

Abstract
BACKGROUND AND PURPOSE
Kyotorphin (KTP; L-Tyr-L-Arg), an endogenous neuropeptide, is potently analgesic when delivered directly to the central nervous system. Its weak analgesic effects after systemic administration have been explained by inability to cross the blood–brain barrier (BBB) and detract from the possible clinical use of KTP as an analgesic. In this study, we aimed to increase the lipophilicity of KTP by amidation and to evaluate the analgesic efficacy of a new KTP derivative (KTP-amide – KTP-NH2).
EXPERIMENTAL APPROACH
We synthesized KTP-NH2. This peptide was given systemically to assess its ability to cross the BBB. A wide range of pain models, including acute, sustained and chronic inflammatory and neuropathic pain, were used to characterize analgesic efficacies of KTP-NH2. Binding to opioid receptors and toxicity were also measured.
KEY RESULTS
KTP-NH2, unlike its precursor KTP, was lipophilic and highly analgesic following systemic administration in several acute and chronic pain models, without inducing toxic effects or affecting motor responses and blood pressure. Binding to opioid receptors was minimal. KTP-NH2 inhibited nociceptive responses of spinal neurons. Its analgesic effects were prevented by intrathecal or i.p. administration of naloxone.
CONCLUSIONS AND IMPLICATIONS
Amidation allowed KTP to show good analgesic ability after systemic delivery in acute and chronic pain models. The indirect opioid-mediated actions of KTP-NH2 may explain why this compound retained its analgesic effects although the usual side effects of opioids were absent, which is a desired feature in next-generation pain medications.
Keywords: kyotorphin, blood–brain barrier, analgesic, pain, neuropeptide
Introduction
Kyotorphin (KTP; L-Tyr-L-Arg) was isolated from bovine brain in 1979 (Takagi et al., 1979a,b) and has subsequently been found in the brain of several mammals and in human cerebrospinal fluid (Kolaeva et al., 2000). Later studies demonstrated the analgesic activity of KTP in animal models, but only following central delivery. KTP was 4.2-fold more potent than endogenous opioids, such as met-enkephalins, after injection into the cisterna magna of mice (Shiomi et al., 1981). However, only a brief activity at a high dose was detected after systemic KTP administration (Chen et al., 1998). The inability of KTP to cross the blood–brain barrier (BBB) was proposed to explain these data and prevent any putative clinical utility of this powerful analgesic.
KTP analgesia is reversed by naloxone, but the in vitro binding to opioid receptors was found to be minimal (Rackham et al., 1982). A distinct receptor for KTP was then proposed (Ueda et al., 1986), having the dipeptide L-Leu-L-Arg as antagonist (Ueda et al., 1989). Despite some experimental evidence (Shiomi et al., 1981), such a specific KTP receptor has never been identified. Furthermore, the flexibility of KTP (Machuqueiro and Baptista, 2007) and the high activity of its isomer L-Tyr-D-Arg also challenge the specific receptor hypothesis. An alternative mechanism is a rapid degradation of KTP, resulting in free L-Arg, the substrate for nitric oxide (NO) synthase (Arima et al., 1997). The NO thus formed would induce analgesia via met-enkephalin release, which is the common final event leading to analgesia in both proposed mechanisms. Whatever the details of the mechanism(s) of action, it is clear that KTP can only become useful after chemical modification to increase its ability to cross the BBB, while preserving most of its structure and chemistry so that the KTP derivative would remain effective and non-toxic.
We started to search for a KTP derivative with minimal differences relative to KTP, but with improved capability to interact with lipids, that is, more likely to cross the BBB. This represented an apparent paradox, as the solubility of KTP in aqueous media should be maintained, while lipid affinity should be increased. This critical feature was the problem of a recent KTP derivative (Lopes et al., 2006). Most of the previously published studies of KTP have used direct administration to the central nervous system. However, from the point of view of drug discovery, only those candidate compounds that are active following systemic administration have pharmacological relevance. Here, we focused on the potential of KTP-NH2 as a new analgesic drug, and the effects, after systemic administration, of this new compound were evaluated in several experimental pain models. Furthermore, insights into the mechanism of action were provided as well as indirect evidence that points towards an increased ability to cross the BBB.
Methods
General synthetic procedures
The peptides were synthesized by a standard solution peptide synthesis using Boc/tert-butyl strategy for the Tyr and minimal side-chain protection (hydrochloride salt) for the Arg-NH2 moieties. Each peptide was analysed for purity on high-performance liquid chromatography (HPLC) and characterized by proton nuclear magnetic resonance (1H-NMR) and high-resolution mass spectra under electrospray ionization (HRMS-ESI).
All commercially available chemicals were used as purchased without further purification. Melting points (capillary tube) were measured with an electrothermal digital melting point apparatus IA 91000 and are uncorrected. 1H NMR spectra were recorded on a Brucker DPX200 Advance spectrometer (Bruker Biospin Corporation, Billerica, MA, USA) at 200 MHz. Spectra recorded in CDCl3 were referenced to residual CHCl3 at 7.26 ppm. Spectra recorded in DMSO-d6 were referenced to residual dimethyl sulphoxide (DMSO) at 2.49 ppm. Coupling constants (J) are given in Hertz (Hz). The following abbreviations were used for spin multiplicity: s = singlet, d = doblet, t = triplet, q = quarter, m = multiplet, dd = double doublet, bs = broad singlet. HRMS were determined under conditions of ESI on a Bucker MicroTof-Q instrument (Bruker Daltonics Inc., Billerica, MA, USA) using a lock-spray source.
HPLC analyses were carried out using a Dionex P680 instrument (Dionex Corporation, Sunnyvale, CA, USA). Separations were achieved on an analytical C18 Kromasil reversed phase column (4.6 mm × 40 mm; 3.5 µm particle size). The peptides were eluted using a linear gradient of 0–100% CH3CN in 0.1% trifluoroacetic acid, at flow rate of 1.0 mL·min−1 over 7 min. The absorbance was measured at 220 nm. The HPLC retention time of each peptide was determined when the peak was at its maximum height. Analytical thin layer chromatography (TLC) was performed on precoated TLC plates, silica gel 60 F254 (Merck, Darmstadt, Germany). The spots on the TLC plates were visualized with UV/visible light (254 nm) and/or stained with a solution of potassium permanganate (1.5 g/100 mL H2O).
Synthesis of KTP-NH2 (Tyr-Arg-NH2)
N-methylmorpholine (NMM) (3.3 mL, 30 mmol) was added to a solution of Boc-Tyr(tBu)-OH (3.374 g, 10 mmol) in N,N-dimethylformamide (DMF) (40 mL), and the resulting mixture was stirred at room temperature for 1 h. Then, benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphoniumfluorophosphate (BOP) (4.42 g, 10 mmol) and H-Arg-NH2 2HCl (2.46 g, 10 mmol) were added. The resulting reaction mixture was stirred overnight at room temperature. Upon completion of the reaction (TLC monitoring), the reaction mixture was filtered. The resulting solution was diluted with ethyl acetate (100 mL) and washed with saturated sodium bicarbonate (3 × 50 mL), water (100 mL), 1 M aqueous potassium hydrogen sulphate (3 × 50 mL) and brine (50 mL). The organic layer was dried over magnesium sulphate, filtered and concentrated in vacuo to afford compound Boc-Tyr(tBu)-Arg-NH2 (2.7 g, 55% yield) as a colourless solid. HPLC: tr (min) = 6.99. M.p. 123–125°C.1H NMR (DMSO-d6, 200 MHz) δ 1.26 (s, 9H, C(CH3)3), 1.29 (s, 9H, C(CH3)3), 1.39–1.78 [m, 4H (-CH2)2], 2.67 (dd, J = 14.0 Hz, J' = 10.6 Hz, 1H of the CH2-βtyrosine), 2.94 (dd, J = 14.0 Hz, J' = 3.6 Hz, 1H of the CH2-βtyrosine), 3.09 (q-apparent, J = 6.0 Hz, 2H, NHCH2), 4.01–4.26 (m, 2H, CH-αtyrosine and CH-αarginine), 6.85 (d, J = 8.4 Hz, 2H, CHaryl), 6.95 (bs, 1H, NH), 6.99 (bs, 1H, NH), 7.11 (bs, 3H, NH), 7.15 (d, J = 8.4 Hz, 2H, CHaryl), 7.35 (bs, 1H, NH), 7.47 (t, J = 6.0 Hz, 1H, NHCH2), 7.87 (d, J = 8.2 Hz, 1H, NH). HRMS (ESI) m/z: calculated for C24H41N6O5[M + H]+ 493.3138; found 493.3139. Boc-Tyr(tBu)-Arg-NH2 (2.6 g, 5.28 mmol) was dissolved in CH2Cl2 (8 mL), and the solution was cooled in an ice bath. TFA (8 mL) was added dropwise, and the resulting mixture was stirred at 0°C for 1–2 h. Upon completion of the reaction (TLC monitoring), the solvent was removed under reduced pressure. The resulting residue was triturated with ether, collected and dried in vacuo. The resulting white solid was dissolved in 1 M aqueous HCl and lyophilized. This process was repeated three times to afford Tyr-Arg-NH2 (1.76 g, 100% yield) as a hydrochloride salt. HPLC: tr (min) = 2.42. M.p. 80–83dec°C.1H NMR (DMSO-d6, 200 MHz) δ 1.55–1.82 [m, 4H (-CH2)2], 2.89 (dd, J = 14.4 Hz, J' = 8.2 Hz, 1H of the CH2-βtyrosine), 3.06–3.23 (m, 2H, NHCH2 and 1H, of the CH2-βtyrosine), 4.09–4.14 (m, 1H, CH-αtyrosine), 4.31–4.38 (m, CH-αarginine), 6.80 (d, J = 8.4 Hz, 2H, CHaryl), 7.14 (d, J = 8.4 Hz, 2H, CHaryl), 7.25 (bs, 4H, NH), 7.51 (bs, 1H, NH), 7.77 (t, J = 5.6 Hz, 1H, NHCH2), 8.13 (bs, 3H, NH), 8.73 (d, J = 8.0 Hz, 1H, NH). HRMS (ESI) m/z: calculated for C15H25N6O3[M + H]+ 337.1983; found 337.1975. Please see the supporting information for details of the synthesis of KTP.
Animals
All animal care and experimental studies were in accordance with the European Community Council Directive (86/609/EEC) and the ethical guidelines for pain research in animals (Zimmermann, 1983). Adult male Wistar rats (Charles River, Lille, France), weighing between 270 and 310 g, were housed in groups (two per cage) at temperature and light-controlled conditions (22 ± 2°C; lights on between 8 a.m. and 8 p.m.), with free access to food and water. In order to induce habituation to the researcher and to minimize stress, the animals used in the tail flick, hot plate and formalin tests were gently handled daily in the test room and devices used to evaluate pain responses, blood pressure or motor behaviour for 30 min during the week that preceded the nociceptive tests and were brought to the same room 1 h before the experiments. In the chronic pain models (monoarthritis and spared nerve injury, SNI), the rats were handled during 14 days after chronic pain induction. The habituation protocols were previously shown to minimize animal stress, by decreasing neuronal activation in brain areas involved in stress and increase of adrenocorticotropic hormone synthesis (Pan et al., 1997). For all experimental groups, group size was ≥6.
Administration of compounds
All administered compounds were dissolved in saline (injectable solution of sodium chloride, 0.9%, from Labesfal, Lisboa, Portugal). Doses were adapted to animals' weight, and the injected volume was kept below 350 µL. The gavage technique was used for oral administrations. Animals were very lightly sedated (isoflurane volatile anaesthesia for 1 min) and intubated with a gastric probe (5 cm length, Fine Science Tools 18061-15, Fine Science Tools Inc., Heidelberg, Germany). After drug delivery into the stomach, the probe was slowly removed. For intrathecal (i.t.) administration of naloxone (Sigma, Lisboa, Portugal), i.t. catheters were implanted into the lumbar subarachnoid space at the L6 spinal cord level under a mixture of ketamine hydrochloride (Ketalar, 0.06 g·kg−1) and medetomidine (Domitor, 0.25 g·kg−1) given intraperitoneally (i.p.). Animals were allowed to recover from surgery for 3 days.
For the experiments involving morphine administration (Labesfal), this analgesic was given in a dose of 5 mg·kg−1 for the acute pain experiments (tail flick and hot plate tests) and 6 mg·kg−1 for chronic pain models (monoarthritis and neuropathic pain). Naloxone was injected i.t. (10 µL; 5 mg·mL−1) or i.p. (5 mg·kg−1), and 10 min later, the peptides or saline were injected i.p.
Models of acute pain
The tail flick test was performed as described previously (D'Amour and Smith, 1941). A radiant heat (infrared) source adjusted to 10 W of a tail flick apparatus (mod. 7360, Ugo Basile, Comerio, Italy) was directed to the ventral surface of the rat's tail. Three trials were performed at each time point in three different parts of the tail (3–5 cm from its tip). The time (in s) required to remove the tail from the heat stimulus was recorded. The tail flick latency was defined as the mean of the three trials. To avoid tissue damage, a cut-off time of 24 s was used. For the hot plate test, a similar procedure was used, but thermal responses were evaluated at the hind paw using the hot plate apparatus (IITC Life Sciences Analgesimeter, San Fernando Valley, CA, USA). The animals were placed in a plexiglass box (12 cm diameter; 22.5 cm height) on a metal surface. The initial temperature was 35°C, and an increasing rate temperature of 9°C per minute was defined. The temperature (in °C) to elicit a hind paw shake was recorded. A cut-off temperature of 52.5°C was defined. In both behavioural tests, the animals were evaluated before KTP, KTP-NH2, morphine or saline drug injection (basal values) and 15, 30, 45, 60 and 90 min after i.p. injections. The behavioural evaluation was extended to 120 min in the case of oral administration. The responses are shown as the difference between values at each time after treatment and the basal values (zero time).
Models of inflammatory and neuropathic pain
The formalin test was performed as described previously (Tjolsen et al., 1992). The animals were injected i.p. with each peptide (32.3 mg·kg−1) or with the same volume of saline. Ten minutes later, 50 µL of 5% neutral formalin was injected s.c. into the dorsal surface of the left hind paw with a 30 G needle. After formalin injection, the rats were placed individually in an open Plexiglas chamber (10 × 20 × 24 cm), and their behaviour was recorded during 60 min. The recordings were analysed using a purpose-made computer programme (Ottoni, 2000). Data are presented as the number of jerks/flinches (named here as paw jerks) of the injected hind paw in 12 successive periods of 5 min each. The animals were killed 2 h after formalin injection for the analysis of c-fos activation, since this is considered the time point of maximal expression of the proto-oncogene (Coggeshall, 2005).
Chronic inflammation (monoarthritis) was induced by injecting the left tibiotarsal joint with 50 µL of a complete Freund's adjuvant (CFA) solution prepared as described previously (Butler et al., 1992), under brief isoflurane anaesthesia (5%; Abbott Laboratories, Amadora, Portugal). After the intra-articular injection, the animals returned to their cages and continued to be handled daily for 14 days for habituation purposes. Thermal nociceptive thresholds were assessed using the radiant-heat paw withdrawal test, as described previously (Butler et al., 1992). Briefly, the inflamed hind paw received a light beam from the tail flick device referred above, and the time (in seconds) the animal took to remove the paw from the stimulus was recorded. The test was performed three times, and the average value was taken as the paw withdraw latency. Cut-off value was defined at 24 s. The test was performed before peptide injection (32.3 mg·kg−1), morphine (6 mg·kg−1) or saline (baseline values) and 15, 30, 45, 60 and 90 min following administration.
An experimental neuropathy was induced using the SNI model, as originally described (Decosterd and Woolf, 2000). Fourteen days after surgery, animals were i.p. injected with 32.3 mg·kg−1 of KTP-NH2, KTP or with the same volume of saline. Nociceptive responses before peptide injection and 30, 60 and 90 min later were collected. Standard tests aimed at assessing mechanical allodynia, cold allodynia and mechanical hyperalgesia were performed by stimulating the lateral plantar surface of the injured hind paw (Decosterd and Woolf, 2000). Mechanical allodynia was assessed with a set of von Frey monofilaments (Somedic, Horby, Sweden) applied in a sequence of increasing forces. The threshold was considered the lowest force that evoked a brisk withdrawal response to at least one out of five stimuli. Cold allodynia was determined by the application of a drop of acetone and by recording the duration of the withdrawal response, with a minimal value of 0.5 s. Mechanical hyperalgesia was determined using the pin prick test, which consists of a brief stimulation with a safety pin at intensity sufficient to indent the skin. The duration of the paw withdrawal was recorded with an arbitrary minimal time of 0.5 s. Additionally, a chronic treatment regimen (daily administration of 32.3 mg·kg−1 for 7 days) was used. Following the seventh injection, the animals were evaluated for the same three sensory modalities. For comparison, in the SNI model, animals received one single administration of morphine (6 mg·kg−1) and were also tested for mechanical allodynia, cold allodynia and mechanical hyperalgesia.
Immunostaining for Fos
Two hours after formalin injection, the animals were transcardially perfused with 200 mL of phosphate-buffered saline (PBS), followed by 1000 mL of 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. Spinal cord segments L4-L5 were removed and post-fixed by immersion for 3 h in fixative. After overnight cryoprotection, at 4°C, in a 30% sucrose solution in 0.1 M PBS, coronal sections (40 µm thick) were cut on a freezing microtome and collected in PBS. One in every three sections obtained from the spinal cord was immunostained for Fos, the protein produced upon c-fos activation, by incubating overnight with a rabbit anti-Fos polyclonal antibody (Ab5, Oncogene, Munich, Germany), at 1:10 000, at room temperature. To decrease background staining, incubation in primary antiserum was preceded by immersion for 2 h in 0.1 M glycine and 10% normal swine serum diluted in a PBS solution containing 0.3% Triton X-100 (PBST). Following repeated washing in PBST, sections were incubated in biotinylated swine anti-rabbit antibody (Dako, Glostrup, Denmark) followed by an ABC solution (Vectorstain Elite, Vector, Burlingame, CA, USA), both for 1 h at 1:200. The DAB chromogen was used (Tavares et al., 1997), and the sections were mounted on gelatin-coated slides, cleared in xylol and coverslipped with Eukitt. Additional spinal sections immune-stained for Fos were counterstained with formol–thionin in order to delimit the spinal dorsal horn (Pinto et al., 2003). Neurons immunoreactive for Fos were counted in the spinal dorsal horn (laminae I–VI) when the immunoreaction for Fos protein was expressed at the nucleus, independently of the staining intensity (Coggeshall, 2005). Mean numbers of Fos-immunoreactive neurons per section were calculated.
In vitro opioid receptor binding
Opioid receptor binding studies were performed by CEREP©, study number 19074. Competitive binding to opioid receptors – human δ2, κ, human µ and the N/OFQ (NOP) receptors – in the presence of radioligands [3H]DADLE, 0.5 nM; [3H]U 69593, 1 nM; [3H]DAMGO, 0.5 nM and [3H]nociceptin, 0.2 nM, respectively, were performed with 150 µM KTP-NH2 and detected by scintillation counting. The source of δ and κ opioid receptors were CHO cells and that for µ opioid and NOP receptors were HEK-293 cells. Receptor nomenclature follows Alexander et al. (2009).
Statistical analyses
Data are represented as the group means ± SEM. The significance of differences in each group was analyzed with Friedman's-test followed by Dunn's multiple comparison test. The significance of differences for each dose and time point compared with the KTP-treated controls was analysed with non-parametric two-tailed Mann–Whitney test. All statistical analyses were calculated with Prism software (GraphPad Software, version 5, La Jolla, CA, USA).
Results
KTP-NH2 analgesia in acute pain models
The analgesic effects of KTP-NH2 were evaluated after i.p. or oral administrations to male Wistar rats. As it is derived from KTP, the doses of KTP-NH2 were based on previous studies using intracisternal injections of KTP (Takagi et al., 1979a,b) and increasing the concentration by about 250-fold for systemic delivery. Following i.p. administration, and unlike the original KTP, a dose- and time-dependent inhibition was detected both in the tail flick and hot plate tests (Figure 1A, B). With the exception of the lower dose (16.7 mg·kg−1), statistically significant increases of response thresholds were obtained with all the KTP-NH2 doses tested. A comparison of the analgesic effects of KTP-NH2 and morphine (Figure 1A, B) showed that the equi-effective dose of KTP-NH2 was about fivefold that of morphine, in terms of mg/kg. In molar terms, for KTP-NH2, 32.3 mg·kg−1= 96 µmol·kg−1 and for morphine, 5 mg·kg−1= 17.5 µmol·kg−1. However, whereas KTP-NH2 induced no detectable side effects on blood pressure (Figure S1), basal temperature or motor performance (Figure S2), morphine appears to induce motor impairment (Patti et al., 2005). Oral administration studies confirmed the analgesic efficacy of KTP-NH2 in the tail flick and hot plate tests (Figure 1C, D). Both the 161 mg·kg−1 and the 200 mg·kg−1 dose also induced a long-lasting inhibition in the tail flick and hot plate tests that lasted until the end of the evaluation period (2 h). No analgesic effects were obtained after i.p. or oral administration of non-amidated KTP (Figure 1A–D) or after saline injections (Figure 1A, B).
Figure 1.
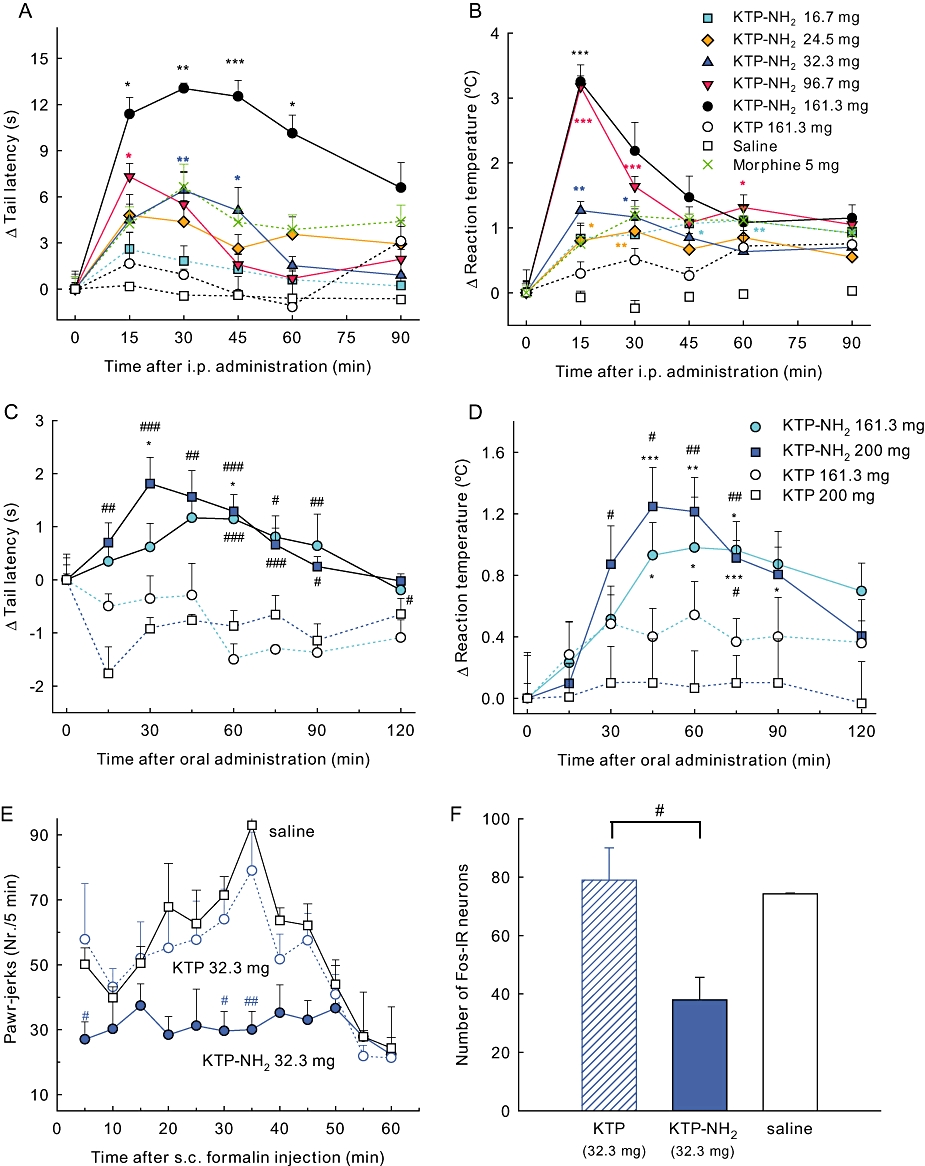
Analgesic profile of KTP-NH2. Analgesia was induced in the tail flick (A,C), hot plate (B,D) and formalin (E) tests after i.p. (A, B, E) or oral (C, D) administration. (A–D) Results expressed as the change in the behavioural responses at each evaluation time point, relative to baseline values (time 0) obtained immediately before injection. The analgesic effects were dose-dependent, starting at 15 min and lasting at least for 30 min. In the formalin test (E), the analgesic effects of an i.p. injection of KTP-NH2 (32.3 mg·kg−1) 10 min before formalin injection were detected in the acute (t < 10 min) and inflammatory (t > 10 min) phases. In panel (F), nociceptive activation of spinal neurons induced by formalin was inhibited by KTP-NH2, as indicated by the lower number of Fos-immunoreactive neurons at the spinal dorsal horn, laminae I–VI, relatively to KTP- or saline-treated animals. Doses shown as mg·kg−1. Data shown are means ± SEM; for all groups n≥ 6. *P < 0.05; **P < 0.01; ***P < 0.001 versus basal response, Friedman test, and #P < 0.05, ##P < 0.01, ###P < 0.001 versus KTP-treated controls, Mann–Whitney test. KTP, kyotorphin.
Inhibition of nociceptive transmission following KTP-NH2 administration
In order to increase pharmacological relevance, we proceeded to investigate the effects of KTP-NH2 in a model of sustained pain, the formalin test, characterized by two distinct pain phases: acute (t < 10 min) and sustained–chronic (t > 10 min). Based on the time course of the effects of the compound in the acute pain tests, KTP-NH2 was given i.p. 10 min before formalin at a dose of 32.3 mg·kg−1. Animals injected with non-amidated KTP (at this dose) or saline showed a pattern of behavioural responses very similar to that of animals injected only with formalin (Figure 1E) (Dubuisson and Dennis, 1977). By contrast, after injection of KTP-NH2, a significant reduction of paw jerks was detected both in the acute and sustained phases of the test, when compared to KTP or saline-treated groups (Figure 1E). The nociceptive activation of spinal dorsal horn neurons laminae I–VI in formalin-injected rats was assessed using immunodetection of the Fos protein, a standard method to evaluate nociceptive activation of neuronal populations at the spinal cord (Pinto et al., 2003). The number of Fos-immunoreactive neurons was lower in the dorsal horn of animals treated with KTP-NH2 (Figure 1F). The higher inhibition of the second phase of the formalin test (Figure 1E), along with the decrease of nociceptive activation induced by KTP-NH2 (Figure 1F), reveals central inhibition of nociceptive transmission.
Efficacy of KTP-NH2 in chronic pain animal models
We next investigated the efficacy of KTP-NH2 in animal models of chronic pain (inflammatory or neuropathic). In the chronic inflammatory pain model – monoarthritis – KTP-NH2 at the dose of 32.3 mg·kg−1 significantly increased the threshold response (Figure 2A), with a profile of action similar to that obtained with acute stimulus. This figure also shows that KTP appeared to decrease withdrawal latencies. This phenomenon was also observed for saline treated animals. Over time, there is an apparent increase in hypersensitivity on the injured paw of the animal. However, none of these effects were statistically significant.
Figure 2.
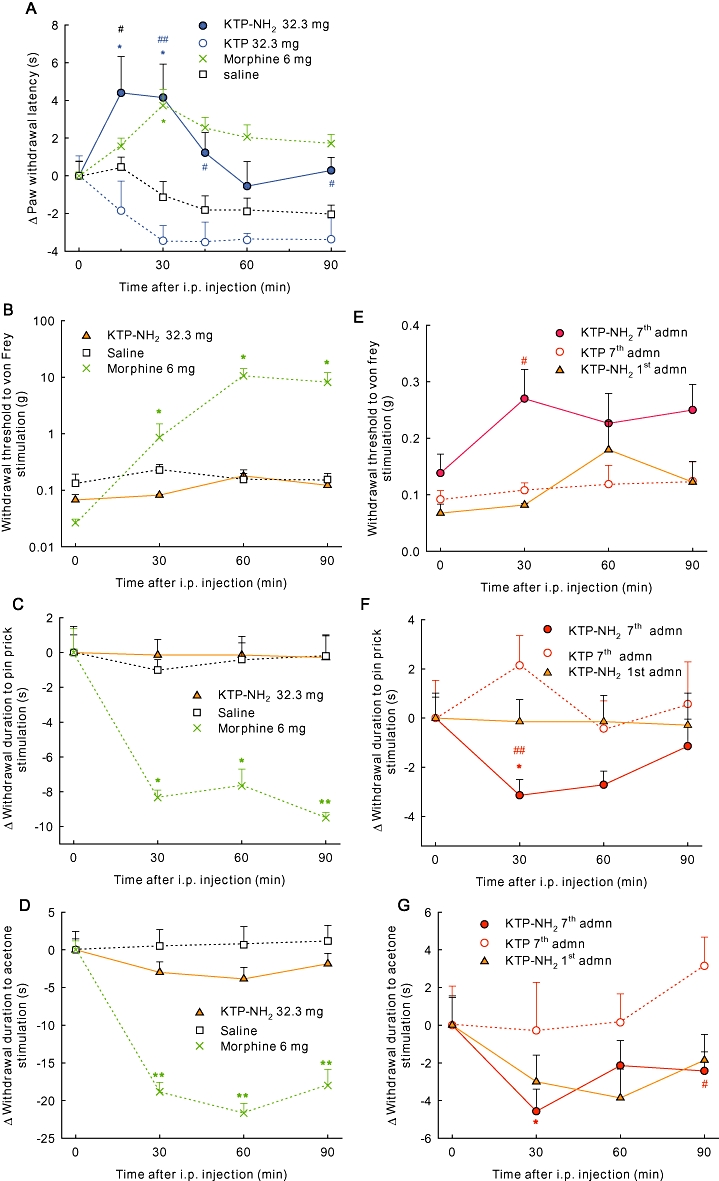
KTP-NH2 suppresses pain behaviour in animal models of chronic pain. (A) KTP-NH2 (32.3 mg·kg−1) significantly increased the threshold response in the monoarthritic model. In the neuropathic pain model (B–G), the administration of a single dose of KTP-NH2 did not induce significant effects (B–D). Daily treatment with KTP-NH2 for 7 days provoked an increase of the weight threshold at 30 min for Von-Frey test (E). In the pin prick (F) and acetone tests (G), a significant decrease of the time of protection was detected 30 min after KTP-NH2 injection. Results are expressed as the change in the behavioural responses of each animal along the course of the experiments. Baseline values (time 0) were obtained immediately before KTP-NH2 injection. Doses shown as mg·kg−1. Data shown are means ± SEM; for all groups n≥ 6. *P < 0.05; **P < 0.01; ***P < 0.001 versus basal response, Friedman test, and #P < 0.05, ##P < 0.01, ###P < 0.001 versus KTP-treated controls, Mann–Whitney test. KTP, kyotorphin.
The SNI model was selected as the model for neuropathic pain. Before the induction of SNI, the animals responded to the 26 g filament in the von Frey test and did not exhibit any behavioural response in the pin-prick or acetone tests, in a manner similar to previous studies (Decosterd and Woolf, 2000; Martins et al., 2010). After the SNI model of neuropathic pain had reached a stable level, a single administration of KTP-NH2 elicited no analgesic effects on the three behavioural tests namely the von Frey (Figure 2B), pin prick (Figure 2C) and acetone (Figure 2D). However, after chronic treatment with KTP-NH2, clear effects were observed. After seven daily injections of KTP-NH2 at 32.3 mg·kg−1, mechanical allodynia (Figure 2E), mechanical hyperalgesia (Figure 2F) and cold allodynia (Figure 2G) were all reversed. This significant analgesia on the three sensory modalities evaluated demonstrated that KTP-NH2 did not induce tolerance. For KTP, and in accordance with the previous results, animals treated for 7 days with the same dose (32.3 mg·kg−1, daily) did not display any inhibition of nociception (Figure 2E–G). In the same model, a single injection of morphine elicited anti-nociception (Figure 2B–D).
Possible opioidergic effects of KTP-NH2
To study the mechanisms of action of KTP-NH2, the animals received i.p. or i.t. injections of naloxone (i.p. 5 mg·kg−1 and i.t. 10 µL; 5 mg·mL−1), 10 min before the i.p. injection of KTP-NH2 (32.3 mg·kg−1), and the effects were evaluated in the tail-flick (Figure 3A, C) and hot-plate tests (Figure 3B, D). Regardless of the route of administration of naloxone, the analgesic effects of KTP-NH2 were reversed in both behavioural tests (Figure 3A–D).
Figure 3.
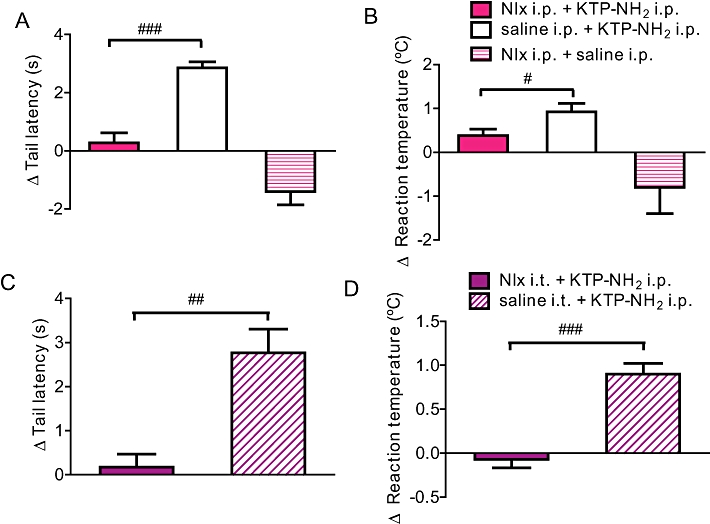
Naloxone reverses analgesia induced by KTP-NH2 in tail flick and hot plate tests. A reversal of KTP-NH2 (32.3 mg·kg−1)-induced analgesia occurred after i.p. (A, B) and i.t. (C, D) administration of naloxone (Nlx; i.p. 5 mg·kg−1 and i.t. 10 µL; 5 mg·mL−1) in tail flick (A, C) and hot plate (B, D) tests. Results are expressed as the change in the behavioural responses at 30 min relative to baseline values. Data shown are means ± SEM; for all groups n≥ 6. ##P < 0.01, ###P < 0.001 versus KTP-treated controls, Mann–Whitney test. (Note: in panel B, # P = 0.05). KTP, kyotorphin.
In order to further understand the involvement of opioid mechanisms in KTP-NH2 mode of action, in vitro binding studies for opioid receptors were performed for δ, κ, µ opioid receptors and the NOP receptor. The percentage of inhibition of control specific binding was 30, 52, 58 and 11, respectively, for a relatively high concentration of KTP-NH2– 150 µM. Although not completely absent, the affinity of KTP-NH2 for opioid receptors was rather weak.
Toxicological evaluation
Toxicological assessments performed after seven daily injections of KTP-NH2 (32.3 mg·kg−1) did not show any histological signs of damage in the liver, kidney or spleen, nor changes in enzymic assays of the hepatic function or antioxidant activity in blood plasma (Table S1). In addition, three standard in vitro cell viability assays – Trypan blue, Crystal Violet and MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay – were performed using three mammalian cell lines: HeLa, Huh-7 and V-79 hamster fibroblasts. For all nine combinations, KTP-NH2 concentrations up to 1000 µM yielded similar results: cytotoxicity was nearly absent (Table S2).
Discussion and conclusions
Amidation proved to be a simple and inexpensive modification that converted kyotorphin into an effective and safe analgesic molecule, very likely able to cross the BBB. Using systemic administration and a wide range of in vivo pain models, we showed that KTP-NH2 was an effective analgesic, with high activity in all the models tested. Thus, derivatization maintained the analgesic potential after systemic administration. Both oral and i.p. injections of KTP-NH2 succeeded in inducing anti-nociception in two acute pain behavioural tests. The doses required to induce analgesia after i.p. administration were lower than those required after oral delivery, by about sixfold, probably due to peptidase degradation in the gastrointestinal tract (Haseto et al., 1994). Furthermore, the results suggested that KTP-NH2 was able to cross the BBB, which may result from an increase in lipophilicity caused by the transformation of the carboxyterminal of the peptide. Central effects are likely to be involved in the analgesic actions of KTP-NH2, as nociceptive activation of the spinal dorsal horn was reversed by KTP-NH2 administration, and this analgesic action was blocked by i.t. delivery of naloxone.
In spite of their differences in the route of effective administration, KTP-NH2 and KTP differ structurally only by the amidation. The present study shown that the analgesic action of KTP-NH2 was reversed by the non-specific opioid receptor antagonist naloxone, and that direct binding of KTP-NH2 to opioid receptors was absent or very weak. Similar results were obtained with the original KTP molecule (Rackham et al., 1982). Additionally, we observed that KTP-NH2 retained its high analgesic efficacy without the side effects of opioids, which is a desired feature in next-generation pain medications. No effects were detected as to motor capacities, blood pressure or tolerance after KTP-NH2 administration. Even after 7 days of daily injection, KTP-NH2 was still analgesic in the SNI neuropathic pain model, which does not occur with similar chronic injections of morphine, as its analgesic efficacy disappears, indicating the development of tolerance (Rougeot et al., 2010). Collectively, our data suggested that opioid pathways are somehow involved in KTP-NH2 activity, although the binding appears to be indirect. Molecularly, morphine interacts preferably with cholesterol-rich membranes (Huidobro-Toro et al., 1976), where opioid receptors are mainly concentrated (Xu et al., 2006). Our preliminary data regarding KTP-NH2 interaction with membrane model systems indicates a preference of this molecule for fluid, negatively charged, membranes (Figure S3), which indicates different profiles of interaction for these two compounds (morphine and KTP-NH2)and supports the hypothesis of distinct molecular modes of action. Therefore, these results suggest that KTP-NH2 may act by the same mechanisms demonstrated for KTP, namely, rapid degradation of KTP-NH2, resulting in the release of L-Arg, the substrate for NO synthase, which would then induce analgesia via met-enkephalin release (Arima et al., 1997). Indirect opioid actions may therefore explain the analgesic effects of KTP-NH2. Drug discovery groups in both academia and industry are struggling to find new pain relieving drugs. As the number of people suffering from pain keeps increasing, few novel molecular entities to fight pain have entered the clinic in the past 50 years, despite the increasing pharmaceutical investment (Woodcock et al., 2007; Melzack, 2008; Kissin, 2010). The remarkable feature about this new peptide, KTP-NH2, is its efficacy in several clinically relevant pain models. There are very few compounds effective in such a broad range of clinical situations. This clearly shows that KTP-NH2 has pharmacological interest for clinical development. Indeed, a patent on the application of KTP-NH2 for the treatment of pain has been already licensed (WO2009/123487 A1).
Acknowledgments
We thank Dr Nuno Oliveira for kindly providing V79 Chinese hamster lung fibroblast cells. The authors thank Dr A. Salomé Veiga, Dr Sónia T. Henriques and Dr Manuel Melo for support and helpful discussions. The authors acknowledge Dr Patrícia Calado and Dr Helena Vieira (Bioalvo, Portugal) for strategic discussions and scientific input provided. M. Martins-Oliveira is also acknowledged for technical support regarding CFA animal models treated with morphine.
Fundação para a Ciência e Tecnologia (Portugal) is acknowledged for funding (SFRH/BD/42158/2007 fellowship to MR and project PTDC/SAU-FCF/69493/2006). Marie Curie Industry-Academia Partnerships and Pathways (European Commission) is also acknowledged for funding (FP7-PEOPLE-2007-3-1-IAPP. project 230654).
Glossary
Abbreviations
- BBB
blood–brain barrier
- BOP
benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphoniumfluorophosphate
- CFA
complete Freund's adjuvant
- DMF
N,N-dimethylformamide
- HRMS-ESI
high-resolution mass spectra under electrospray ionization
- i.t.
intrathecal
- KTP
kyotorphin
- KTP-NH2
kyotorphin-amide
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NMM
N-methylmorpholine
- NOP
N/OFQ opioid receptor
- PBS
phosphate-buffered saline
- SNI
spared nerve injury
Conflicts of interest
MMB Ribeiro, M Pinto, M Heras, A Correia, E Bardaji, I Tavares and MARB Castanho are named as inventors in the patent WO2009/123487 A1.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 KTP-NH2 administration induced no blood pressure alterations.
Figure S2 KTP-NH2 effect on motor coordination.
Figure S3 KTP-NH2 interaction with membrane model systems.
Table S1 Absent toxicity following a one week KTP-NH2 daily administration (3.23 mg/100 bw)
Table S2 Effects of KTP-NH2 on HeLa, Huh-7 and V-79 cell viability
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (4th edn.) 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima T, Kitamura Y, Nishiya T, Taniguchi T, Takagi H, Nomura Y. Effects of kyotorphin (L-tyrosyl-L-arginine) ON[3H]NG-nitro-L-arginine binding to neuronal nitric oxide synthase in rat brain. Neurochem Int. 1997;30:605–611. doi: 10.1016/s0197-0186(96)00098-8. [DOI] [PubMed] [Google Scholar]
- Butler SH, Godefroy F, Besson JM, Weil-Fugazza J. A limited arthritic model for chronic pain studies in the rat. Pain. 1992;48:73–81. doi: 10.1016/0304-3959(92)90133-V. [DOI] [PubMed] [Google Scholar]
- Chen P, Bodor N, Wu WM, Prokai L. Strategies to target kyotorphin analogues to the brain. J Med Chem. 1998;41:3773–3781. doi: 10.1021/jm970715l. [DOI] [PubMed] [Google Scholar]
- Coggeshall RE. Fos, nociception and the dorsal horn. Prog Neurobiol. 2005;77:299–352. doi: 10.1016/j.pneurobio.2005.11.002. [DOI] [PubMed] [Google Scholar]
- D'Amour F, Smith D. A method for determining loss of pain sensation. J Pharmacol Exp Ter. 1941;72:74–79. [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Dubuisson D, Dennis SG. The formalin test: a quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain. 1977;4:161–174. doi: 10.1016/0304-3959(77)90130-0. [DOI] [PubMed] [Google Scholar]
- Haseto S, Ouchi H, Isoda T, Mizuma T, Hayashi M, Awazu S. Transport of low and high molecular peptides across rabbit Peyer's patches. Pharm Res. 1994;11:361–364. doi: 10.1023/a:1018948617587. [DOI] [PubMed] [Google Scholar]
- Huidobro-Toro JP, Canessa M, Fischer S. Interaction of morphine with cholesterol monolayers. Biochim Biophys Acta. 1976;436:237–241. doi: 10.1016/0005-2736(76)90234-0. [DOI] [PubMed] [Google Scholar]
- Kissin I. The development of new analgesics over the past 50 years: a lack of real breakthrough drugs. Anesth Analg. 2010;110:780–789. doi: 10.1213/ANE.0b013e3181cde882. [DOI] [PubMed] [Google Scholar]
- Kolaeva SG, Semenova TP, Santalova IM, Moshkov DA, Anoshkina IA, Golozubova V. Effects of L-thyrosyl – L-arginine (kyotorphin) on the behavior of rats and goldfish. Peptides. 2000;21:1331–1336. doi: 10.1016/s0196-9781(00)00275-8. [DOI] [PubMed] [Google Scholar]
- Lopes SC, Soares CM, Baptista AM, Goormaghtigh E, Cabral BJ, Castanho MA. Conformational and orientational guidance of the analgesic dipeptide kyotorphin induced by lipidic membranes: putative correlation toward receptor docking. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2006;110:3385–3394. doi: 10.1021/jp053651w. [DOI] [PubMed] [Google Scholar]
- Machuqueiro M, Baptista AM. The pH-dependent conformational states of kyotorphin: a constant-pH molecular dynamics study. Biophys J. 2007;92:1836–1845. doi: 10.1529/biophysj.106.092445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins I, Costa-Araujo S, Fadel J, Wilson SP, Lima D, Tavares I. Reversal of neuropathic pain by HSV-1-mediated decrease of noradrenaline in a pain facilitatory area of the brain. Pain. 2010;151:137–145. doi: 10.1016/j.pain.2010.06.027. [DOI] [PubMed] [Google Scholar]
- Melzack R. The future of pain. Nat Rev Drug Discov. 2008;7:629. doi: 10.1038/nrd2640. [DOI] [PubMed] [Google Scholar]
- Ottoni EB. EthoLog 2.2: a tool for the transcription and timing of behavior observation sessions. Behav Res Methods Instrum Comput. 2000;32:446–449. doi: 10.3758/bf03200814. [DOI] [PubMed] [Google Scholar]
- Pan B, Castro-Lopes JM, Coimbra A. Chemical sensory deafferentation abolishes hypothalamic pituitary activation induced by noxious stimulation or electroacupuncture but only decreases that caused by immobilization stress. A c-fos study. Neuroscience. 1997;78:1059–1068. doi: 10.1016/s0306-4522(96)00661-6. [DOI] [PubMed] [Google Scholar]
- Patti CL, Frussa-Filho R, Silva RH, Carvalho RC, Kameda SR, Takatsu-Coleman AL, et al. Behavioral characterization of morphine effects on motor activity in mice. Pharmacol Biochem Behav. 2005;81:923–927. doi: 10.1016/j.pbb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Pinto M, Lima D, Castro-Lopes J, Tavares I. Noxious-evoked c-fos expression in brainstem neurons immunoreactive for GABAB, mu-opioid and NK-1 receptors. Eur J Neurosci. 2003;17:1393–1402. doi: 10.1046/j.1460-9568.2003.02586.x. [DOI] [PubMed] [Google Scholar]
- Rackham A, Wood PL, Hudgin RL. Kyotorphin (tyrosine-arginine): further evidence for indirect opiate receptor activation. Life Sci. 1982;30:1337–1342. doi: 10.1016/0024-3205(82)90017-0. [DOI] [PubMed] [Google Scholar]
- Rougeot C, Robert F, Menz L, Bisson JF, Messaoudi M. Systemically active human opiorphin is a potent yet non-addictive analgesic without drug tolerance effects. J Physiol Pharmacol. 2010;61:483–490. [PubMed] [Google Scholar]
- Shiomi H, Ueda H, Takagi H. Isolation and identification of an analgesic opioid dipeptide kyotorphin (Tyr-Arg) from bovine brain. Neuropharmacology. 1981;20:633–638. doi: 10.1016/0028-3908(81)90109-x. [DOI] [PubMed] [Google Scholar]
- Takagi H, Shiomi H, Ueda H, Amano H. Morphine-like analgesia by a new dipeptide, L-tyrosyl-L-arginine (Kyotorphin) and its analogue. Eur J Pharmacol. 1979a;55:109–111. doi: 10.1016/0014-2999(79)90154-7. [DOI] [PubMed] [Google Scholar]
- Takagi H, Shiomi H, Ueda H, Amano H. A novel analgesic dipeptide from bovine brain is a possible Met-enkephalin releaser. Nature. 1979b;282:410–412. doi: 10.1038/282410a0. [DOI] [PubMed] [Google Scholar]
- Tavares I, Almeida A, Albino-Teixeira A, Lima D. Lesions of the caudal ventrolateral medulla block the hypertension-induced inhibition of noxious-evoked c-fos expression in the rat spinal cord. Eur J Pain. 1997;1:149–160. doi: 10.1016/s1090-3801(97)90073-2. [DOI] [PubMed] [Google Scholar]
- Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- Ueda H, Yoshihara Y, Misawa H, Fukushima N, Katada T, Ui M, et al. The kyotorphin (tyrosine-arginine) receptor and a selective reconstitution with purified Gi, measured with GTPase and phospholipase C assays. J Biol Chem. 1989;264:3732–3741. [PubMed] [Google Scholar]
- Ueda H, Yoshihara Y, Takagi H. A putative met-enkephalin releaser, kyotorphin enhances intracellular Ca2+ in the synaptosomes. Biochem Biophys Res Commun. 1986;137:897–902. doi: 10.1016/0006-291x(86)91164-2. [DOI] [PubMed] [Google Scholar]
- Woodcock J, Witter J, Dionne RA. Stimulating the development of mechanism-based, individualized pain therapies. Nat Rev Drug Discov. 2007;6:703–710. doi: 10.1038/nrd2335. [DOI] [PubMed] [Google Scholar]
- Xu W, Yoon SI, Huang P, Wang YL, Chen CG, Chong PLG, et al. Localization of the kappa opioid receptor in lipid rafts. J Pharmacol Exp Ther. 2006;317:1295–1306. doi: 10.1124/jpet.105.099507. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.