

Abstract
BACKGROUND AND PURPOSE
Proteinase-activated receptor-2 (PAR2) is widely expressed in the CNS under normal physiological conditions. However, its potential role in modulating neuronal excitability and synaptic transmission remains to be determined. Here, we have investigated whether PAR2 activation modulates synaptic activity in the hippocampus.
EXPERIMENTAL APPROACH
PAR2 activation and its effect on the hippocampus were examined in rat primary cultures and acute slices using whole cell patch clamp and standard extracellular recordings, respectively.
KEY RESULTS
PAR2 activation leads to a depolarization of hippocampal neurones and a paradoxical reduction in the occurrence of synaptically driven spontaneous action potentials (APs). PAR2-induced neuronal depolarization was abolished following either the inhibition of astrocytic function or antagonism of ionotropic glutamate receptors whilst the PAR2-induced decrease in AP frequency was also reduced when astrocytic function was inhibited. Furthermore, when examined in acute hippocampal slices, PAR2 activation induced a profound long-term depression of synaptic transmission that was dependent on NMDA receptor activation and was sensitive to disruption of astrocytic function.
CONCLUSIONS AND IMPLICATIONS
These novel findings show that PAR2 activation indirectly inhibits hippocampal synaptic activity and indicate that these receptors may play an active role in modulating normal physiological CNS function, in addition to their role in pathophysiological disorders.
Keywords: proteinase-activated receptor-2, hippocampus, astrocyte, NMDA, neuronal excitability, synaptic transmission
Introduction
Proteinase-activated receptors (PARs) are a novel class of G-protein coupled receptors that are unique in their activation, whereby the cleavage of the N-terminus by a serine proteinase liberates a sequence that acts as a ‘tethered ligand’. The ‘tethered ligand’ then binds to the second extracellular loop of the receptor, leading to the activation of the receptor. To date, four members of the PAR family have been cloned (PAR1–4; nomenclature follows Alexander et al., 2009) with PAR1, 3 & 4 preferentially activated by thrombin, whereas trypsin and trypsin-like proteinases preferentially activate PAR2 (see Ossovskaya and Bunnett, 2004; Bushell, 2007; Luo et al., 2007). Within the CNS, the endogenous activators for PAR2 and the physiological significance of its activation remain unclear but potential candidates include tryptase, trypsinogen IV and neurotrypsin (Bushell, 2007).
Our knowledge of PAR function in the brain is largely based on work with PAR1 and these results have revealed astrocytic, microglial and neuronal actions (Luo et al., 2007). PAR1 receptors are involved in a range of pathophysiological conditions including HIV encephalitis, Parkinson's disease and stroke (Luo et al., 2007), whilst disruption of PAR1 affects emotional learning in mice (Almonte et al., 2007). In contrast to our knowledge of PAR1 function in the brain and to our extensive understanding of PAR2 function in the periphery, the functional role of PAR2 actions in the brain remains largely unexplored. Recent evidence from human tissue has implicated PAR2 in CNS disorders including Alzheimer's disease, HIV dementia and multiple sclerosis (Noorbakhsh et al., 2006; 2007; Afkhami-Goli et al., 2007). In each of these studies, an increase in PAR2 expression, similar to that observed in certain peripheral disease states, was evident with the receptor being proposed to play either a protective (neuronal) or degenerative (glial) role depending on the cell type in which increased expression occurs (Bushell, 2007). However, PAR2 is constitutively expressed, albeit at low levels, in both the human and rodent CNS under normal conditions (D'Andrea et al., 1998; Striggow et al., 2001; Riek-Burchardt et al., 2002; Bushell et al., 2006; Lohman et al., 2008), although it remains to be elucidated what role it plays in this situation. We have previously shown that PAR2 activation in hippocampal cultures elicits elevations in intracellular calcium ([Ca2+]i) through the Gq/PLC pathway (Bushell et al., 2006) and, recently, PAR2 activation has been shown to modulate epileptiform activity in vivo (Lohman et al., 2008) and produce deficits in motivational learning (Lohman et al., 2009). Therefore, the aim of the current study was to assess, at the cellular and synaptic level, the consequences of PAR2 activation in the CNS.
Methods
Primary hippocampal culture
All animal care and experimental procedures were in accordance with UK Home Office guidelines and approved by the University Ethics Committee. Primary hippocampal cultures were prepared as described previously (Greenwood et al., 2007). Briefly, 1- to 2-day-old Sprague Dawley rats were killed by cervical dislocation and decapitated. The hippocampi were removed and triturated, and the resulting cells were plated at a density of 3 × 105 cells·mL−1 onto 13 mm poly-L-lysine coated coverslips. Cultures were incubated in culture medium consisting of Neurobasal-A Medium (Invitrogen, UK) supplemented with 2% (v/v) B-27 (Invitrogen, UK) and 2 mM L-glutamine and maintained in a humidified atmosphere at 37°C/5% CO2 for 13–16 days in vitro (DIV). After 5 DIV, cytosine-D-arabinofuranoside (10 µM) was added to inhibit glial cell proliferation. Both electrophysiology and Ca2+ imaging experiments were performed on cells taken from at least three separate cultures obtained from different rats.
Hippocampal slice preparation
Hippocampal slices were prepared as previously described (McNair et al., 2006). Juvenile Wistar rats (16–24 days old) were killed by cervical dislocation and decapitation. Brains were rapidly removed and transferred into ice-cold (0–3°C), oxygenated (95% O2/5% CO2) ACSF of the following composition (in mM): NaCl 124, KCl 3, NaHCO3 26, NaH2PO4 1.25, MgSO4 1, d-glucose 10 and CaCl2 2. Parasagittal whole brain slices (400 µm) were cut using a vibratome and hippocampal regions dissected free from surrounding brain tissue before being transferred to a humidified interface-type holding chamber containing ACSF continuously bubbled with 95% O2/5% CO2. Slices were initially incubated at 35°C for 30 min, and then allowed to equilibrate for 1 h at room temperature before use.
Electrophysiology
Cultured neurones
Hippocampal cultures (DIV 13–16) were perfused with HEPES-buffered saline (HBS) composed of the following (in mM): NaCl 140, KCl 5, MgCl2 2, CaCl2 2, HEPES 10, d-glucose 10, pH 7.4, 310 mOsm. Glass microelectrodes of 3–5 MΩ resistance were pulled, fire-polished and filled with intracellular solution containing (in mM): KCl 150, MgCl2 1, CaCl2 1, HEPES 10, EGTA 0.5, Mg-ATP 3, GTP 0.3, pH 7.2, 290 mOsm. Spontaneous synaptic activity was recorded at room temperature from cultured hippocampal neurones using whole cell patch clamp in current clamp mode using an Axopatch 200B amplifier (Molecular Devices, USA), connected to a personal computer interfaced with Digidata 1322A interface (Molecular Devices, USA) and captured using pClamp9.0 software (Molecular Devices, USA). Neurones with an initial membrane potential more positive than −55 mV were not used and the membrane potential of all other recorded cells held at −65 mV. No capacitance and series resistance compensation were applied. All drugs were applied via the perfusate. Off-line analysis was performed using Mini analysis software (Synaptosoft, USA) and pClamp9.2 software (Molecular Devices, USA).
Acute slices
Slices were transferred into a semi-submerged recording chamber in a humidified and oxygenated (95% O2/5% CO2) atmosphere maintained at 30°C. Slices were continuously perfused with ACSF at a flow rate at 1–2 mL·min−1 with a bipolar stimulating electrode and recording electrode positioned in the CA1 stratum radiatum of the Schaffer-collateral pathway. Field excitatory postsynaptic potentials (fEPSP) were recorded in response to 0.05 Hz stimulation, with responses initially set to approximately 50% of maximum. All drugs were administered via addition to the perfusate. To induce long-term potentiation (LTP), 15 min of stable baseline was obtained before high-frequency stimulation (HFS, 100 Hz, 1 s) was applied. Recordings were made with an Axoclamp 2B amplifier (Molecular Devices, USA), with the fEPSP slope monitored and analysed online and re-analysed off-line using LTP ver0.96 (http://www.winltp.com, Anderson and Collingridge, 2007). For all slice experiments, n represents one slice per animal.
Ca2+ imaging
All imaging experiments were performed on a digital epifluorescence imaging system (WinFluor, J. Dempster, University of Strathclyde) mounted on a Nikon TE300 microscope using a 40× objective. Hippocampal cultures (DIV 13–16) were loaded with fura-2 AM (5 µM, 45–60 min, room temperature) prior to experiments. Ratiometric images (340/380 nm) were obtained from the somata of astrocytes every 300 ms. Experiments were performed on cultures continually perfused (1–2 mL·min−1) with HBS at room temperature. All compounds investigated were added via the perfusate. Cells were identified as astrocytes based on their morphological characteristics and their lack of response to high extracellular potassium (25 mM). Data were calculated as changes in fluorescence ratio and expressed as ΔF/F0.
Statistics
All data are expressed as mean ± SEM. Data were compared by paired or unpaired Student's t-tests, or one-way analysis of variance with Tukey's comparison as appropriate. Differences were considered significant when P < 0.05.
Materials
All salts were obtained from VWR (UK). (S)-dihydroxyphenylglycine [(S)-DHPG], racemic (2R)-amino-5-phosphonovaleric acid [(DL)-AP5], (S)-α-methyl-4-carboxyphenylglycine [(S)-MCPG], 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) and tetrodotoxin (TTX) were obtained from Ascent Scientific (UK), sodium fluoroacetate (FAc) from Sigma-Aldrich (UK) and Ro-25–6981 from Tocris (UK). The PAR peptides, SLIGRL, LRGILS and 2-furoyl-LIGRL (2f-LIGRL) were obtained from both the Peptide Synthesis Facility at the University of Calgary (Canada) (http://wcm2.ucalgary.ca/peptides) or Peptide Synthetics (UK) (http://www.peptidesynthetics.co.uk) with all three peptides from both sources used in experiments. These peptides were carboxy terminal amidated but are presented solely as the amino acid sequence for clarity.
Results
PAR2 activation depolarizes cultured hippocampal neurones and reduces synaptically driven spontaneous action potential frequency
Application of the selective PAR2 activating peptide, SLIGRL (1–100 µM), a concentration range previously shown to induce a near maximal intracellular Ca2+ increase in neurones and astrocytes (Bushell et al., 2006), induced a reversible and concentration-dependent depolarization of cultured hippocampal neurones (1 µM, 0.7 ± 0.7 mV, n = 13; 10 µM, 1.7 ± 0.3 mV, n = 11; 100 µM, 4.7 ± 0.9 mV, n = 16, P < 0.05, Figure 1A–C). The effect of SLIGRL reveals an unusually steep concentration–response relationship but is similar to that seen previously in our laboratory in Ca2+ imaging studies (Bushell et al., 2006). In contrast to SLIGRL, application of the PAR2 inactive control peptide, LRGILS (100 µM) produced no significant change in membrane potential in all neurones tested (0.9 ± 0.03 mV, n = 8, P > 0.05, Figure 1B,C).
Figure 1.

Proteinase-activated receptor-2 (PAR2) activation depolarizes neuronal membrane potential in hippocampal cultures. (A) Representative trace illustrating the effect of PAR2 activation by SLIGRL (100 µM). (B) SLIGRL depolarized the membrane potential of cultured hippocampal neurones in a concentration-dependent manner whereas the reverse peptide was without effect. (C) Summary of the effect of PAR2 activation on neuronal membrane potential in hippocampal cultures. *P < 0.05.
Recent studies have indicated that the release of glutamate from astrocytes may account for the neuronal effects observed following PAR1 activation. Hence we investigated whether activation of ionotropic glutamate receptors is involved in the SLIGRL-induced depolarization. In the presence of (DL)-AP5 (100 µM) alone, SLIGRL induced a significant but reduced depolarization (3.0 ± 0.4 mV, n = 5, P > 0.05 compared to SLIGRL alone, Figure 1C) whereas in the presence of (DL)-AP5 (100 µM) and NBQX (20 µM), which completely prevented the occurrence of spontaneous action potentials (APs), the SLIGRL-induced depolarization was abolished (1.0 ± 0.8 mV, n = 8, P < 0.05 compared to SLIGRL alone, Figure 1C). However, when applied in the presence of TTX (1 µM), which also abolished the occurrence of spontaneous APs, SLIGRL still induced a significant depolarization (3.3 ± 0.6 mV, n = 8, P > 0.05 compared to SLIGRL alone, Figure 1C). In addition to the observed depolarization, a reversible reduction of spontaneous AP frequency was also observed upon SLIGRL (100 µM) application (70.4 ± 5.0% decrease, n = 13, P < 0.01, Figure 2A,B). LRGILS (100 µM) was without effect (7.7 ± 9.9% decrease, n = 8, P > 0.05, Figure 2B) whereas in the presence of (DL)-AP5 (100 µM) SLIGRL induced a reduced but significant reduction in AP frequency (47.8 ± 8.6% decrease, n = 5, P > 0.05 compared to SLIGRL alone, Figure 2B). In addition, manual manipulation of the membrane potential by current injection to levels similar to that observed in the presence of SLIGRL (−65 mV to −60 mV) resulted in an increase in AP frequency (120.9 ± 24.9% increase, n = 15, P < 0.001, Figure 2B).
Figure 2.

Proteinase-activated receptor-2 (PAR2) activation reduces action potential frequency in hippocampal cultures. (A) SLIGRL (100 µM) significantly reduces spontaneous action potential (AP) frequency whereas no reduction is observed following LRGILS application. (B) Summary of the effect of PAR2 activation in the absence and presence of (DL)-AP5 and manual depolarization to −60mV from −65mV on AP frequency. **P < 0.01; ***P < 0.001.
Astrocytic function and PAR2-induced neuronal effects
As glutamate antagonists abolished the PAR2-induced depolarization, we investigated further the source of the glutamate utilizing FAc, which impairs astrocytic function by being taken up and metabolized by astrocytes and inhibiting the Krebs cycle enzyme aconitase (Fonnum et al., 1997). This glial toxin has been widely employed to specifically reduce astrocytic function in a range of studies (Zhang et al., 2003; Shigetomi et al., 2008; Okada-Ogawa et al., 2009; Henneberger et al., 2010). We thus investigated the role of glia in the SLIGRL-mediated effects by pretreating cultures with FAc (10 µM) for 3 h as has been described previously (Shigetomi et al., 2008). Under these conditions, the spontaneous AP frequency was unaffected when compared to control cultures (control 1.23 ± 0.08 Hz, n = 16, FAc treated 1.30 ± 0.10 Hz, n = 8, P > 0.05) strongly indicating that FAc is not toxic to the cultures per se. However, the SLIGRL (100 µM)-induced depolarization was abolished (0.5 ± 0.4 mV, n = 6, P < 0.05 compared to no FAc, Figure 3A,C) in hippocampal cultures pretreated with FAc (10 µM, 3 h). In contrast, application of SLIGRL still produced a reduction in spontaneous AP frequency (44.3 ± 8.1% decrease, n = 8, P < 0.05; Figure 3B,C) but this was significantly reduced compared to the suppression seen in the absence of FAc treatment (P < 0.01 compared to no FAc). Using calcium imaging to monitor intracellular calcium levels ([Ca2+]i), SLIGRL produced a pronounced rise in astrocytic [Ca2+]i (149.4 ± 8.3% peak increase, n = 22, P < 0.001, Figure 3D) consistent with previous reports (Wang et al., 2002; Bushell et al., 2006). To confirm that FAc reduced astrocytic function, Ca2+ imaging experiments were performed on hippocampal cultures treated with FAc (10 µM, 3 h). Under these conditions, SLIGRL-induced elevations in astrocytic [Ca2+]i were significantly reduced, compared with control (76.1 ± 12.1% peak increase, n = 25, P < 0.001 compared to no FAc, Figure 3D). In contrast, SLIGRL-induced increases in astrocytic [Ca2+]i were not significantly altered in the presence of the glutamate antagonists mentioned above (126 ± 10% increase, P > 0.05 compared to no antagonists).
Figure 3.

Impairment of astrocytic activity reduced the modulatory effects of proteinase-activated receptor-2 (PAR2) activation. (A) Pre-incubation of cultures with fluoroacetate (FAc; 10 µM, 3 h) abolished the SLIGRL (100 µM)-induced depolarization compared with control. (B) FAc pre-incubation reduced the decrease in action potential (AP) frequency observed following PAR2 activation compared to control. (C) Summary of the effects of FAc on neuronal membrane potential and AP frequency. (D) FAc pre-incubation significantly reduced the SLIGRL-induced increase in [Ca2+]i in astrocytes compared with control. *P < 0.05; **P < 0.01.
Activation of PAR2 induces a long-lasting reduction in synaptic transmission in hippocampal slices
Having established the consequences of SLIGRL application in hippocampal cultures, we next investigated whether PAR2 activation modulates synaptic transmission in acute hippocampal slices. Surprisingly, SLIGRL (100 µM, 30 min) induced a profound long-term depression (LTD) of synaptic transmission at the Schaffer-collateral to CA1 pyramidal cells synapse (19.3 ± 2.5%, n = 12, P < 0.01, Figure 4A) whilst application of LRGILS (100 µM, Figure 4A) was without effect. Furthermore, another activator of PAR2, 2f-LIGRL (100 µM, 30 min), also induced an LTD of synaptic transmission (24.1 ± 4.9%, n = 9, P < 0.05, Figure 4A). The robust SLIGRL-induced form of synaptic depression was unlikely to be due to pathological changes as no alteration in fibre volley amplitude was observed (Figure 4A). Furthermore, the depression of synapses could be reversed by subsequent delivery of HFS (149.4 ± 2.8%, n = 10), the relative amplitude and time course of which was indistinguishable from responses to HFS seen in untreated naïve slices (147.9 ± 3.9%, n = 9, Figure 4B). With astrocytes being shown to contribute to the PAR2-mediated effects seen in hippocampal cultures, their role in SLIGRL-induced LTD was investigated. Exposure of hippocampal slices to FAc (10 µM, 3 h) had no significant effect on basal synaptic transmission when investigated using input–output curves (Figure 5B), which, in agreement with that found in cultures, indicates that FAc is not toxic to the slices under our experimental conditions. However, following the exposure of hippocampal slices to FAc (10 µM, 3 h), SLIGRL (100 µM) induced a small (7.6 ± 1.5%, n = 6), but significantly attenuated long-lasting reduction in synaptic transmission (LTD) compared with untreated slices (P < 0.05 compared to untreated slices, Figure 5A,D). As NMDA receptors are required for LTD induced by low-frequency stimulation at this synapse, we investigated their contribution to SLIGRL-induced LTD. The NMDA receptor antagonist (DL)-AP5 (100 µM) abolished SLIGRL-induced LTD [1.1 ± 1.3%, n = 9, P < 0.01 compared with no (DL)-AP5, Figure 5A,D]. Moreover, the GluN2B-selective, NMDA receptor antagonist, Ro 25-6981 (3 µM) also abolished the SLIGRL-induced LTD (0.5 ± 3.1%, n = 5, P < 0.05 compared to no Ro 25-6981, Figure 5D). In contrast, SLIGRL application induced robust LTD (25.7 ± 2.2%, n = 8, P < 0.01) in the presence of the metabotropic glutamate (mGlu) receptor antagonist (S)-MCPG (500 µM, Figure 5A,D), suggesting a lack of mGlu receptor involvement in the PAR2 response. Furthermore, induction of LTD with the group I mGlu receptor agonist DHPG, (50 µM, 10 min, 43.8 ± 1.9%, n = 4, Figure 5C) did not prevent further LTD induced by subsequent application of SLIGRL (100 µM, 19.8 ± 1.5%, n = 4, Figure 5C,D). This depression was additive to that induced by DHPG and not significantly different from that seen in naïve slices (P > 0.05 compared with SLIGRL-LTD in naïve slices).
Figure 4.

Proteinase-activated receptor-2 (PAR2) activation induced long-term depression in acute hippocampal slices. (A) PAR2 activation by SLIGRL (100 µM) and 2f-LIGRL (100 µM) leads to a significant long-lasting reduction in synaptic transmission with no effect on fibre volley amplitude. Representative traces for SLIGRL application are shown for the time points indicated. (B) High-frequency stimulation (at arrow) readily induced long-term potentiation in slices following SLIGRL application, which was comparable with the long-term potentiation induced in naïve slices.
Figure 5.
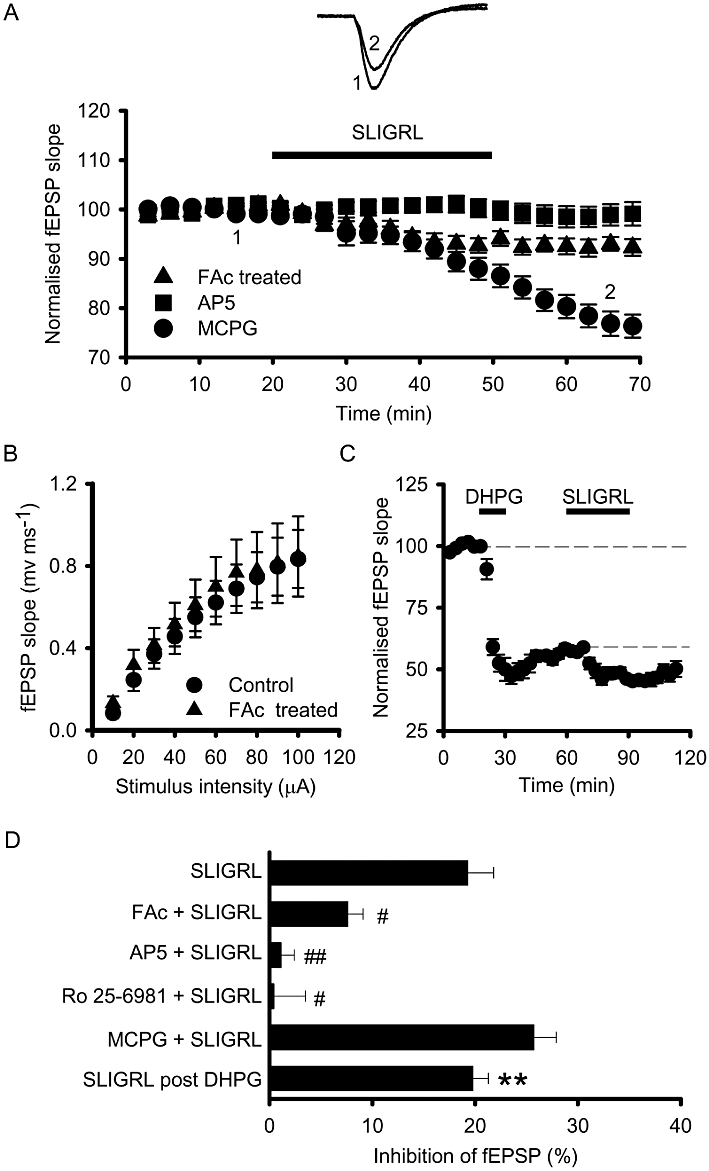
SLIGRL-induced long-term depression (LTD) involves astrocytic and NMDA receptor activation. (A) SLIGRL (100 µM)-induced LTD was abolished by (DL)-AP5 and reduced by FAc pretreatment but unaffected by (S)-MCPG. (B) Input–output curves are unaffected by pretreatment with FAc (10 µM, 3 h). (C) DHPG (50 µM)-induced LTD did not occlude SLIGRL-induced LTD. (D) Summary of the effects of proteinase-activated receptor-2 activation in acute hippocampal slices. **P < 0.01, #P < 0.05 compared to SLIGRL alone, ##P < 0.01 compared to SLIGRL alone.
Discussion
In the present study, we show for the first time that activation of PAR2 lead to changes in neuronal excitability and synaptic transmission in both hippocampal cultures and acute slices. Furthermore, we provide evidence that NMDA receptors contributs to these PAR2-mediated effects and that the effects were sensitive to the disruption of astrocytic function.
Dual action of PAR2 activation
We have shown that PAR2 activation causes a significant depolarization of hippocampal neurones as well as a profound suppression of evoked glutamatergic synaptic transmission. The apparent paradoxical effect in the neuronal culture system whereby a depolarization is associated with a reduction in the occurrence of spontaneous APs is explained by the fact that the majority of neuronal discharge in this preparation is driven by ongoing synaptic activity. Thus, at a network level, the suppression of synaptic transmission dampens network activity and thus constitutes the dominant response. Furthermore, whilst the stimulation of PAR2 by the activating peptide produced a relatively transient depolarization of hippocampal neurones, the same activating peptide induced an enduring suppression of synaptic transmission which appeared as a form of long-lasting synaptic depression (LTD). Using ionotropic glutamate receptor antagonists and by impairing astrocytic activity with FAc, we showed that intact astrocytic function and the activation of glutamate receptors are required for the full expression of these PAR2-mediated effects. The reduction in synaptic transmission observed in both primary cultures and acute slices following SLIGRL application would be expected to protect against glutamate toxicity under conditions of hyperexcitability or trauma. Indeed, this is supported by our recent finding that PAR2 activation protects against neurotoxicity induced by kainic acid (Greenwood and Bushell, 2010). Furthermore, PAR2 activation also protects against seizures and epileptogenesis (Lohman et al., 2008). In addition to regulating pathological processes, PAR2-mediated inhibition of synaptic transmission and alteration of network dynamics may also contribute to physiological processes such as the recently described regulation of motivational learning (Lohman et al., 2009). With recent evidence implicating changes in PAR2 expression in CNS disorders including Alzheimer's disease, HIV dementia and multiple sclerosis (Noorbakhsh et al., 2006; 2007; Afkhami-Goli et al., 2007), the findings of the present study suggest that further investigation is required to identify the exact role of PAR2 in CNS function under both physiological as well as pathophysiological conditions. Whilst changes in the expression of PAR2 are known to occur in the latter (see Bushell, 2007), key unanswered questions remain where, when and under what conditions PAR2 are activated.
PAR2 activation may involve complex signalling
PAR2 receptors have been shown to be expressed in both astrocytes and neurones (D'Andrea et al., 1998; Striggow et al., 2001; Riek-Burchardt et al., 2002; Bushell et al., 2006; Lohman et al., 2008) and couple through a number of second messenger pathways. In addition to this complexity, however, the current findings suggest an important role of ionotropic glutamate receptor activation in both the membrane depolarization and in the suppression of synaptic transmission. Specifically, these data suggest an essential role for the NMDA subtype of receptor with PAR2-mediated effects being reduced in cultures and slices by (DL)-AP5 and the subunit selective antagonist Ro 25-6981, suggesting a role for receptors containing GluN2B subunits in particular. Although the role of NMDA subtypes in LTD induction is controversial (MacDonald et al., 2006; Li et al., 2007; Kollen et al., 2008; Brigman et al., 2010; Peng et al., 2010), it is clear that GluN2B-containing NMDA receptors played a key role in the PAR2-induced form of LTD reported here. In addition to the essential requirement for NMDA receptor activation, the finding that SLIGRL-induced LTD was insensitive to mGlu receptor antagonism (or mGlu agonist occlusion) is important in distinguishing the enduring synaptic depression induced by PAR2 activation from other forms of long-lasting synaptic depression at this synapse (Gladding et al., 2009; Lüscher and Huber, 2010).
So what is the source of the glutamate? One possibility is that a modest depolarization of the principal neurones by PAR2 activation produces a release of glutamate through recurrent circuits which then results in the activation of NMDA receptors and the suppression of synaptic transmission. We believe this is unlikely due to a number of reasons. Firstly, the depolarization is itself dependent on glutamatergic transmission (Figure 1) and likely to be indirect. Secondly, the experiments with TTX suggests that the requirement for glutamate is independent of fast APs and thus is unlikely to be neuronal in origin (Figure 1). Together, these factors suggest that PAR2 activation mediates its neuronal actions indirectly via activation of astrocytic PAR2 and the subsequent AP-independent release of glutamate. Astrocytic localization of PAR2 is well established in both culture preparations (Ubl et al., 1998; Wang et al., 2002; Bushell et al., 2006) and native tissues neuropil (Bushell et al., 2006; Hermann et al., 2009) and white matter (Noorbakhsh et al., 2006; Afkhami-Goli et al., 2007). Moreover, previous work has shown that another member of the PAR family, PAR1, mediates some of its effects indirectly via astrocytic signalling (Lee et al., 2007; Shigetomi et al., 2008). Our finding that impairment of astrocytic function using the established (Zhang et al., 2003; Shigetomi et al., 2008; Okada-Ogawa et al., 2009; Henneberger et al., 2010) glial toxin FAc significantly reduced the effects of SLIGRL both in terms of astrocytic calcium response, neuronal depolarization and altered in AP frequency (Figure 4) as well as synaptic depression (Figure 5) strongly indicates a similar role of astrocytic signalling in mediating the actions of PAR2. Moreover, that FAc did not affect AP frequency in cultured neurones or the stimulus intensity–response curves in acute slice experiments would argue against any confounding FAc neurotoxicity under our experimental conditions. Astrocytic activation via G-protein coupled receptors leading to the release of gliotransmitters, including glutamate, is well established (see Araque, 2008; Fiacco et al., 2009; Santello and Volterra, 2009) but the exact role of these gliotransmitters on neuronal function remains an area of debate (see Fiacco et al., 2007; Lee et al., 2007; Shigetomi et al., 2008; Agulhon et al., 2010). Whilst the current data support a role for astrocytes and NMDA receptors in mediating the actions of PAR2 activation, a direct effect of SLIGRL on neurones could potentially account for some of the effects observed here. However, the cultured neurones form a highly active network making it difficult to scrutinize the action of PAR2 activation on intrinsic properties. In the absence of input resistance data, it is not possible to determine whether the activation of depolarizing currents or the suppression of hyperpolarizing currents are responsible for the transient intrinsic response. Nevertheless, the transient nature of the cellular level depolarization compared to the enduring synaptic depression suggests that different mechanisms are at play.
In conclusion, we have demonstrated for the first time that PAR2 activation leads to robust long-lasting suppression of synaptic transmission and provide evidence that these effects are mediated, at least in part, by indirect astrocytic and NMDA receptor activation. These data, alongside recent in vivo studies (Lohman et al., 2008; 2009) provide strong evidence that PAR2 activation may be a novel target for the regulation of neuronal function not only in pathological disease states but under normal physiological conditions.
Acknowledgments
This work was funded by grants from Tenovus Scotland, Medical Research Scotland and the Wellcome Trust to TJB. JG was funded by a Synergy Scholarship and Overseas Research Student award (ORSAS award) by the Universities of Glasgow and Strathclyde.
Glossary
Abbreviations
- (DL)-AP5
racemic (2R)-amino-5-phosphonovaleric acid
- (S)-MCPG
(S)-α-methyl-4-carboxyphenylglycine
- 2f-LIGRL
2-furoyl-LIGRL
- AP
action potential
- DHPG
dihydroxyphenylglycine
- DIV
days in vitro
- FAc
sodium fluoroacetate
- fEPSP
field excitatory postsynaptic potentials
- HBS
HEPES-buffered saline
- HFS
high-frequency stimulation
- LTD
long-term depression
- LTP
long-term potentiation
- NBQX
2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione
- PAR
proteinase-activated receptor
- TTX
tetrodotoxin
Conflicts of interest
To the best of our knowledge, there are no conflicts of interest regarding the content of this manuscript.
Supporting Information
Teaching Materials; Figs 1–5 as PowerPoint slide.
References
- Afkhami-Goli A, Noorbakhsh F, Keller AJ, Vergnolle N, Westaway D, Jhamandas JH, et al. Proteinase-activated receptor-2 exerts protective and pathogenic cell type-specific effects in Alzheimer's disease. J Immunol. 2007;179:5493–5503. doi: 10.4049/jimmunol.179.8.5493. [DOI] [PubMed] [Google Scholar]
- Agulhon C, Fiacco TA, McCarthy KD. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signalling. Science. 2010;327:1250–1254. doi: 10.1126/science.1184821. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almonte AG, Hamill CE, Chhatwal JP, Wingo TS, Barber JA, Lyuboslavsky PN, et al. Learning and memory deficits in mice lacking protease activated receptor-1. Neurobiol Learn Mem. 2007;88:295–304. doi: 10.1016/j.nlm.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson WW, Collingridge GL. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J Neurosci Methods. 2007;162:346–356. doi: 10.1016/j.jneumeth.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Araque A. Astrocytes process synaptic information. Neuron Glia Biol. 2008;4:3–10. doi: 10.1017/S1740925X09000064. [DOI] [PubMed] [Google Scholar]
- Brigman JL, Wright T, Talani G, Prasad-Mulcare S, Jinde S, Seabold GK, et al. Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J Neurosci. 2010;30:4590–4600. doi: 10.1523/JNEUROSCI.0640-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell TJ. The emergence of proteinase-activated receptor-2 as a novel target for the treatment of inflammation-related CNS disorders. J Physiol. 2007;581:7–16. doi: 10.1113/jphysiol.2007.129577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell TJ, Plevin R, Cobb S, Irving AJ. Characterization of proteinase-activated receptor 2 signalling and expression in rat hippocampal neurons and astrocytes. Neuropharmacology. 2006;50:714–725. doi: 10.1016/j.neuropharm.2005.11.024. [DOI] [PubMed] [Google Scholar]
- D'Andrea MR, Derian CK, Leturcq D, Baker SM, Brunmark A, Ling P, et al. Characterization of protease-activated receptor-2 immunoreactivity in normal human tissues. J Histochem Cytochem. 1998;46:57–64. doi: 10.1177/002215549804600204. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, Taves SR, Petravicz J, Casper KB, Dong X, et al. Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron. 2007;54:611–626. doi: 10.1016/j.neuron.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, McCarthy KD. Sorting out astrocyte physiology from pharmacology. Annu Rev Pharmacol Toxicol. 2009;49:151–174. doi: 10.1146/annurev.pharmtox.011008.145602. [DOI] [PubMed] [Google Scholar]
- Fonnum F, Johnsen A, Hassel B. Use of fluorocitrate and fluoroacetate in the study of brain metabolism. Glia. 1997;21:106–113. [PubMed] [Google Scholar]
- Gladding CM, Fitzjohn SM, Molnár E. Metabotropic glutamate receptor-mediated long-term depression: molecular mechanisms. Pharmacol Rev. 2009;61:395–412. doi: 10.1124/pr.109.001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood SM, Bushell TJ. Astrocytic activation and an inhibition of MAP kinases are required for proteinase-activated receptor-2-mediated protection from neurotoxicity. J Neurochem. 2010;113:1471–1480. doi: 10.1111/j.1471-4159.2010.06737.x. [DOI] [PubMed] [Google Scholar]
- Greenwood SM, Mizielinska SM, Frenguelli BG, Harvey J, Connolly CN. Mitochondrial dysfunction and dendritic beading during neuronal toxicity. J Biol Chem. 2007;282:26235–26244. doi: 10.1074/jbc.M704488200. [DOI] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of d-serine from astrocytes. Nature. 2010;463:232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Van Meter MJ, Rood JC, Rogers RC. Proteinase-activated receptors in the nucleus of the solitary tract: evidence for glial–neural interactions in autonomic control of the stomach. J Neurosci. 2009;29:9292–9300. doi: 10.1523/JNEUROSCI.6063-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollen M, Dutar P, Jouvenceau A. The magnitude of hippocampal long term depression depends on the synaptic location of activated NR2-containing N-methyl-D-aspartate receptors. Neuroscience. 2008;154:1308–1317. doi: 10.1016/j.neuroscience.2008.04.045. [DOI] [PubMed] [Google Scholar]
- Lee CJ, Mannaioni G, Yuan H, Woo DH, Gingrich MB, Traynelis SF. Astrocytic control of synaptic NMDA receptors. J Physiol. 2007;581:1057–1081. doi: 10.1113/jphysiol.2007.130377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Huang FS, Abbas AK, Wigström H. Role of NMDA receptor subtypes in different forms of NMDA-dependent synaptic plasticity. BMC Neurosci. 2007;8:55. doi: 10.1186/1471-2202-8-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman RJ, O'Brien TJ, Cocks TM. Protease-activated receptor-2 regulates trypsin expression in the brain and protects against seizures and epileptogenesis. Neurobiol Dis. 2008;30:84–93. doi: 10.1016/j.nbd.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Lohman RJ, Jones NC, O'Brien TJ, Cocks TM. A regulatory role for protease-activated receptor-2 in motivational learning in rats. Neurobiol Learn Mem. 2009;92:301.9. doi: 10.1016/j.nlm.2009.03.010. [DOI] [PubMed] [Google Scholar]
- Luo W, Wang Y, Reiser G. Protease-activated receptors in the brain: receptor expression, activation, and functions in neurodegeneration and neuroprotection. Brain Res Rev. 2007;56:331–345. doi: 10.1016/j.brainresrev.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JF, Jackson MF, Beazely MA. Hippocampal long-term synaptic plasticity and signal amplification of NMDA receptors. Crit Rev Neurobiol. 2006;18:71–84. doi: 10.1615/critrevneurobiol.v18.i1-2.80. [DOI] [PubMed] [Google Scholar]
- McNair K, Davies CH, Cobb SR. Plasticity-related regulation of the hippocampal proteome. Eur J Neurosci. 2006;23:575–580. doi: 10.1111/j.1460-9568.2005.04542.x. [DOI] [PubMed] [Google Scholar]
- Noorbakhsh F, Vergnolle N, McArthur JC, Silva C, Vodjgani M, Andrade-Gordon P, et al. Proteinase-activated receptor-2 induction by neuroinflammation prevents neuronal death during HIV infection. J Immunol. 2006;174:7320–7329. doi: 10.4049/jimmunol.174.11.7320. [DOI] [PubMed] [Google Scholar]
- Noorbakhsh F, Tsutsui S, Vergnolle N, Boven LA, Shariat N, Vodjgani M, et al. Proteinase-activated receptor 2 modulates neuroinflammation in experimental autoimmune encephalomyelitis and multiple sclerosis. J Exp Med. 2007;203:425–435. doi: 10.1084/jem.20052148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada-Ogawa A, Suzuki I, Sessle BJ, Chiang CY, Salter MW, Dostrovsky JO, et al. Astroglia in medullary dorsal horn (trigeminal spinal subnucleus caudalis) are involved in trigeminal neuropathic pain mechanisms. J Neurosci. 2009;29:11161–11171. doi: 10.1523/JNEUROSCI.3365-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev. 2004;84:579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- Peng Y, Zhao J, Gu QH, Chen RQ, Xu Z, Yan JZ, et al. Distinct trafficking and expression mechanisms underlie LTP and LTD of NMDA receptor-mediated synaptic responses. Hippocampus. 2010;20:646–658. doi: 10.1002/hipo.20654. [DOI] [PubMed] [Google Scholar]
- Riek-Burchardt M, Striggow F, Henrich-Noack P, Reiser G, Reymann KG. Increase of prothrombin-mRNA after global cerebral ischemia in rats, with constant expression of protease nexin-1 and protease-activated receptors. Neurosci Lett. 2002;329:181–184. doi: 10.1016/s0304-3940(02)00645-6. [DOI] [PubMed] [Google Scholar]
- Santello M, Volterra A. Synaptic modulation by astrocytes via Ca2+-dependent glutamate release. Neuroscience. 2009;158:253–259. doi: 10.1016/j.neuroscience.2008.03.039. [DOI] [PubMed] [Google Scholar]
- Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci. 2008;28:6659–6663. doi: 10.1523/JNEUROSCI.1717-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striggow F, Riek-Burchardt M, Kiesel A, Schmidt W, Henrich-Noack P, Breder J, et al. Four different types of protease-activated receptors are widely expressed in the brain and are up-regulated in hippocampus by severe ischemia. Eur J Neurosci. 2001;14:595–608. doi: 10.1046/j.0953-816x.2001.01676.x. [DOI] [PubMed] [Google Scholar]
- Ubl JJ, Vöhringer C, Reiser G. Co-existence of two types of [Ca2+]i-inducing protease-activated receptors (PAR1 and PAR2) in rat astrocytes and C6 glioma cells. Neuroscience. 1998;86:597–609. doi: 10.1016/s0306-4522(97)00686-6. [DOI] [PubMed] [Google Scholar]
- Wang H, Ubl JJ, Reiser G. Four subtypes of protease-activated receptors, co-expressed in rat astrocytes, evoke different physiological signalling. Glia. 2002;37:53–63. doi: 10.1002/glia.10012. [DOI] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, et al. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;4:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.