

Abstract
BACKGROUND AND PURPOSE
Nitric oxide (NO) plays an important role in endothelial function, and impaired NO production is involved in hypertension. Therefore, compounds that regulate endothelial NO synthase (eNOS) may be of therapeutic benefit. A novel, low molecular weight compound AVE3085 is a recently developed compound with the ability to enhance eNOS transcription. The present study investigated the effects of AVE3085 in endothelial dysfunction associated with hypertension.
EXPERIMENTAL APPROACH
Spontaneously hypertensive rats (SHRs) were treated with AVE 3085 (10 mg·kg·day−1, orally) for 4 weeks. Isometric force measurement was performed on rings of isolated aortae in organ baths. Protein expression of eNOS, phosphorylated-eNOS and nitrotyrosine in the aortae were examined by Western blotting. mRNA for eNOS in rat aortae were examined by reverse-transcriptase polymerase chain reaction (RT-PCR).
KEY RESULTS
AVE3085 greatly improved endothelium-dependent relaxations in the aortae of SHRs. This functional change was accompanied by up-regulated expression of eNOS protein and mRNA, enhanced eNOS phosphorylation and decreased formation of nitrotyrosine. Furthermore, AVE3085 treatment reduced the blood pressure in SHR without affecting that of hypertensive eNOS−/− mice.
CONCLUSIONS AND IMPLICATIONS
The eNOS-transcription enhancer AVE3085 restored impaired endothelial function in a hypertensive model. The present study provides a solid basis for the potential development of eNOS-targeting drugs to restore down-regulated eNOS, as a new strategy in hypertension.
Keywords: cardiovascular diseases, endothelium, nitric oxide, nitric oxide synthase, SHR
Introduction
The vascular endothelium plays a pivotal role in the pathophysiology of the cardiovascular system. Nitric oxide (NO), generated by endothelial NO synthase (eNOS), is physiologically important in vascular homeostasis. NO is a key regulator of endothelial cell growth (Ziche et al., 1994), migration (Murohara et al., 1999), vascular remodelling (Rudic et al., 1998) and angiogenesis (Ziche et al., 1994). In addition, NO protects against mitochondrial oxidative stress (Borniquel et al., 2006), and a crucial role is played by eNOS in the functional activity of stem and progenitor cells (Aicher et al., 2003).
eNOS is constitutively present in the endothelium, and its expression is regulated by various biophysical, biochemical and hormonal stimuli, under both physiological and pathological conditions. Physiological stimuli which up-regulate the eNOS expression include shear stress, growth factors and hormones such as oestrogens (Li et al., 2002; Searles, 2006; Chan et al., 2010). Stimulation of β-adrenoceptors can also increase the eNOS activity in cultured endothelial cells (Kou and Michel, 2007). Other compounds in clinical use are also able to increase the eNOS expression and activity, including statins (Laufs et al., 1998), angiotensin-converting enzyme inhibitors (Linz et al., 1999), angiotensin AT1 receptor antagonists and dihydropyridine calcium channel blockers (Ding and Vaziri, 1998; Leung et al., 2006).
Endothelial dysfunction is closely associated with disturbed NO production and eNOS regulation in vascular pathogenesis. For example, hypertension is associated with a decreased NO formation (Huang et al., 1995), while impaired bioavailability of NO is a characteristic found in patients with atherosclerosis (Zeiher et al., 1993). Serum amyloid A induces endothelial dysfunction involving eNOS down-regulation (Wang et al., 2008). Tumour necrosis factor (TNF)-α decreases eNOS expression at the post-transcriptional level (Yan et al., 2008). Likewise, soluble CD40 ligand (CD40L), a trimeric transmembrane protein and a member of the TNF family, also reduces the levels of eNOS mRNA and protein, stability of eNOS mRNA, eNOS activity and cellular NO content (Chen et al., 2008). Other pathological situations involving endothelial dysfunction include HIV-1 protein expression (Kline et al., 2008) and homocysteine imbalance (Zhou et al., 2006), together with the actions of variety of endogenous factors including the chemokine CXCL1 (growth-related oncogene-α;GRO-α), a member of the CXC chemokine family involved in the development of atherosclerosis (Bechara et al., 2007), C-reactive protein (Schwartz et al., 2007) and Rho-kinase (Shin et al., 2007). In some rare conditions such as chronic alcohol-induced hypertension (Husain et al., 2007) and neonatal persistent pulmonary hypertension (Gien et al., 2007), eNOS expression is also decreased.
AVE compounds were developed to enhance eNOS activity. AVE9488 has been reported to increase eNOS activity (Sasaki et al., 2006), and the combination of an up-regulated eNOS expression and a reversal of eNOS uncoupling are probably responsible for the observed anti-atherosclerotic action of AVE9488 (Wohlfart et al., 2008). Another new compound, AVE3085, was also found to increase eNOS levels and reverse vascular dysfunction and inflammation in the hindlimbs in a rat model of diabetes (Riad et al., 2008). In addition, we have demonstrated that AVE3085 is effective in protecting coronary artery endothelium against hypoxia–reoxygenation injury during cardioplegic exposure (Xue et al., 2010).
The present study aimed to investigate the actions of AVE3085 in ameliorating endothelial dysfunction in hypertension.
Methods
All animal care and experimental procedures were in accordance with institutional guidelines and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85–23, revised 1996).
Animals and drug treatment
Adult male spontaneously hypertensive rats (SHR) and normotensive Wistar Kyoto rats (WKY) (6 months old) were purchased from the Laboratory Animal Service Center, Chinese University of Hong Kong. Adult male eNOS−/− mice (4 months old) were from Johannes Gutenberg University, Mainz, Germany. Rats were assigned to four groups: SHR + AVE, SHR + vehicle, WKY + AVE and WKY + vehicle. In the AVE-treated groups, AVE3085 was administrated to rats by oral gavage once daily for 4 weeks (10 mg·kg·day−1), whilst in the non-treatment groups, SHR or WKY rats received vehicle (5% methylcellulose) daily for 4 weeks. Male eNOS−/− mice (n = 7) were treated for up to 8 weeks with AVE3085 in chow at a concentration of 150 mg·kg−1, resulting in a daily dose of about 30 mg·kg−1. A higher dose of AVE3085 was used for mice than in rats (10 mg·kg−1) because of the higher metabolic rate in mice than in rats (Rucker and Storms, 2002). Pilot studies showed that similar plasma exposure profiles were achieved when using the selected doses: 10 mg·kg−1 for rats and 30 mg·kg−1 for mice.
Systolic blood pressure measurement
Systolic blood pressure was measured by a non-invasive tail-cuff method. A pressure signal from the tail artery was detected by a pulse transducer relayed via a NIBP controller and a Powerlab, and recorded by Chart software (all from AD Instruments, Sydney, Australia). Pressure measurements were measured five times for each rat to obtain an average value.
Blood vessel preparation
The thoracic aorta was dissected out, cleaned of surrounding connective tissue and placed in freshly prepared ice-cold and oxygenated Krebs–Henseleit solution containing (mmol·L−1): 119 NaCl, 4.7 KCl, 2.5 CaCl2, 1 MgCl2, 25 NaHCO3, 1.2 KH2PO4 and 11 d-glucose, and then cut into ring segments (∼3 mm in length). Each ring was suspended between two stainless steel hooks in a 10 mL organ bath filled with Krebs solution. Bathing solution was continuously bubbled with 95% O2 and 5% CO2 and maintained at 37°C (pH of 7.3–7.5). An optimal baseline tone of 2 g was applied to all rings. Relaxations in response to acetylcholine (ACh) and to sodium nitroprusside (SNP) were examined in phenylephrine-contracted rings.
Primary culture of rat aortic endothelial cells
Aortae of WKY and SHR were dissected in sterilized phosphate-buffered saline (PBS). After digestion by 0.2% collagenase for 15 min at 37°C, RPMI-1640 (Gibco, Grand Island, NY, USA) was added, and endothelial cells were collected by centrifugation at 57×g for 5 min. Thereafter, the pellet was gently resuspended in RPMI-1640 supplemented with 10% fetal bovine serum and cultured in a 75 mm culture flask. To remove other cell types, the medium was changed after 1 h incubation and maintained until 70% confluence before use (Chan et al., 2000).
Western blot analysis of eNOS, phosphorylated-eNOS (p-eNOS) and nitrotyrosine
Aortae were isolated and frozen in liquid nitrogen and homogenized in an ice-cold radioimmunoprecipitation assay lysis buffer that contained 1 µg·mL−1 leupeptin, 5 µg·mL−1 aprotinin, 100 µg·mL−1 phenylmethylsulphonyl fluoride, 1 mmol·L−1 sodium orthovanadate, 1 mmol·L−1 EDTA, 1 mmol·L−1 EGTA, 1 mmol·L−1 sodium fluoride and 2 µg·mL−1β-glycerolphosphate. Tissue or cell lysates were centrifuged at 20 000×g for 20 min. The supernatants were collected, and protein concentrations were analyzed using the Lowry method (Bio-rad, Hercules, CA). Protein samples were separated by electrophoresis on a 10% SDS-poly-acrylamide gel and transferred onto an immobilon-P polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA). Non-specific binding sites were blocked by 5% non-fat milk or 1% BSA in 0.05% Tween-20 phosphate-buffered saline (PBST), then incubated overnight at 4°C with primary antibodies against p-eNOS(Ser1177) (1:1000; Upstate Biotechnology, Lake Placid, NY, USA), total eNOS (1:500; BD Transduction Laboratories, Lexington, KY, USA), or nitrotyrosine (1:1000; Upstate Biotechnology) and GAPDH (1:3000; Ambion, Austin, TX, USA), followed by a horseradish peroxidase-conjugated swine anti-rabbit or anti-mouse IgG (DakoCytomation, Carpinteria, CA), developed with an enhanced chemiluminescence detection system (ECL reagents, Amersham Pharmacia, Uppsala, Sweden) and finally exposed to X-ray films. Equal protein loading was verified with use of a housekeeping anti-GAPDH antibody. Densitometry was performed using a documentation programme.
Reverse-transcriptase polymerase chain reaction (RT-PCR) analysis of eNOS mRNA
RT-PCR was performed with the primers: eNOS, 5′-TGGCCGTGGAACAACTGGA-3′ (sense) and 5′-TGAGCTGACAGAGTAGTACC-3′ (antisense); GAPDH, 5′-TATGATGACATCAAGAAGGTGG-3′ (sense) and 5′-CACCACCCTGTTGCTGTA-3′ (antisense). The arterial rings were snap-frozen in liquid nitrogen and homogenized, and mRNA was extracted using the Aurum total RNA Mini kit (Bio-Rad, Hercules, CA, USA) according to manufacturer's instructions. The extracted RNA was reverse transcribed using the iScript™ cDNA synthesis kit (Bio-Rad). RNA templates were mixed with iScript reaction mix, iScript reverse transcriptase and nuclease-free water, and incubated for 5 min at 25°C, 30 min at 42°C, 5 min at 85°C and stopped at 4°C. PCR was performed with Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA) with thermal cycles of 5 min at 95°C, 30 cycles of 1 min at 95°C, 1 min at 60°C, 1 min at 72°C, finally followed by 7 min at 72°C. PCR product was then fractionated electrophoretically on a 1% agarose gel containing 0.5 mg·mL−1 ethidium bromide. Sizes of the fragments were 350 bp for eNOS and 210 bp for GAPDH. Densitometry was performed using a documentation program (FluorChem, Alpha Innotech Corp., San Leandro, CA). Data were normalized with GAPDH and compared with WKY.
Statistical analysis
Results were expressed as mean ± s.e.mean on rings from a number (n) of different rats. The cumulative concentration-response relationship was analyzed with a non-linear curve fitting (GraphPad Prism, Version 4.0, La Jolla, CA, USA). Protein expression was normalized to GAPDH and then expressed relative to control. Student's t-test was used and concentration–response curves were analysed by one-way anova followed by Bonferroni post hoc tests. P < 0.05 indicates significant difference.
Materials
Phenylephrine, acetylcholine and SNP were from Sigma (St. Louis, MO, USA). Aliskiren was obtained from Novartis (Basel, Switzerland). AVE3085 (CAS no. 450348–85-3, empirical formula C17H13F2NO3) was provided by Sanofi-Aventis (Frankfurt, Germany) (Wohlfart et al., 2008).
Results
Effect of 4 week AVE3085 treatment on endothelium-dependent and -independent relaxations
Treatment with AVE3085 for 4 weeks significantly improved ACh-induced endothelium-dependent relaxations in the aortae of SHRs (Emax= 58.0 ± 3.1% vs. 33.2 ± 3.0%, P < 0.01; Figure 1A) without affecting those in WKY aortae (Figure 1B). By contrast, SNP-induced endothelium-independent relaxations in WKY and SHR aortae were not affected by treatment with AVE3085 (Figure 1C, D).
Figure 1.
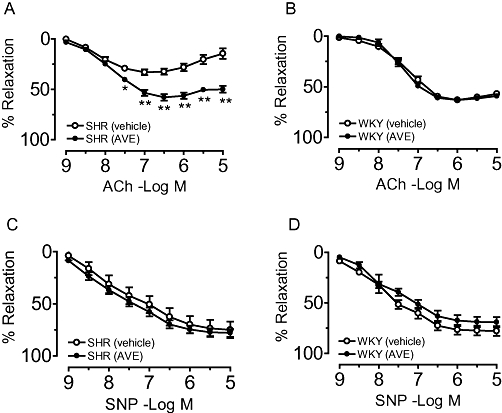
Effect of treatment with AVE3085 for 4 weeks on endothelium-dependent and -independent relaxations in aortae of SHRs. AVE3085 treatment (4 weeks) significantly improved acetylcholine (ACh)-induced endothelium-dependent relaxations in the aortae of SHRs (A) without affecting those in WKY aortae (B). Sodium nitroprusside (SNP)-induced relaxations were comparable in WKY and SHR aortae treated with or without AVE3085 (C and D). Data are mean ± SEM of six to eight rats. **P < 0.01 versus SHR.
Effect of AVE3085 treatment on eNOS and p-eNOS levels
In the aortae of SHR, the protein level of both eNOS and p-eNOS was reduced compared with those in WKY aortae; AVE3085 treatment for 4 weeks increased levels of p-eNOS (Figure 2A, B) and eNOS (Figure 2A, C) in SHR aortae without affecting levels of eNOS and p-eNOS in WKY aortae (Figure 2A–C).
Figure 2.
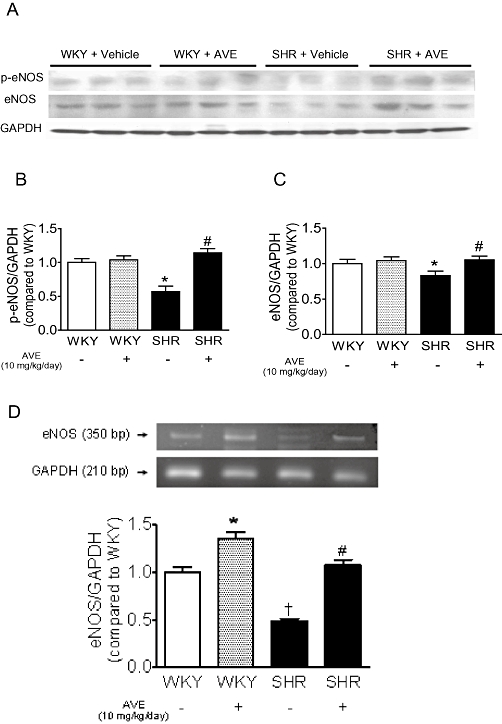
Effect of 4 weeks of AVE3085 treatment on eNOS and p-eNOS expressions and eNOS mRNA in SHR aortae. Representative images (A) and summarized diagram (B and C) showing that AVE3085 treatment for 4 weeks increased levels of p-eNOS (B) and eNOS (C) protein in SHR aortae without affecting those of WKY aortae. (D) AVE3085 treatment for 4 weeks enhanced eNOS mRNA expression in both SHR and WKY aortae. Data are mean ± SEM of four rats. *P < 0.05 versus WKY, #P < 0.05 versus SHR and †P < 0.05 versus WKY.
Effect of AVE3085 treatment on eNOS mRNA expression
Compared with WKY aortae, SHR aortae showed significantly reduced eNOS mRNA expression (Figure 2D). Such down-regulation was reversed by 4 week treatment with AVE3085. eNOS mRNA expression was also up-regulated in WKY aortae by AVE3085 treatment (Figure 2D).
Effect of AVE3085 treatment on nitrotyrosine formation
Nitrotyrosine (60 kDa) formation significantly increased in SHR aortae, and this increase was reduced by 4 week AVE3085 treatment (Figure 3).
Figure 3.
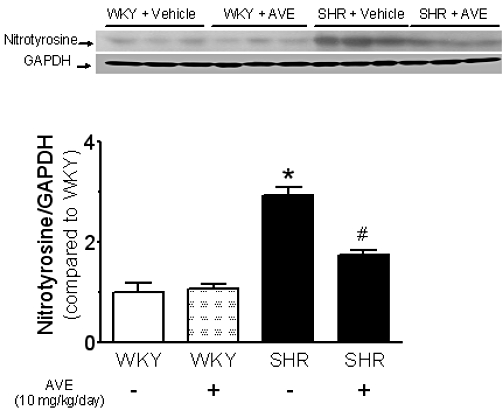
Effect of 4 week AVE3085 treatment on nitrotyrosine formation in SHR aortae. The formation of nitrotyrosine (60 kDa) was significantly increased in SHR aortae and was reduced by treatment with AVE3085 for 4 weeks. Data are mean ± SEM of four rats. *P < 0.05 versus WKY and #P < 0.05 versus SHR.
Effect of 2 h incubation with AVE3085 on endothelium-dependent and -independent relaxations
Aortae from untreated SHR or WKY rats were incubated ex vivo with AVE3085 (10 µmol·L−1) for 2 h. This in vitro treatment markedly increased ACh-induced relaxations in SHR aortae (50.2 ± 4.5% vs. 26.9 ± 4.4%, P < 0.05; Figure 4A) without affecting those in WKY aortae (Figure 4B). Again, there was no difference in SNP-induced relaxations in WKY and SHR aortae with or without AVE3085 treatment (Figure 4C, D).
Figure 4.
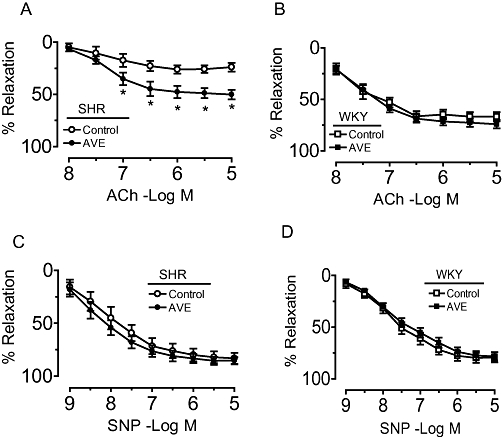
Effect of 2 h AVE3085 incubation on endothelium-dependent and -independent relaxations in SHR aortae. Two-hour incubation with AVE3085 (10 µmol·L−1) increased acetylcholine (ACh)-induced endothelium-dependent relaxations in SHR aortae (A) without affecting those in WKY aortae (B). No differences were observed in the sodium nitroprusside (SNP)–induced relaxations in WKY and SHR aortae with and without AVE3085 treatment (C and D). Data are mean ± SEM of six to eight rats. *P < 0.05 versus SHR.
Effect of 2 h AVE3085 treatment on eNOS and p-eNOS levels
Exposure to AVE3085 (10 µmol·L−1) for 2 h increased p-eNOS (Figure 5A) and eNOS levels (Figure 5B) in SHR aortae without affecting those in WKY aortae (Figure 5A, B).
Figure 5.
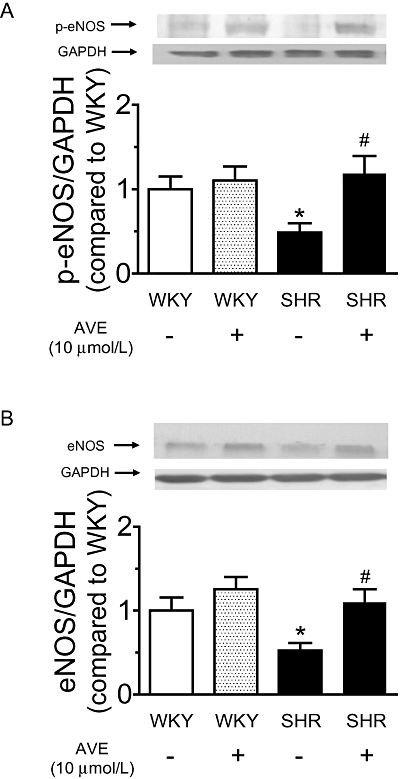
Effect of 2 h AVE3085 treatment on eNOS and p-eNOS levels in SHR aortae. AVE3085 (10 µmol L–1) incubation for 2 h increased the levels of p-eNOS (A) and eNOS protein (B) in SHR aortae. Data are mean ± SEM of four rats. *P < 0.05 versus WKY and #P < 0.05 versus SHR.
Effect of the removal of endothelium on eNOS expression after AVE3085 treatment
Incubation with AVE3085 (10 µmol·L−1) for 12 h significantly increased the eNOS expression in SHR aortae with intact endothelium (Figure 6B). AVE3085 also increased the eNOS expression in WKY aortae (Figure 6A). The level of eNOS was very low in both WKY (Figure 6A) and SHR (Figure 6B) aortae after mechanical removal of the endothelium, and AVE3085 did not increase the eNOS expression in aortae without endothelium (Figure 6A, B).
Figure 6.
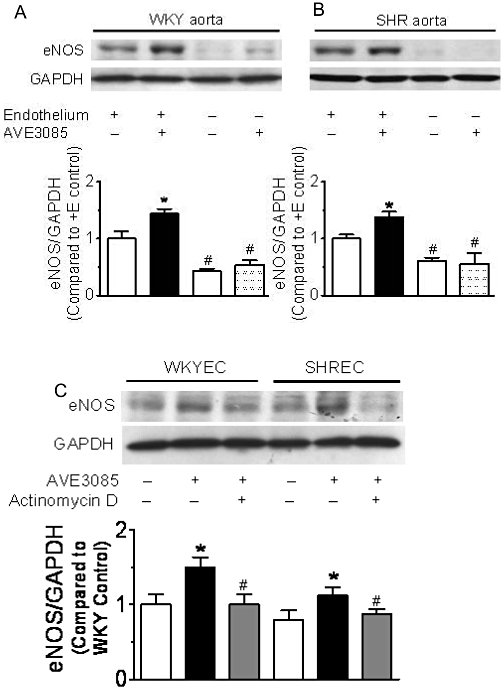
Effect of 12 h AVE3085 treatment on eNOS expression in aortae with or without endothelium and in primary endothelial cells. AVE3085 (10 µmol·L−1, 12 h) increased eNOS expression in SHR (B) aortae with endothelium. eNOS expression was very low in both WKY and SHR aortae after removal of endothelium. Data are mean ± SEM of six rats. *P < 0.05 versus control with endothelium, #P < 0.05 versus control with endothelium. In primary endothelial cells from both WKY (WKY EC) and SHR (SHR EC) aortae, AVE3085 (10 µmol·L−1, 12 h) increased eNOS expression, which was inhibited by co-incubation with the transcription inhibitor actinomycin D (0.1 µmol·L−1) (C). Data are mean ± SEM of four rats. *P < 0.05 versus control, #P < 0.05 versus AVE3085 from each group.
Effect of AVE3085 on primary endothelial cells from WKY and SHR aortae
AVE3085 (10 µmol·L−1) incubation for 12 h increased the eNOS expression in primary cultures of endothelial cells from both WKY and SHR aortae, and this increase was inhibited by co-incubation with transcription inhibitor actinomycin D (0.1 µmol·L−1) (Figure 6C).
Anti-hypertensive effect of AVE3085 in SHR
Oral administration of AVE3085 at 10 mg·kg·day−1 for 4 weeks reduced systolic blood pressure in SHRs (151.8 ± 1.8 vs. 170.0 ± 4.0 mmHg, P < 0.001). The anti-hypertensive effect of AVE3085 was comparable with that of aliskiren (10 mg·kg·day−1 for 4 weeks), a renin inhibitor (147.7 ± 1.3 mmHg). By contrast, 4 week AVE3085 treatment had no effect on blood pressure in WKY rats (114.8 ± 2.3 vs. 121.5 ± 4.2 mmHg) (Figure 7A). Male eNOS−/− mice of 4 month old had a significantly higher systolic blood pressure than the wild-type C57BL/6J mice (113.4 ± 1.4 vs. 91.1 ± 2.3 mmHg, P < 0.05), which is in accordance with previously published values (Wohlfart et al., 2008). Treatment of eNOS−/− mice with AVE3085 (30 mg·kg·day−1) up to 8 weeks had no effect on blood pressure (Figure 7B).
Figure 7.
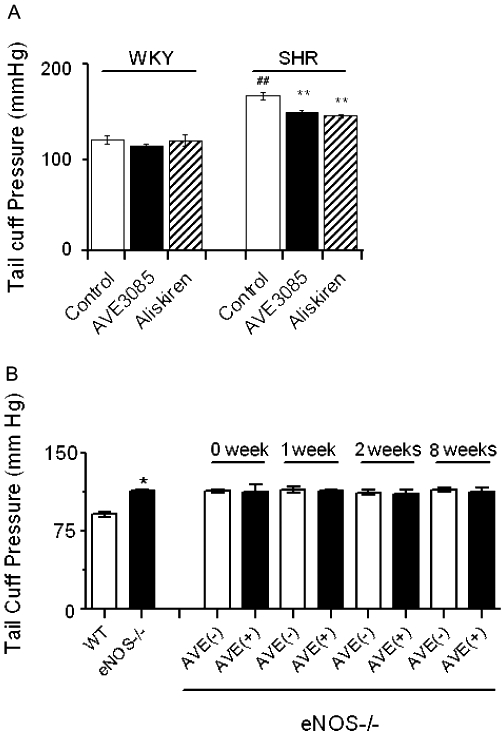
Anti-hypertensive effects of AVE3085 in SHR. In (A) oral administration of AVE3085 for 4 weeks reduced systolic blood pressure in SHR but had no effects on systolic blood pressure in WKY rats. In panel (B), male eNOS−/− mice (4 months old) had a significantly higher systolic blood pressure than the wild-type C57BL/6J mice, but treatment of eNOS−/− mice with AVE3085 for up to 8 weeks had no effect on blood pressure. Data are mean ± SEM of five to seven animals. WT: wild-type mice, AVE(+): with AVE3085 treatment, AVE(–): without AVE3085 treatment. ##P < 0.001 versus WKY control, **P < 0.001 versus control, *P < 0.05 versus WT.
Discussion
The present study demonstrated that oral administration of AVE3085 for 4 weeks reduced blood pressure in SHR, and that treatment with AVE3085 either acutely for 2 h or chronically for 4 weeks markedly augmented endothelium-dependent relaxations in SHR aortae in vitro. We suggest that the effect of AVE3085 is causally associated with the increase in eNOS expression, and this is supported by the fact that AVE3085 treatment did not reduce blood pressure in eNOS-deficient mice.
Although it still remains unclear whether endothelial dysfunction is a primary abnormality, or such dysfunction arises as a consequence of elevated blood pressure, it is clear that the endothelium undoubtedly participates in the pathogenesis of hypertension. In the present study, we showed that AVE3085 is effective in lowering blood pressure in hypertensive rats. AVE3085 restored the impaired endothelium-dependent relaxations in the aortae of SHRs, and hence, the improved endothelial function might be related to its anti-hypertensive effect.
Hypertension is associated with a decreased NO production (Huang et al., 1995). Currently, one of the strategies of treating hypertension is to use vasodilators. By targeting endogenous eNOS production instead of directly relaxing vascular smooth muscle, the present results may suggest an alternative therapeutic approach to the traditional treatment strategy by preventing the pathological changes in endothelial cells in hypertension, that is, the eNOS down-regulation that results in a reduced generation of NO. We have clearly shown that this strategy is useful in SHRs. However, whether this approach can be applied to hypertensive patients still requires further investigation.
The key role of oxidative stress is recognized in the pathological mechanisms of endothelial dysfunction associated with atherosclerosis, coronary artery disease, diabetes and hypertension (Li and Shah, 2004). As a marker of NO-dependent oxidative stress, the increased formation of nitrotyrosine in SHR aortae, as shown in the present study, further suggests a positive role of oxidative stress in the pathogenesis of vascular dysfunction in hypertension. Furthermore, the decreased nitrotyrosine formation observed after AVE3085 treatment suggests that this compound may have potential in inhibiting vascular oxidative stress.
Since AVE3085 enhances eNOS expression, this raises the possibility of eNOS uncoupling after treatment with this eNOS enhancer. However, our previous study showed that AVE9488, a structurally similar eNOS enhancer, possesses a capacity to up-regulate eNOS expression and facilitates the re-coupling of oxygen reduction and NO synthesis in eNOS (Wohlfart et al., 2008). AVE3085 may also be able to reverse eNOS uncoupling although further studies are warranted.
Acknowledgments
This work was supported by grants from the Research Grant Council of Hong Kong (CUHK4651/07M&4789/09M), Direct allocation 2041457&2041561, CUHK Li Ka Shing Institute of Health Sciences and focused Investment Scheme, Hong Kong; Tianjin Municipal Science & Technology Commission (09ZCZDSF04200 & 10JCYBJC26400), Ministry of Science & Technology, China (2009DFB30560, 2010CB529502), and the Providence St Vincent Medical Foundation, Portland, OR, USA.
Glossary
Abbreviations
- CD40L
CD40 ligand
- eNOS
endothelial nitric oxide synthase
- GRO-α
growth-related oncogene-α
- RT-PCR
reverse-transcriptase polymerase chain reaction
- SNP
sodium nitroprusside
- SHR
spontaneously hypertensive rat
- TNF
tumour necrosis factor
- WKY
Wistar Kyoto rat
Conflict of interest
None.
Supporting Information
Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- Bechara C, Wang X, Chai H, Lin PH, Yao Q, Chen C. Growth-related oncogene-alpha induces endothelial dysfunction through oxidative stress and downregulation of eNOS in porcine coronary arteries. Am J Physiol Heart Circ Physiol. 2007;293:H3088–H3095. doi: 10.1152/ajpheart.00473.2007. [DOI] [PubMed] [Google Scholar]
- Borniquel S, Valle I, Cadenas S, Lamas S, Monsalve M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1alpha. FASEB J. 2006;20:1889–1891. doi: 10.1096/fj.05-5189fje. [DOI] [PubMed] [Google Scholar]
- Chen C, Chai H, Wang X, Jiang J, Jamaluddin MS, Liao D, et al. Soluble CD40 ligand induces endothelial dysfunction in human and porcine coronary artery endothelial cells. Blood. 2008;112:3205–3216. doi: 10.1182/blood-2008-03-143479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan W, Yao X, Ko W, Huang Y. Nitric oxide mediated endothelium-dependent relaxation induced by glibenclamide in rat isolated aorta. Cardiovasc Res. 2000;46:180–187. doi: 10.1016/s0008-6363(99)00423-x. [DOI] [PubMed] [Google Scholar]
- Chan YC, Leung FP, Wong WT, Tian XY, Yung LM, Lau CW, et al. Therapeutically relevant concentrations of raloxifene dilate pressurized rat resistance arteries via calcium-dependent endothelial nitric oxide synthase activation. Arterioscler Thromb Vasc Biol. 2010;30:992–999. doi: 10.1161/ATVBAHA.110.203935. [DOI] [PubMed] [Google Scholar]
- Ding Y, Vaziri ND. Calcium channel blockade enhances nitric oxide synthase expression by cultured endothelial cells. Hypertension. 1998;32:718–723. doi: 10.1161/01.hyp.32.4.718. [DOI] [PubMed] [Google Scholar]
- Gien J, Seedorf GJ, Balasubramaniam V, Markham N, Abman SH. Intrauterine pulmonary hypertension impairs angiogenesis in vitro: role of vascular endothelial growth factor nitric oxide signaling. Am J Respir Crit Care Med. 2007;176:1146–1153. doi: 10.1164/rccm.200705-750OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- Husain K, Vazquez-Ortiz M, Lalla J. Down regulation of aortic nitric oxide and antioxidant systems in chronic alcohol-induced hypertension in rats. Hum Exp Toxicol. 2007;26:427–434. doi: 10.1177/0960327106072993. [DOI] [PubMed] [Google Scholar]
- Kline ER, Kleinhenz DJ, Liang B, Dikalov S, Guidot DM, Hart CM, et al. Vascular oxidative stress and nitric oxide depletion in HIV-1 transgenic rats are reversed by glutathione restoration. Am J Physiol Heart Circ Physiol. 2008;294:H2792–H2804. doi: 10.1152/ajpheart.91447.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou R, Michel T. Epinephrine regulation of the endothelial nitric-oxide synthase: roles of RAC1 and beta3-adrenergic receptors in endothelial NO signalling. J Biol Chem. 2007;282:32719–32729. doi: 10.1074/jbc.M706815200. [DOI] [PubMed] [Google Scholar]
- Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- Leung HS, Yao X, Leung FP, Ko WH, Chen ZY, Gollasch M, et al. Cilnidipine, a slow-acting Ca2+ channel blocker, induces relaxation in porcine coronary artery: role of endothelial nitric oxide and [Ca2+]i. Br J Pharmacol. 2006;147:55–63. doi: 10.1038/sj.bjp.0706450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wallerath T, Forstermann U. Physiological mechanisms regulating the expression of endothelial-type NO synthase. Nitric Oxide. 2002;7:132–147. doi: 10.1016/s1089-8603(02)00127-1. [DOI] [PubMed] [Google Scholar]
- Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–R1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- Linz W, Wohlfart P, Scholkens BA, Malinski T, Wiemer G. Interactions among ACE, kinins and NO. Cardiovasc Res. 1999;43:549–561. doi: 10.1016/s0008-6363(99)00091-7. [DOI] [PubMed] [Google Scholar]
- Murohara T, Witzenbichler B, Spyridopoulos I, Asahara T, Ding B, Sullivan A, et al. Role of endothelial nitric oxide synthase in endothelial cell migration. Arterioscler Thromb Vasc Biol. 1999;19:1156–1161. doi: 10.1161/01.atv.19.5.1156. [DOI] [PubMed] [Google Scholar]
- Riad A, Westermann D, Van Linthout S, Mohr Z, Uyulmaz S, Becher PM, et al. Enhancement of endothelial nitric oxide synthase production reverses vascular dysfunction and inflammation in the hindlimbs of a rat model of diabetes. Diabetologia. 2008;51:2325–2332. doi: 10.1007/s00125-008-1159-9. [DOI] [PubMed] [Google Scholar]
- Rucker R, Storms D. Interspecies comparisons of micronutrient requirements: metabolic vs. absolute body size. J Nutr. 2002;132:2999–3000. doi: 10.1093/jn/131.10.2999. [DOI] [PubMed] [Google Scholar]
- Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest. 1998;101:731–736. doi: 10.1172/JCI1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Heeschen C, Aicher A, Ziebart T, Honold J, Urbich C, et al. Ex vivo pretreatment of bone marrow mononuclear cells with endothelial NO synthase enhancer AVE9488 enhances their functional activity for cell therapy. Proc Natl Acad Sci USA. 2006;103:14537–14541. doi: 10.1073/pnas.0604144103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz R, Osborne-Lawrence S, Hahner L, Gibson LL, Gormley AK, Vongpatanasin W, et al. C-reactive protein downregulates endothelial NO synthase and attenuates reendothelialization in vivo in mice. Circ Res. 2007;100:1452–1459. doi: 10.1161/01.RES.0000267745.03488.47. [DOI] [PubMed] [Google Scholar]
- Searles CD. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am J Physiol Cell Physiol. 2006;291:C803–C816. doi: 10.1152/ajpcell.00457.2005. [DOI] [PubMed] [Google Scholar]
- Shin HK, Salomone S, Potts EM, Lee SW, Millican E, Noma K, et al. Rho-kinase inhibition acutely augments blood flow in focal cerebral ischemia via endothelial mechanisms. J Cereb Blood Flow Metab. 2007;27:998–1009. doi: 10.1038/sj.jcbfm.9600406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chai H, Wang Z, Lin PH, Yao Q, Chen C. Serum amyloid A induces endothelial dysfunction in porcine coronary arteries and human coronary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2008;295:H2399–H2408. doi: 10.1152/ajpheart.00238.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlfart P, Xu H, Endlich A, Habermeier A, Closs EI, Hubschle T, et al. Antiatherosclerotic effects of small-molecular-weight compounds enhancing endothelial nitric-oxide synthase (eNOS) expression and preventing eNOS uncoupling. J Pharmacol Exp Ther. 2008;325:370–379. doi: 10.1124/jpet.107.128009. [DOI] [PubMed] [Google Scholar]
- Xue HM, He GW, Huang JH, Yang Q. New strategy of endothelial protection in cardiac surgery: use of enhancer of endothelial nitric oxide synthase. World J Surg. 2010;34:1461–1469. doi: 10.1007/s00268-010-0520-6. [DOI] [PubMed] [Google Scholar]
- Yan G, You B, Chen SP, Liao JK, Sun J. Tumor necrosis factor-alpha downregulates endothelial nitric oxide synthase mRNA stability via translation elongation factor 1-alpha 1. Circ Res. 2008;103:591–597. doi: 10.1161/CIRCRESAHA.108.173963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiher AM, Drexler H, Saurbier B, Just H. Endothelium-mediated coronary blood flow modulation in humans. Effects of age, atherosclerosis, hypercholesterolemia, and hypertension. J Clin Invest. 1993;92:652–662. doi: 10.1172/JCI116634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Chai H, Courson A, Lin PH, Lumsden AB, Yao Q, et al. Ginkgolide A attenuates homocysteine-induced endothelial dysfunction in porcine coronary arteries. J Vasc Surg. 2006;44:853–862. doi: 10.1016/j.jvs.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, et al. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P. J Clin Invest. 1994;94:2036–2044. doi: 10.1172/JCI117557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.