-AMC intracellularly was assessed using a fluorescent plate reader. The cleavage was inhibited by incubation of the parasites with antipain, leupeptin or AEBSF (IC50 values of 9.3 nM, 48.1 nM and 14.4 μM, respectively, compared to Ki values for recombinant OPB of 69 nM for antipain, 26 nM for leupeptin and a 90% reduction of OPB activity for AEBSF). E-64d, K11777, 1,10 phenanthroline, EDTA, pepstatin A and PMSF were unable to inhibit the cleavage of Bz-R-AMC in the living promastigotes. The inhibition profile of the cleavage in live cells correlates well with the inhibition profile of the recombinant enzyme, which is a serine peptidase [38].
The two Δopb clones were found to have, respectively, 0.7% and 1.4% of the activity of wild type parasites towards Bz-R-AMC (Fig. 4A and data not shown). Whilst the level of OPB protein in the re-expresser lines was greater than in wild type parasites (Fig. 2C), the respective re-expresser lines for the two clones displayed 25% and 38% of the cleavage activity relative to wild type parasites (Fig. 4A and data not shown). In contrast, the Bz-R-AMC activity in the WT[OPB] promastigotes was 20 times the enzymatic activity of the wild type cells, WT[pNUS] or the WT[OPBS577G] (Fig. 4B). Western immunoblotting carried out with anti-OPB antibody on supernatant and pellet fractions of L. major wild type revealed that the protein is soluble; EF-1α was used as a soluble control protein, whilst GP63 was used as a membrane control protein (Fig. 4C). Depletion of OPB from L. major promastigote lysates by immunoprecipitation using the purified anti-OPB specific antibody reduced Bz-R-AMC activity in cell lysates, to a similarly low level as that found in Δopb null mutants (5% ± 0.7, relative to wild type). As the OPB antibody does not cross-react with the 104 kDa OPB2 (Fig. 1), this confirms that the activity detected was due to OPB. OPB bound to the anti-OPB antibody was unable to cleave Bz-R-AMC, indicating the antibody blocked the active site of the enzyme (data not shown).
Fig. 4.

Reduction of serine peptidase activity in Δopb
A: Effect of deletion and re-expression of OPB in L. major promastigotes on the cleavage of 5 μM Bz-R-AMC. Relative activity was calculated compared to WT for each experiment. For each assay, 6 × 106 cells were used, with the mean of between 5 and 11 experiments shown ± SEM.
B: Effect of over-expression of OPB in L. major promastigotes on the cleavage of 5 μM Bz-R-AMC. Relative activity was calculated compared to WT for each experiment. For each assay, 6 × 106 cells were used, with the mean of between 4 and 11 experiments shown ± SEM.
C: Fractionation of OPB. Western immunoblot showing partition of OPB to either soluble or membrane fraction of L. major. Lane 1; soluble fraction, 2; pelleted fraction. Purified anti-OPB antibody was used, with controls of anti-EF-1α and anti-GP63 antibodies.
3.3 Mapping of the OPB2 active site
OPB2 (LmjF06.0340) is the clan SC, family S9 serine peptidase with the closest sequence identity to OPB and we hypothesized that it potentially could compensate for some of the functions of OPB in the Δopb mutants. As no physiological substrates for OPB or OPB2 are known, we approached this hypothesis by investigating if OPB2 and OPB might have similar substrate specificity, based on modelling of the OPB2 active site. Residues important to the substrate specificity of OPB were based on the high-resolution crystal structure of L. major OPB in complex with antipain [38]. The residues involved in binding the inhibitor in OPB are summarized in Table 2. To investigate the conservation of these residues in OPB2, a primary sequence alignment of the two enzymes was carried out. To achieve a more accurate result, a multiple sequence alignment was performed with L. major OPB and OPB2 (minus the C-terminal extension [31]), plus OPB homologues from T. cruzi, T. brucei, T. evansi, L. infantum, L. amazonensis and L. braziliensis using CLUSTALW [45]. The relevant data were then extracted and used to calculate sequence identities between L. major OPB and OPB2 and to produce Fig. 5. In this region the two proteins are 24.6% identical with the catalytic domain showing 30.6% identity. The catalytic triad and all of the residues found to be important in binding antipain in the active site of OPB are identical in OPB2 (Fig. 5), showing that it is likely that the two enzymes have similar specificity towards small peptide substrates.
Table 2.
Important residues found interacting with antipain in the structure of OPB [38]. The catalytic triad ( ), residues involved in the S1 (
), residues involved in the S1 ( ,
, ), S2 (
), S2 ( ,
, ) and S3 (
) and S3 ( ) binding sites are highlighted and correspond to Figure 5. These residues are all conserved in OPB2.
) binding sites are highlighted and correspond to Figure 5. These residues are all conserved in OPB2.
| LmOPB Residue | AIP residue | Interaction | Importance |
|---|---|---|---|
|
| |||
|
Ser-577-OG |
P1-C | Covalent | Catalytic triad/S1 binding site |
|
Asp-662 |
n/a | n/a | Catalytic triad |
|
His-697 |
n/a | n/a | Catalytic triad |
|
Tyr-496-OH |
P1-O | Hydrogen-bond | Oxyanion/S1 binding site |
|
Ala-578-N |
P1-O | Hydrogen-bond | Oxyanion/S1 binding site |
|
Glu-621-OE1 |
P1-NE/NH2 | Hydrogen-bond | S1 binding site |
|
Arg-664-O |
P1-NH1 | Hydrogen-bond | S1 binding site |
|
Tyr-499-OH |
P1-N | Hydrogen-bond | S1 binding site |
|
Phe-603 |
P1 | pi-cation | S1 binding site |
|
Val-665 |
P1 | Hydrophobic | S1 binding site |
|
Arg-664-NH1 |
P2-O | Hydrogen-bond | S2-binding site |
|
Tyr-499 |
P2 | Hydrophobic | S2 binding site (predicted) |
|
Leu-617 |
P3 | Hydrophobic | S3-binding site |
Fig. 5.

Sequence alignment of L. major OPB and OPB2.
Alignment based on a multiple sequence alignment using eight OPB homologues performed by CLUSTALW. The catalytic domain of OPB is underlined in red and the propeller domain left blank. Predicted inserts of OPB2 into the OPB sequence are shown in pink. Identical residues are shaded, with those involved in the catalytic triad in red. Red, green and blue stars highlight all of the residues involved in the S1-S3 binding pockets, respectively. Solid stars represent hydrogen-bonding interactions while the hollow stars denote pi-cation or hydrophobic interactions. This shows that all of the important residues involved in the binding of antipain in OPB are conserved in OPB2.
4. Discussion
This study has revealed that OPB is expressed at similar levels in the three main life cycle stages of L. major (Fig. 1), whereas previous reports indicated that OPB is upregulated in amastigotes of L. braziliensis and L. donovani compared with promastigotes [17, 18]. The higher expression reported for L. braziliensis amastigotes was based on RNA levels detected using differential display methodology [17]; interestingly, OPB was not one of the genes found to have higher expression in L. major amastigotes in another differential display analysis [19]. Microarray analysis of RNA expressed at differential levels between promastigotes and amastigotes of L. mexicana similarly revealed no change for OPB [20]. The analysis of L. donovani expression levels was completed using proteomic techniques and axenic amastigotes [18]. Thus the differences in data could reflect either species or material variations, but our evidence suggests that the enzyme has a constitutive role in L. major.
Our data show clearly that OPB is not essential for the viability of promastigotes of L. major, as we obtained two independent null mutant clones (Fig. 2). Analysis of these null mutants demonstrated that OPB is responsible for most of the intracellular cleavage of Bz-R-AMC in living promastigotes, as there was only some 1% of hydrolysis of the substrate in the mutants compared with the wild-type parasites. A gene-dose response was found, since the heterozygote had approximately half of the activity of the wild type parasites. This finding is in agreement with the dose response in enzymatic activity found when OPB was knocked out in T. cruzi [24]. In wild-type parasites, OPB was localised to the soluble fraction by cellular fractionation (Fig. 4D). A soluble protein is consistent with the cytosolic location found for other kinetoplastid OPBs [23, 25]. Western blot analysis confirmed that OPB was highly over-expressed in the WT[OPB] cell line (Fig. 2D) and this correlated with a 20-fold increase in activity towards Bz-R-AMC in comparison to wild type cells (Fig. 4C). Over expression of the active site mutant, however, led to a drop in activity relative to the wild type, confirming that the S577 residue is essential for catalytic activity, as was predicted by the structure of OPB [38]. OPB protein levels were higher in the Δopb re-expressor than wild type (Fig 2D), yet activity towards Bz-R-AMC was lower (Fig 4A). This was not the case for the over-expression cell lines, possibly indicating that OPB can fold into an active conformation more effectively in wild type cells than Δopb. These differences do not appear to affect the viability of the various cell lines, as all grew normally in culture.
Metacyclic promastigotes are acknowledged as the infective stage of Leishmania [46] and so a decreased number of metacyclic promastigotes may lead to a reduced ability to infect and survive in macrophages. We have shown that there was a reduction in the number of metacyclic promastigotes in the Δopb populations while the number of metacyclic promastigotes was at a similar level as with the wild type parasites in the populations of cells re-expressing OPB (Fig. 3A). When isolated metacyclic promastigotes were used to infect murine macrophages it was found that there was indeed a reduction in the ability of Δopb to infect macrophages and proliferate within them compared with both wild type parasites and the line re-expressing OPB (Fig. 3B and C). This suggests that the lack of OPB is responsible not only for a defect in the ability to transform to the metacyclic promastigote stage, but also to have a reduced ability to further transform to amastigotes and to survive and thrive as this stage. However, when isolated metacyclic promastigotes were used to infect BALB/c mice, there was no significant difference in the development of footpad lesions (Fig. 3D). BALB/c mice, the strain which was used in this study, are particularly susceptible to infection with Leishmania [47], so it possible that OPB-deficient mutants may be less virulent in other mice strains or in the natural host. In addition, it may be that initiating infection of mice with numbers of promastigotes closer to those naturally egested upon bloodfeeding of sand flies could lead to a different result. Up to around 1,000 L. major promastigotes have been shown to be egested by Phlebotomus papatasi [48], whereas 50,000 promastigotes were injected into the BALB/c mice in this study.
The role of OPB in metacyclogenesis, or differentiation to amastigotes, remains to be elucidated. The role could be akin to the defect in metacyclogenesis caused by a lack of autophagy [49], a self-digestion process whereby cells degrade proteins and organelles in the cytoplasm [50]. As members of the POP family are known to be restricted to the cleavage of peptides [5], it is possible that OPB aids metacyclogenesis or differentiation through degradation of peptides in the cytosol, providing material for the remodelling of the parasite undertaken at this time, analogous to the degradation of proteins in the lysosome through autophagy. Alternatively, OPB could be involved in the processing of propeptides or other regulatory peptides involved in a cascade leading to differentiation to infective parasite forms. The virulence of OPB null mutants in an animal model suggests that the differentiation defect is transitory and that once the parasite has established in the mammalian host it can replicate normally. OPB would thus appear not to be a suitable drug target, despite its absence from mammals.
The inhibitory profile of the hydrolysis of Bz-R-AMC was determined using standard peptidase and proteasome inhibitors. The inhibition by antipain, leupeptin and AEBSF is indicative of a serine peptidase, which matches the inhibitory profile of the recombinant enzyme [38]. Leishmania contain a second OPB-like gene, named OPB2 [31], which is likely to have a similar enzymatic activity to OPB. OPB2 has a low overall sequence identity with OPB (<20%), but is more conserved in the catalytic domain (31%), which contains the serine, aspartic acid, histidine triad. Mapping of the active site of OPB2, based on the recently determined crystal structure of OPB [38], allowed us to predict that all the important residues for substrate binding in the S1-S3 pockets were the same (Fig. 5). Residues involved in the S1 and S2 binding pockets of OPB have been predicted previously and highlighted two pairs of acidic residues [9]. Multiple sequence alignments of OPB and OPB2 suggest that the S1 subsite of OPB2 contains two Glu residues but that the S2 subsite differed from what was expected (Fig. 5). Our findings show that one of the conserved Glu residues in L. major OPB (Glu-621) and several other important residues in the S1 binding site are conserved between OPB and OPB2. In addition, residues found in the S2 and S3 binding pockets of L. major OPB are also conserved in OPB2 (Table 2). This suggests that OPB2 might have similar substrate specificity to OPB and might compensate for some of the functions of OPB in the null mutants, allowing their continued survival and virulence. No apparent hydrolysis of Bz-R-AMC by OPB2 was observed in OPB null mutants, possibly due to inefficient hydrolysis of such a short peptide by the enzyme, or perhaps OPB2 is present in a compartment that is not accessible to Bz-R-AMC. As no physiological substrates of OPB or OPB2, or recombinant OPB2, are available, experiments are underway to attempt to generate L. major OPB2-deficient parasites and OPB/OPB2 double mutants to test these hypotheses.
ACKNOWLEDGEMENTS
This work was supported by the Medical Research Council (G0700127).
REFERENCES
- 1.Herwaldt BL. Leishmaniasis. Lancet. 1999;354:1191–1199. doi: 10.1016/S0140-6736(98)10178-2. [DOI] [PubMed] [Google Scholar]
- 2.Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004;27:305–318. doi: 10.1016/j.cimid.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Murray HW, Berman JD, Davies CR, Saravia NG. Advances in leishmaniasis. The Lancet. 2005;366:1561–1577. doi: 10.1016/S0140-6736(05)67629-5. [DOI] [PubMed] [Google Scholar]
- 4.Kamhawi S. Phlebotomine sand flies and Leishmania parasites: friends or foes? Trends Parasitol. 2006;22:439–445. doi: 10.1016/j.pt.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 5.Polgar L. The prolyl oligopeptidase family. Cell Mol Life Sci. 2002;59:349–362. doi: 10.1007/s00018-002-8427-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coetzer THT, Goldring JPD, Huson LEJ. Oligopeptidase B: a processing peptidase involved in pathogenesis. Biochimie. 2008;90:336–344. doi: 10.1016/j.biochi.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 7.Rea D, Fulop V. Structure-function properties of prolyl oligopeptidase family enzymes. Cell Biochem Biophys. 2006;44:349–365. doi: 10.1385/CBB:44:3:349. [DOI] [PubMed] [Google Scholar]
- 8.Polgar L. A potential processing enzyme in prokaryotes: oligopeptidase B, a new type of serine peptidase. Proteins. 1997;28:375–379. [PubMed] [Google Scholar]
- 9.Morty RE, Fulop V, Andrews NW. Substrate recognition properties of oligopeptidase B from Salmonella enterica serovar Typhimurium. J Bacteriol. 2002;184:3329–3337. doi: 10.1128/JB.184.12.3329-3337.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hemerly JP, Oliveira V, Del Nery E, et al. Subsite specificity (S3, S2, S1′, S2′ and S3′) of oligopeptidase B from Trypanosoma cruzi and Trypanosoma brucei using fluorescent quenched peptides: comparative study and identification of specific carboxypeptidase activity. Biochem J. 2003;373:933–939. doi: 10.1042/BJ20030342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanatani A, Masuda T, Shimoda T, et al. Protease II from Escherichia coli: sequencing and expression of the enzyme gene and characterization of the expressed enzyme. J Biochem (Tokyo) 1991;110:315–320. doi: 10.1093/oxfordjournals.jbchem.a123577. [DOI] [PubMed] [Google Scholar]
- 12.de Matos Guedes HL, Carneiro MP, Gomes DC, Rossi-Bergmanmn B, de Simone SG. Oligopeptidase B from L. amazonensis: molecular cloning, gene expression analysis and molecular model. Parasitol Res. 2007;101:853–863. doi: 10.1007/s00436-007-0552-5. [DOI] [PubMed] [Google Scholar]
- 13.Ivens AC, Peacock CS, Worthey EA, et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peacock CS, Seeger K, Harris D, et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 2007;39:839–847. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morty RE, Authie E, Troeberg L, Lonsdale-Eccles JD, Coetzer TH. Purification and characterisation of a trypsin-like serine oligopeptidase from Trypanosoma congolense. Mol Biochem Parasitol. 1999;102:145–155. doi: 10.1016/s0166-6851(99)00097-3. [DOI] [PubMed] [Google Scholar]
- 16.Silverman JM, Chan SK, Robinson DP, et al. Proteomic analysis of the secretome of Leishmania donovani. Genome Biol. 2008;9:R35. doi: 10.1186/gb-2008-9-2-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gamboa D, van Eys G, Victoir K, et al. Putative markers of infective life stages in Leishmania (Viannia) braziliensis. Parasitology. 2007;134:1689–1698. doi: 10.1017/S003118200700306X. [DOI] [PubMed] [Google Scholar]
- 18.Rosenzweig D, Smith D, Opperdoes F, Stern S, Olafson RW, Zilberstein D. Retooling Leishmania metabolism: from sand fly gut to human macrophage. The FASEB Journal. 2008:22. doi: 10.1096/fj.07-9254com. [DOI] [PubMed] [Google Scholar]
- 19.Ouakad M, Chenik M, Ben Achour-Chenik Y, Louzir H, Dellagi K. Gene expression analysis of wild Leishmania major isolates: identification of genes preferentially expressed in amastigotes. Parasitol Res. 2007;100:255–264. doi: 10.1007/s00436-006-0277-x. [DOI] [PubMed] [Google Scholar]
- 20.Holzer TR, McMaster WR, Forney JD. Expression profiling by whole-genome interspecies microarray hybridization reveals differential gene expression in procyclic promastigotes, lesion-derived amastigotes, and axenic amastigotes in Leishmania mexicana. Mol Biochem Parasitol. 2006;146:198–218. doi: 10.1016/j.molbiopara.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 21.Burleigh BA, Woolsey AM. Cell signalling and Trypanosoma cruzi invasion. Cellular Microbiology. 2002;4:701–711. doi: 10.1046/j.1462-5822.2002.00226.x. [DOI] [PubMed] [Google Scholar]
- 22.Burleigh BA, Andrews NW. A 120-kDa alkaline peptidase from Trypanosoma cruzi is involved in the generation of a novel Ca2+-signaling factor for mammalian cells. J Biol Chem. 1995;270:5172–5180. doi: 10.1074/jbc.270.10.5172. [DOI] [PubMed] [Google Scholar]
- 23.Burleigh BA, Caler EV, Webster P, Andrews NW. A cytosolic serine endopeptidase from Trypanosoma cruzi is required for the generation of Ca2+ signaling in mammalian cells. J Cell Biol. 1997;136:609–620. doi: 10.1083/jcb.136.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caler EV, Vaena de Avalos S, Haynes PA, Andrews NW, Burleigh BA. Oligopeptidase B-dependent signaling mediates host cell invasion by Trypanosoma cruzi. EMBO J. 1998;17:4975–4986. doi: 10.1093/emboj/17.17.4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morty RE, Pelle R, Vadasz I, Uzcanga GL, Seeger W, Bubis J. Oligopeptidase B from Trypanosoma evansi. A parasite peptidase that inactivates atrial natriuretic factor in the bloodstream of infected hosts. J Biol Chem. 2005;280:10925–10937. doi: 10.1074/jbc.M410066200. [DOI] [PubMed] [Google Scholar]
- 26.Troeberg L, Pike RN, Morty RE, Berry RK, Coetzer TH, Lonsdale-Eccles JD. Proteases from Trypanosoma brucei brucei. Purification, characterisation and interactions with host regulatory molecules. Eur J Biochem. 1996;238:728–736. doi: 10.1111/j.1432-1033.1996.0728w.x. [DOI] [PubMed] [Google Scholar]
- 27.Morty RE, Lonsdale-Eccles JD, Mentele R, Auerswald EA, Coetzer TH. Trypanosome-derived oligopeptidase B is released into the plasma of infected rodents, where it persists and retains full catalytic activity. Infect Immun. 2001;69:2757–2761. doi: 10.1128/IAI.69.4.2757-2761.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ndung’u JM, Wright NG, Jennings FW, Murray M. Changes in atrial natriuretic factor and plasma renin activity in dogs infected with Trypanosoma brucei. Parasitol Res. 1992;78:553–556. doi: 10.1007/BF00936451. [DOI] [PubMed] [Google Scholar]
- 29.Morty RE, Troeberg L, Pike RN, et al. A trypanosome oligopeptidase as a target for the trypanocidal agents pentamidine, diminazene and suramin. FEBS Lett. 1998;433:251–256. doi: 10.1016/s0014-5793(98)00914-4. [DOI] [PubMed] [Google Scholar]
- 30.Morty RE, Troeberg L, Powers JC, Ono S, Lonsdale-Eccles JD, Coetzer TH. Characterisation of the antitrypanosomal activity of peptidyl alpha-aminoalkyl phosphonate diphenyl esters. Biochem Pharmacol. 2000;60:1497–1504. doi: 10.1016/s0006-2952(00)00459-7. [DOI] [PubMed] [Google Scholar]
- 31.de Matos Guedes HL, de Carvalho RSN, de Oliveira Gomes DC, Rossi-Bergmanmn B, de Simone SG. Oligopeptidase B-2 from Leishmania amazonensis with an unusual C-terminal extension. Acta Parasitologica. 2008;53:197–204. [Google Scholar]
- 32.Bastos IM, Grellier P, Martins NF, et al. Molecular, functional and structural properties of the prolyl oligopeptidase of Trypanosoma cruzi (POP Tc80), which is required for parasite entry into mammalian cells. Biochem J. 2005;388:29–38. doi: 10.1042/BJ20041049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grellier P, Vendeville S, Joyeau R, et al. Trypanosoma cruzi prolyl oligopeptidase Tc80 is involved in nonphagocytic mammalian cell invasion by trypomastigotes. J Biol Chem. 2001;276:47078–47086. doi: 10.1074/jbc.M106017200. [DOI] [PubMed] [Google Scholar]
- 34.Bastos IM, Motta FN, Charneau S, et al. Prolyl oligopeptidase of Trypanosoma brucei hydrolyzes native collagen, peptide hormones and is active in the plasma of infected mice. Microbes Infect. 2010;12:457–466. doi: 10.1016/j.micinf.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 35.Mottram JC, Frame MJ, Brooks DR, et al. The multiple cpb cysteine proteinase genes of Leishmania mexicana encode isoenzymes that differ in their stage regulation and substrate preferences. J Biol Chem. 1997;272:14285–14293. doi: 10.1074/jbc.272.22.14285. [DOI] [PubMed] [Google Scholar]
- 36.Sacks DL, Hieny S, Sher A. Identification of cell surface carbohydrate and antigenic changes between noninfective and infective developmental stages of Leishmania major promastigotes. J Immunol. 1985;135:564–569. [PubMed] [Google Scholar]
- 37.Hart DT, Vickerman K, Coombs GH. A quick, simple method for purifying Leishmania mexicana amastigotes in large numbers. Parasitology. 1981;82:345–355. doi: 10.1017/s0031182000066889. [DOI] [PubMed] [Google Scholar]
- 38.McLuskey K, Paterson NG, Bland ND, Isaacs NW, Mottram JC. Crystal structure of Leishmania major oligopeptidase B gives insight into the enzymatic properties of a trypanosomatid virulence factor. J Biol Chem. 285:39249–39259. doi: 10.1074/jbc.M110.156679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flinn HM, Rangarajan D, Smith DF. Expression of a hydrophilic surface protein in infective stages of Leishmania major. Mol Biochem Parasitol. 1994;65:259–270. doi: 10.1016/0166-6851(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 40.Button LL, Reiner NE, McMaster WR. Modification of GP63 genes from diverse species of Leishmania for expression of recombinant protein at high levels in Escherichia coli. Mol Biochem Parasitol. 1991;44:213–224. doi: 10.1016/0166-6851(91)90007-s. [DOI] [PubMed] [Google Scholar]
- 41.Misslitz A, Mottram JC, Overath P, Aebischer T. Targeted integration into a rRNA locus results in uniform and high level expression of transgenes in Leishmania amastigotes. Mol Biochem Parasitol. 2000;107:251–261. doi: 10.1016/s0166-6851(00)00195-x. [DOI] [PubMed] [Google Scholar]
- 42.Tetaud E, Lecuix I, Sheldrake T, Baltz T, Fairlamb AH. A new expression vector for Crithidia fasciculata and Leishmania. Mol Biochem Parasitol. 2002;120:195–204. doi: 10.1016/s0166-6851(02)00002-6. [DOI] [PubMed] [Google Scholar]
- 43.Jacobsen W, Christians U, Benet LZ. In vitro evaluation of the disposition of a novel cysteine protease inhibitor. Drug Metab Dispos. 2000;28:1343–1351. [PubMed] [Google Scholar]
- 44.Alexander J, Satoskar AR, Russell DG. Leishmania species: models of intracellular parasitism. J Cell Sci. 1999;112(Pt 18):2993–3002. doi: 10.1242/jcs.112.18.2993. [DOI] [PubMed] [Google Scholar]
- 45.Chenna R, Sugawara H, Koike T, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.da Silva R, Sacks DL. Metacyclogenesis is a major determinant of Leishmania promastigote virulence and attenuation. Infect Immun. 1987;55:2802–2806. doi: 10.1128/iai.55.11.2802-2806.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen L, Zhang ZH, Watanabe T, et al. The involvement of neutrophils in the resistance to Leishmania major infection in susceptible but not in resistant mice. Parasitol Int. 2005;54:109–118. doi: 10.1016/j.parint.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Warburg A, Schlein Y. The effect of post-bloodmeal nutrition of Phlebotomus papatasi on the transmission of Leishmania major. Am J Trop Med Hyg. 1986;35:926–930. doi: 10.4269/ajtmh.1986.35.926. [DOI] [PubMed] [Google Scholar]
- 49.Besteiro S, Williams RA, Morrison LS, Coombs GH, Mottram JC. Endosome sorting and autophagy are essential for differentiation and virulence of Leishmania major. J Biol Chem. 2006;281:11384–11396. doi: 10.1074/jbc.M512307200. [DOI] [PubMed] [Google Scholar]
- 50.Reggiori F, Klionsky DJ. Autophagosomes: biogenesis from scratch? Curr Opin Chem Biol. 2005;17:415–422. doi: 10.1016/j.ceb.2005.06.007. [DOI] [PubMed] [Google Scholar]