

Abstract
BACKGROUND AND PURPOSE
Roscovitine, a cyclin-dependent kinase (CDK) inhibitor that induces tumour cell death, is under evaluation as an anti-cancer drug. By triggering leukocyte apoptosis, roscovitine can also enhance the resolution of inflammation. Beyond death-inducing properties, we tested whether roscovitine affects leukocyte-endothelial cell interaction, a vital step in the onset of inflammation.
EXPERIMENTAL APPROACH
Leukocyte-endothelial cell interactions were evaluated in venules of mouse cremaster muscle, using intravital microscopy. In primary human endothelial cells, we studied the influence of roscovitine on adhesion molecules and on the nuclear factor-κB (NF-κB) pathway. A cellular kinome array, in vitro CDK profiling and RNAi methods were used to identify targets of roscovitine.
KEY RESULTS
In vivo, roscovitine attenuated the tumour necrosis factor-α (TNF-α)-induced leukocyte adherence to and transmigration through, the endothelium. In vitro, roscovitine strongly inhibited TNF-α-evoked expression of endothelial adhesion molecules (E-selectin, intercellular cell adhesion molecule, vascular cell adhesion molecule). Roscovitine blocked NF-κB-dependent gene transcription, but not the NF-κB activation cascade [inhibitor of κB (IκB) kinase activity, IκB-α degradation, p65 translocation]. Using a cellular kinome array and an in vitro CDK panel, we found that roscovitine inhibited protein kinase A, ribosomal S6 kinase and CDKs 2, 5, 7 and 9. Experiments using kinase inhibitors and siRNA showed that the decreased endothelial activation was due solely to blockade of CDK5 and CDK9 by roscovitine.
CONCLUSIONS AND IMPLICATIONS
Our study highlights a novel mode of action for roscovitine, preventing endothelial activation and leukocyte-endothelial cell interaction by inhibition of CDK5 and 9. This might expand its usage as a promising anti-inflammatory compound.
Keywords: inflammation, roscovitine, cyclin-dependent kinases, endothelium, leukocytes
Introduction
Cyclin-dependent kinases (CDKs) are key regulators of cell cycle progression. Interference with the cell cycle by means of small molecule CDK inhibitors has been of great interest as a novel therapeutic approach against cancer. The substituted purine analogue, R-roscovitine, also known as seliciclib or CYC202, was first synthesized in 1997 and represents an orally bioavailable, second generation, CDK inhibitor. It exhibits cytotoxic effects in a variety of cancer cells in vitro (Aldoss et al., 2009). In vivo, its anti-tumour activity was extensively evaluated in numerous human tumour xenografts (Blagden and de Bono, 2005). Currently, roscovitine is being tested in a phase I clinical trial together with sapacitabine in patients with advanced solid tumours (http://www.clinicaltrials.gov) and in two phase II trials for its efficacy on non-small cell lung cancer and nasopharyngeal carcinoma (Aldoss et al., 2009).
Besides its anti-cancer properties, roscovitine has been found to exert anti-inflammatory actions in several inflammatory in vivo models, such as glomerulonephritis, pleurisy or arthritis (Leitch et al., 2009). These actions were ascribed to its influence on immune cells: roscovitine inhibits proliferation and evokes apoptosis of leukocytes, thus enhancing the resolution of inflammation (Rossi et al., 2006). The extravasation of leukocytes from the blood into the inflammation-affected tissue is a vital process that triggers and maintains inflammation and is mediated by a sequence of interactions between circulating leukocytes and endothelial cells. Surprisingly, effects of roscovitine on leukocyte-endothelial cell interaction and, in particular, on endothelial activation have not been studied to date. We hypothesized that roscovitine, beyond its proliferation-inhibiting and cell death-inducing actions, would affect leukocyte recruitment and/or diapedesis by directly influencing endothelial signalling processes. Therefore, we analysed the influence of roscovitine on leukocyte-endothelial cell interaction in vivo and in vitro and investigated the action of roscovitine on endothelial cells activated by inflammatory stimuli.
Methods
Animals
All animal care and experimental procedures were in accordance with the local animal protection legislation (Government of Upper Bavaria). C57BL/6 mice were purchased from Charles River (Sulzfeld, Germany). All experiments were performed with male mice of 6–8 week old.
Intravital microscopical analysis of leukocyte-endothelial cell interaction in the mouse cremaster muscle
The surgical preparation was performed as originally described by (Baez, 1973) with minor modifications. Mice were anaesthetized by intraperitoneal injection of ketamine (100 mg·kg−1) and xylazine (10 mg·kg−1). The left femoral artery was cannulated in a retrograde manner for administration of drugs and microspheres (2 µm diameter; Molecular Probes/Invitrogen, Karlsruhe, Germany). Surgical preparation of cremaster muscles and intravital microscopy were performed as previously described (Mempel et al., 2003). One group of mice (n = 6) were intraarterially injected with roscovitine (3.5 µg in 100 µL PBS), the other group (n = 6) only received the vehicle. Concurrently, leukocyte recruitment to the cremaster muscle was induced by intrascrotal injection of 500 ng recombinant murine tumour necrosis factor-α (TNF-α) (R&D Systems, Wiesbaden, Germany) in 400 µL PBS. After 240 min, five vessel segments were randomly chosen in a central area of the spread-out cremaster muscle among those that were at least 150 µm away from neighboring postcapillary venules and did not branch over a distance of at least 150 µm. After having obtained recordings of migration parameters, centreline blood flow velocity was determined by measuring the distance between several images of one microsphere under stroboscopic illumination. Blood samples were collected by cardiac puncture for the determination of systemic leukocyte counts using a Coulter ACT Counter (Coulter, Miami, FL, USA). Anesthetized animals were then killed by exsanguination. For offline analysis of parameters describing the sequential steps of leukocyte extravasation, the Cap-Image analysis software was used (Zeintl, Heidelberg, Germany). Rolling leukocytes were defined as those moving slower than the associated blood flow and quantified for 30 s. Individual rolling velocity was determined from video recordings by randomly choosing 5 leukocytes per venule and measuring the time necessary to travel a distance of 100 µm. Firmly adherent cells were determined as those resting in the associated blood flow for more than 30 s and related to the luminal surface per 100 µm vessel length. Transmigrated cells were counted in regions of interest covering 75 µm on both sides of a vessel over 100 µm vessel length.
Analysis of leukocyte apoptosis in vivo
One group of mice (n = 5) received 3.5 µg roscovitine in 100 µL PBS via injection into the tail vein. Control mice (n = 5) only received the vehicle. Two hundred and forty minutes later, mice were killed by cervical dislocation. Five minutes before killing, all mice were injected intraperitoneally with 100 µL heparin solution (Heparin-Na 25000 I.E./5 mL; Braun, Melsungen, Germany). Blood was collected from the heart, centrifuged, and the buffy coat was removed. After lysis of erythrocytes with distilled water (1 min on ice), the leukocyte suspension (in 0.9% NaCl) was centrifuged and stained with Annexin-V/propidium iodide (PI) by using the Annexin-V Apoptosis Detection Kit FITC (eBioscience/NatuTec, Frankfurt am Main, Germany) according to the manufacturer's protocol. Leukocytes were analysed on a FACSCanto II flow cytometer (Becton Dickinson, Heidelberg, Germany).
Isolation and culture of primary human umbilical vein endothelial cells
Human umbilical vein endothelial cells (HUVECs) were isolated by digestion of umbilical veins with 0.1 g·L−1 collagenase A (Roche; Mannheim, Germany). HUVECs were cultured in Endothelial Cell Growth Medium (Promocell; Heidelberg, Germany) supplemented with 10% heat-inactivated fetal calf serum (PAA; Pasching, Austria). All experiments were performed with cells at passage 3 (splitting ratio 1:3). Cells were routinely tested for contamination with mycoplasms using the PCR detection kit VenorGeM (Minerva Biolabs, Berlin, Germany).
Isolation of human granulocytes
Peripheral blood was obtained from healthy volunteers (25 mL, containing 400 µL EDTA 50 mg·mL−1). After centrifugation, the buffy coat was carefully removed and incubated with CD15 MicroBeads (Miltenyi, Bergisch Gladbach, Germany). Granulocytes were purified from the buffy coat by magnetic cell sorting using the MiniMACS Separator (Miltenyi).
Granulocyte adhesion assay
Confluent HUVECs in 24-well plates were used. Control cells were left untreated. Cells were exposed to 3, 10 and 30 µM roscovitine. After 30 min, TNF-α (10 ng·mL−1) was applied. Twenty-four hours later, freshly isolated granulocytes (105 cells) were centrifuged onto the HUVEC monolayer and allowed to adhere for 30 min. HUVECs were gently washed twice with PBS (containing Ca2+ and Mg2+) to remove non-adherent granulocytes. Adhered leukocytes were quantified by a myeloperoxidase (MPO) assay. Conversion of the MPO substrate dianisidine hydrochloride (Sigma-Aldrich)/H2O2 was assessed photometrically (540 nm) using a SpectraFluor Plus plate reader (Tecan, Crailsheim, Germany).
Flow cytometric analysis
Oxidative burst in granulocytes was assessed by measuring the intracellular oxidation of dihydrorhodamine-123 (DHR; Invitrogen, Karlsruhe, Germany). Freshly isolated granulocytes were primed with DHR (1 µM) for 10 min at 37°C. Cells were pretreated for 30 min with 10 or 20 µM roscovitine and then activated with 10−7 M formyl-methionyl-leucyl-phenylalanine (fMLP; Sigma-Aldrich) for 15 min. The reaction was stopped on ice and cells were analysed by flow cytometry (FACSCalibur, BD Biosciences, Heidelberg, Germany).
For evaluation of CD11b surface expression, freshly isolated granulocytes were pretreated for 30 min with 10 or 20 µM roscovitine and then activated with 10−7 M fMLP for 15 min. Subsequently, cells were fixed with 4% buffered formalin (Sigma-Aldrich), washed, incubated with FITC-labelled anti-CD11b antibody (AbD Serotec, Düsseldorf, Germany) and analysed by flow cytometry (FACSCalibur).
For analysis of the expression of adhesion molecules in endothelial cells, confluent HUVECs were either left untreated (control) or pretreated for 30 min with roscovitine (0.1, 1, 10 and 30 µM) before TNF-α (10 ng·mL−1) was applied [for intercellular cell adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 for 24 h; for E-selectin for 6 h]. HUVECs were washed twice with PBS, trypsinized and fixed in 4% buffered formalin. After washing, cells were incubated with fluorescence-labelled antibodies against ICAM-1 (Biozol, Eching, Germany), E-selectin (BD) or VCAM-1 (BD) and analysed by flow cytometry (FACSCalibur).
Quantitative RT-PCR
Confluent HUVECs were pretreated with roscovitine (10 or 30 µM) for 30 min and subsequently activated with TNF-α (10 ng·mL−1) for 4 h. RNA was isolated using the RNeasy Mini Kit according to the manufacturer's instructions (Qiagen, Hilden, Germany). Amounts of RNA were quantified with a NanoDrop spectrophotometer (Thermo Scientific/Peqlab, Erlangen, Germany) and integrity of the RNA was controlled on agarose gels. In total, 2 µg of RNA were reversely transcribed using the cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Quantitative real-time PCR was performed on an ABI 7300 RealTime PCR System using the TaqMan Universal PCR Mastermix (Applied Biosystems). ICAM-1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primers were designed using the Primer Express 2.0 software (Applied Biosystems). The following primers were used: human ICAM-1 (forward, 5′-GCA GAC AGT GAC CAT CTA CAG CTT-3′; reverse, 5′-CTT CTG AGA CCT GTG GCT TCG T-3′; probe, 6-FAM-5′-CCG GCG CCC AAC GTG ATT CT-3′-BHQ-1) and human GAPDH (forward, 5′-GGG AAG GTG AAG GTC GGA GT-3′; reverse, 5′-TCC ACT TTA CCA GAG TTA AAA GCA G-3′; probe, 6-FAM-5′-ACC AGG CGC CCA ATA CGA CCA A-3′-TAMRA). Calculation of relative mRNA amount was carried out according to (Pfaffl, 2001).
Quantification of endothelial apoptosis
Quantification of apoptosis was carried out as described by Nicoletti et al. (1991) by counting the nuclei with subdiploid DNA content after staining with propidium iodide (PI; Sigma-Aldrich). Confluent HUVECs were either left untreated (control) or treated with roscovitine (0.1, 1, 10 and 30 µM) for 24 or 48 h. After treatment, the supernatant of each well was collected to include cells that underwent anoikis. Cells were trypsinized, resuspended in the supernatant and centrifuged. Subsequently, cells were incubated in a buffer containing Triton X-100 (Merck, Darmstadt, Germany) and PI. After incubation overnight, cells were analysed by flow cytometry (FACSCalibur). Events left of the G1/G0 peak in the histogram were considered apoptotic cells.
Dual luciferase nuclear factor-κB (NF-κB) reporter gene assay
Human umbilical vein endothelial cells were transiently cotransfected with the firefly luciferase reporter vector pGL4.32[luc2P/NF-κB-RE/Hygro] and the Renilla luciferase reporter vector pGL4.74[hRluc/TK] from Promega (Mannheim, Germany) at a ratio of 1:10 with the Amaxa HUVEC Nucleofactor Kit (Lonza, Cologne, Germany), seeded and cultured for 24 h. Then cells were pretreated with roscovitine (0.3, 3, 10, 20 or 30 µM) for 30 min. Subsequently, TNF-α (10 ng·mL−1) was applied for 6 h. Cells were harvested in lysis buffer and the reporter gene activity assay was performed using the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. Luciferase activity was measured using a Berthold Orion II (Berthold Detection Systems, Pforzheim, Germany) luminometer. The NF-κB-dependent luciferase activity was normalized to the activity of the internal control (Renilla luciferase).
In vitro IκB kinase β activity assay
The effect of roscovitine on recombinant inhibitor of κB (IκB) kinase (IKK) β activity in vitro was determined using the HTScan® IKKβ Kinase Assay Kit (Cell Signaling), which includes active IKKβ kinase (GST fusion protein), biotinylated peptide substrate and a phospho-serine antibody. IKKβ was pretreated with roscovitine (3, 10, 20 or 30 µM) or the positive control staurosporine (100 µM) for 5 min prior to adding the substrate peptide. The assay was performed in 96-well high-binding streptavidin-coated plates. The absorbance of each well was measured at 450 nm using a Tecan Sunrise microplate absorbance reader. The percentage inhibition of IKKβ kinase activity was calculated as follows: % inhibition = 1 –[(Δinhibited –Δblank)/(Δuninhibited –Δblank)]× 100.
Western blot analysis
Confluent cells were lysed in RIPA buffer additionally containing phenylmethylsulphonyl fluoride (PMSF), Na3VO4, NaF, sodium pyrophosphate, sodium β-glycerophosphate, H2O2 and Complete (Roche, Mannheim, Germany). Lysates were centrifuged and supernatants were used for protein quantification (Bradford assay). Standard Laemmli sample buffer was added and lysates were boiled for 5 min at 95°C. SDS polyacrylamide gel electrophoresis (PAGE) was performed and separated proteins were transferred onto nitrocellulose membranes by semi-dry electroblotting. Membranes were blocked with non-fat dry milk powder or bovine serum albumin (BSA). Primary antibodies: the rabbit polyclonal anti-phospho-IKKα/β (1:500), mouse monoclonal anti-CDK7 (1:1000) and rabbit polyclonal anti-phospho(Ser2/5) Rpb1 CTD RNA polymerase II (1:1000) antibodies were from Cell Signaling/New England Biolabs (Frankfurt am Main, Germany). The mouse monoclonal anti-β-actin antibody (1:1000) was from Chemicon (Millipore, Billerica, MA, USA). The rabbit polyclonal anti-IκBα (1:1000), rabbit polyclonal anti-CDK2 (1:1000), mouse monoclonal anti-CDK5 antibody (1:1000) and mouse monoclonal anti-CDK9 (1:1000) antibodies were from Santa Cruz (Heidelberg, Germany). For detection, secondary antibodies were labelled with either horseradish-peroxidase or infrared dyes (Li-Cor Biosciences, Lincoln, NE, USA).
NF-κB p65 translocation
Human umbilical vein endothelial cells were cultured until confluence on collagen-treated slides (ibidi, Martinsried, Germany). Cells were pretreated with roscovitine (10 µM) for 30 min before TNF-α (10 ng·mL−1) was applied for 5 or 30 min. Cell were washed, fixed with 4% buffered formaldehyde (Sigma-Aldrich), permeabilized by 0.2% Triton X-100 (Merck) and blocked in 0.2% BSA. The anti-p65 NF-κB antibody (1:100) was from Santa Cruz, the Alexa 488 goat anti-rabbit (1:400) was from Molecular Probes/Invitrogen. Confocal laser scanning microcopy was performed on a Zeiss LSM 510 Meta (Zeiss, Oberkochen, Germany).
Preparation of nuclear extracts and electrophoretic mobility shift assay
Confluent HUVECs were pretreated with roscovitine (10 µM) for 30 min. TNF-α (10 ng·mL−1) was applied for 0.5, 1, 6 or 16 h. Cells were lysed in buffer A (HEPES pH 7.9 10 mM, KCl 10 mM, EDTA 0.1 mM, EGTA 0.1 mM, dithiothreitol (DTT) 1.0 mM, PMSF 0.5 mM, glycerol 25%). Nonidet P-40 (0.06%) was added. After vigorous vortexing, lysates were centrifuged, supernatants removed and pellets incubated in buffer B (HEPES pH 7.9 20 mM, NaCl 0.4 mM, EDTA 0.1 mM, EGTA 0.1 mM, DTT 1.0 mM, PMSF 0.5 mM). After centrifugation, supernatants (nuclear extract) were collected and protein concentration was determined (Bradford assay). The NF-κB consensus sequence 5′-AGT TGA GGG GAC TTT CCC AGG C-3′ (Promega, Mannheim, Germany) was 5′ end-labelled with [γ-32P]-ATP using the T4 polynucleotide kinase (Promega). Nuclear extracts were incubated with poly(dIdC) and the radioactive oligonucleotide. Protein-oligonucleotide complexes were separated by non-denaturating PAGE and afterwards exposed to Cyclone Storage Phosphor Screens (Canberra-Packard, Schwandorf, Austria) followed by analysis with a Cyclone Storage Phosphor Imager Station (Canberra-Packard).
Kinome chip analysis (PepChip)
Confluent HUVECs in 4 × 100 mm dishes were used. Cells were treated with TNF-α for 15 or 30 min with 10 µM roscovitine prior to 15 min incubation with TNF-α. Cells were washed and lysed with M-PER Mammalian Protein Extraction Reagent (Pierce, Rockford, IL, USA), containing 2.5 mM sodium pyrophosphate, 2 mM sodium β-glycerophosphate, 1 mM Na3VO4 and 1 mM NaF. Lysates were centrifuged and supernatants were used for kinome analysis: 70 µL lysates per array was mixed with 10 µL activation solution (20 µCi [γ-33P] ATP, 50% glycerol, 5 mM DTT, 50 mM MgCl2, 50 mM MnCl2, 250 µg·mL−1 PEG 8000 and 250 µg·mL−1 BSA) and centrifuged. Next, 70 µL of the supernatant was loaded onto the array and incubated for 2 h at 37°C in saturated humidity. On the PepChip, 1152 different peptides with specific phosphorylation motifs for the respective kinases were spotted in triplicates (van Baal et al., 2006). Chips were washed in two cycles, first 5 min in 2 M NaCl containing 1% Triton X-100, followed by 5 min in PBS containing 1% Triton X-100. Afterward, chips were rinsed three times with distilled water and then air-dried. Phosphor-storage screens were exposed to the chip for 24 h to determine and to quantify the phosphorylation status of peptides (i.e. kinase substrates), which gave information about the activity of the associated upstream kinase. The phosphorylation status of the chips was compared spot by spot. The results were ranked by extent of inhibition of phosphorylation (residual activity). The most strongly inhibited kinases according to this score are shown in Table 1.
Table 1.
Kinome array
| Peptide | Kinase | Residual activity |
|---|---|---|
| KKKKGSLDSDN | PKA | 17% |
| KSEPISPPRDR | CDK5 | 39% |
| DDRHDSGLDSM | RS6K | 56% |
A cellular kinome array (see Methods for details) was performed on lysates of endothelial cells that had been treated with either TNF-α (10 ng·mL−1, 15 min) alone or roscovitine (10 µM, 30 min pretreatment) and TNF-α (10 ng·mL−1, 15 min). Kinases most strongly inhibited by roscovitine in endothelial cells are indicated in Table 1.
CDK, cyclin-dependent kinase; PKA, protein kinase A; RS6K, ribosomal S6 kinase; TNF-α, tumour necrosis factor-α.
In vitro kinase panel
The respective protein kinases (CDK1, 2, 4, 7, 9) were expressed in Sf9 insect cells or E. coli as human recombinant GST-fusion or as His-tagged proteins. The kinases were purified by affinity chromatography with GSH- or Ni-NTH-agarose. The purity of the kinases was checked by SDS-PAGE/silver staining and the identity was verified by mass spectroscopy. A proprietary protein kinase assay (33PanQinase® Activity Assay; ProQinase, Freiburg, Germany) was used for measuring kinase activity. The assays were performed in 96-well FlashPlates from Perkin Elmer/NEN (Boston, MA, USA). The assay contained 60 mM HEPES-NaOH (pH 7.5), 3 mM MgCl2, 3 mM MnCl2, 3 mM sodium orthovanadate, 1.2 mM DTT, 50 mg·mL−1 PEG 20000, 1 mM [γ-33P]-ATP (approximately 5 × 105 cpm per well). The reaction cocktails were incubated at 30°C for 80 min. The reaction was stopped with 2% H3PO4, and plates were aspirated and washed two times with 0.9% NaCl. Incorporation of 33Pi was determined with a microplate scintillation counter (Microbeta; Wallac/PerkinElmer, Waltham, MA, USA). Nine concentrations of roscovitine in the range from 10−5 to 10−8 M were tested. IC50 values were calculated with Quattro Workflow V2.0.2.2 (Quattro Research GmbH, Munich, Germany). The model used was ‘sigmoidal response (variable slope)’ with parameters ‘top’ fixed at 100% and ‘bottom’ at 0%.
The influence of roscovitine on the activity of CDK5 was measured by Cerep (Celle l'Evescault, France) using a homogenous time-resolved fluorescence (HTRF)-based kinase assay. Eight concentrations of roscovitine in the range from 3 × 10−9 to 10−5 were tested. Human recombinant CDK5 was expressed in insect cells. Substrates: ATP and ATF-2 (80 nM). Incubation: 45 min at 22°C. Reaction product: phospho-ATF-2. The results were expressed as per cent of control specific activity [(measured specific activity/control specific activity) × 100] and as per cent inhibition of control specific activity [100 – (measured specific activity/control specific activity) × 100] obtained in the presence of roscovitine. The IC50 value was determined by a software developed by Cerep (Hill software).
Gene silencing
Cyclin-dependent kinase 2 and CDK7 were silenced in HUVECs by transfecting cells with the Amaxa HUVEC Nucleofactor kit (Lonza). ON-TARGET plus siRNA from Dharmacon (Lafayette, CO, USA) was used to silence CDK2 (J-003236-11/12) and CDK7 (J-003241-09/10/11/12). ON-TARGET plus siCONTROL non-targeting siRNA was used as control. Twenty-four hours after transfection, cells were stimulated with TNF-α (10 ng·mL−1) for 24 h. Then, cells were trypsinized and collected for flow cytometry and Western blot analysis (see above).
Cyclin dependent kinase 5 and CDK9 silencing in HUVECs was performed by Sirion Biotech (Martinsried, Germany). HUVECs were provided by the authors and treated with infectious adenoviruses encoding short hairpin RNA (non-targeting, CDK5 and CDK9 shRNA) by Sirion. Seventy-two hours after infection, cells were incubated with TNF-α (10 ng·mL−1) for 24 h, trypsinized and collected for flow cytometry and Western blot analysis (see above).
Statistical analysis
Statistical analysis was performed with Prism version 3.03 (GraphPad). Unpaired Student's t-test was used to compare two groups. To compare three or more groups, one-way analysis of variance (anova) followed by Newman–Keuls post hoc test was used. Data from the cremaster muscle model were analysed with the Mann–Whitney rank sum test. Data are expressed as mean ± SEM. P-values < 0.05 were considered as statistically significant.
Materials
Roscovitine [(R)-roscovitine, also known as seliciclib or CYC202] and rapamycin were from Sigma-Aldrich (Taufkirchen, Germany), the protein kinase A (PKA) inhibitor, PKI (6-22)-amide and cAMPS-Rp triethylammonium salt were from Biotrend (Cologne, Germany), recombinant human TNF-α was from PeproTech (Hamburg, Germany). Calyculin was from Millipore (Schwalbach, Germany). Staurosporine was from New England Biolabs (Frankfurt am Main, Germany).
Results
Roscovitine attenuated leukocyte-endothelial cell interaction in vivo and in vitro
The interaction of leukocytes with the endothelium in venules of the mouse cremaster muscle was analysed by using intravital microscopy. Roscovitine (3.5 µg, i.a.) decreased TNF-α-evoked leukocyte adhesion by 40% (Figure 1A) and transmigration by 26% (Figure 1B). Leukocyte rolling along the endothelium was unaltered (9.1/30 s ± 3.5/30 s in TNF-α-treated mice; 8.7/30 s ± 4.7/30 s in roscovitine- and TNF-α-treated mice), whereas leukocyte rolling velocity was slightly, but not significantly, increased by roscovitine (Figure 1C). The total count of systemic leukocytes was not altered significantly (6400·µL−1± 1400·µL−1 in TNF-α-treated mice; 5000·µL−1± 1100·µL−1 in roscovitine- and TNF-α-treated animals). Moreover, leukocytes isolated from animals treated with roscovitine alone (3.5 µg, i.v.) did not exhibit signs of apoptosis or cell death (Annexin-V/propidium iodide staining) as shown in Figure 1D. In vitro, we measured the firm adherence of freshly isolated human granulocytes on HUVECs. As shown in Figure 1E, roscovitine concentration-dependently inhibited the TNF-α-evoked leukocyte adhesion.
Figure 1.
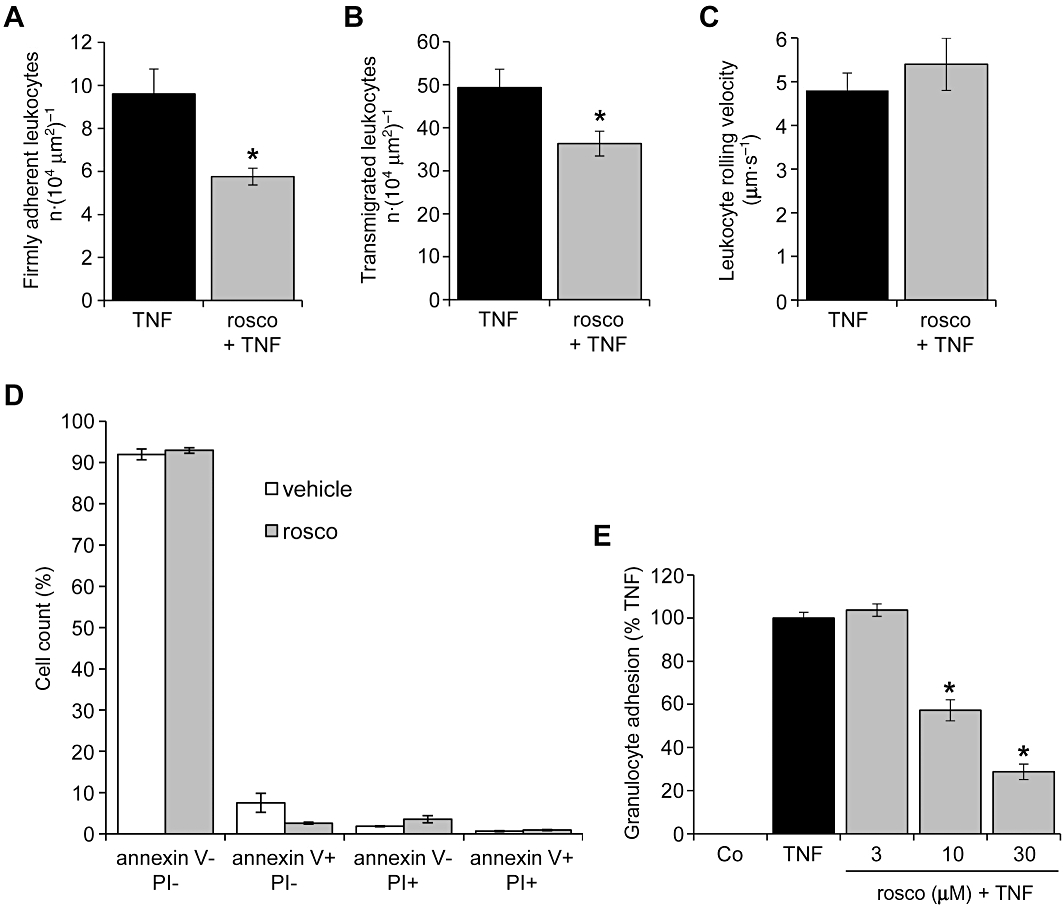
Roscovitine attenuates leukocyte-endothelial cell interaction in vivo and in vitro. (A–C) Mice were treated with TNF-α (500 ng intrascrotally) for 4 h alone or in combination with roscovitine (rosco; 3.5 µg, i.a.). n = 6 per group. *P < 0.05 versus TNF-α. Numbers of firmly adherent (A) and transmigrated leukocytes (B) as well as the leukocyte rolling velocity (C) were quantified via intravital microscopy of mouse cremaster venules. (D) Mice were treated with roscovitine (3.5 µg, i.a.) or with vehicle for 4 h. Leukocytes were isolated and stained with Annexin-V/propidium iodide (PI) to assess cell death. n = 5 per group. (E) HUVECs were either left untreated (Co) or pretreated with roscovitine for 30 min. TNF-α (10 ng·mL−1) was applied for 24 h before freshly isolated human granulocytes were added for 30 min. Firmly adherent granulocytes were quantified by measuring myeloperoxidase (MPO) activity. n = 3. *P < 0.05 versus TNF-α. HUVECs, human umbilical vein endothelial cells; TNF-α, tumour necrosis factor-α.
Roscovitine slightly decreased leukocyte activation and strongly inhibited endothelial cell adhesion molecule expression
The influence of roscovitine on the inflammatory activation of leukocytes and endothelial cells was investigated by analysing ROS production and CD11b expression and levels of endothelial cell adhesion molecules respectively. In human granulocytes, roscovitine did not influence the fMLP-induced oxidative burst (Figure 2A), but decreased the surface levels of CD11b (Figure 2B). In human endothelial cells, the TNF-α-evoked surface protein expression of the adhesion molecules ICAM-1 (Figure 2C), E-selectin (Figure 2D) and VCAM-1 (Figure 2E) was concentration-dependently inhibited by roscovitine with IC50 values of ∼1 µM for all three adhesion molecules. Also mRNA expression of ICAM-1 was decreased by roscovitine (Figure 2F), suggesting that roscovitine might affect transcriptional processes. To exclude the possibility that these effects are simply caused by cytotoxicity, we analysed the apoptosis-inducing action of roscovitine (in the absence of TNF-α) in endothelial cells and found that apoptosis rates were not altered at concentrations up to 30 µM applied for 24 h and 48 h (data not shown). In summary, these results indicated that leukocyte activation might be slightly inhibited by roscovitine but that the inhibition of endothelial activation was much more pronounced, at even lower concentrations. Thus, we decided to proceed with an in-depth analysis of the mechanisms utilized by roscovitine to interfere with endothelial activation pathways.
Figure 2.
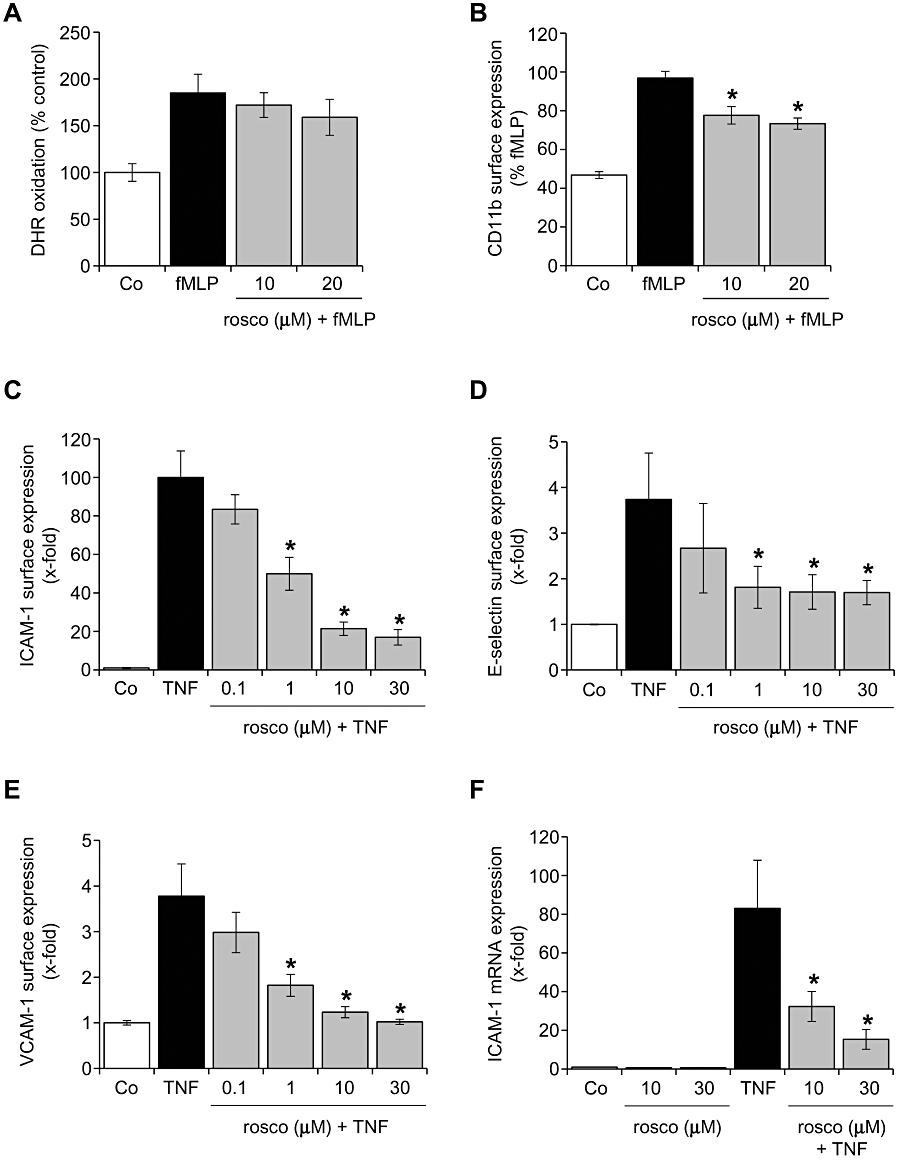
Roscovitine slightly decreased leukocyte activation and strongly inhibited endothelial cell adhesion molecule expression. (A, B) Freshly isolated human granulocytes were either left untreated (Co) or pretreated with roscovitine (rosco) for 30 min. fMLP (100 nM) was applied for 15 min. (A) Granulocytes were loaded with 10 µM dihydrorhodamine (DHR) and analysed by flow cytometry. n = 3. (B) CD11b surface expression was measured by flow cytometry. n = 3. *P < 0.05 versus fMLP. (C–F) HUVECs were either left untreated (Co) or pretreated with roscovitine (rosco) for 30 min. TNF-α (10 ng·mL−1) was applied for 24 h (C, E), 6 h (D) or 4 h (F). Surface protein expression of ICAM-1 (C), E-selectin (D), VCAM-1 (E) was determined by flow cytometry. Expression of mRNA was measured by quantitative RT-PCR (F). n = 3, each. *P < 0.05 versus TNF-α. fMLP, formyl-methionyl-leucyl-phenylalanine; HUVECs, human umbilical vein endothelial cells; ICAM, intercellular cell adhesion molecule; TNF-α, tumour necrosis factor-α; VCAM, vascular cell adhesion molecule.
Roscovitine blocked NF-κB-dependent gene expression, but did not influence the NF-κB activation cascade
Expression of adhesion molecules on the endothelium is largely governed by the transcription factor NF-κB. In a dual luciferase reporter gene assay, roscovitine strongly attenuated NF-κB-dependent reporter gene expression induced by TNF-α (Figure 3A). Consequently, we hypothesized that roscovitine interfered with the NF-κB activation cascade. However, we could detect any influence of roscovitine on neither the activity of recombinant IKKβ (Figure 3B) nor TNF-α-induced cellular IKKα/β activation (Figure 3C). For a satisfactory detection of phospho-IKK in Figure 3C, cells were additionally treated with the phosphatase inhibitor calyculin (1 nM). Also the TNF-α-evoked degradation of the NF-κB inhibitor IκB-α (Figure 3D), the translocation of the NF-κB subunit p65 into the nucleus (Figure 3E) and the DNA-binding activity of NF-κB (Figure 3F) were unaltered after exposure of HUVECs to roscovitine.
Figure 3.
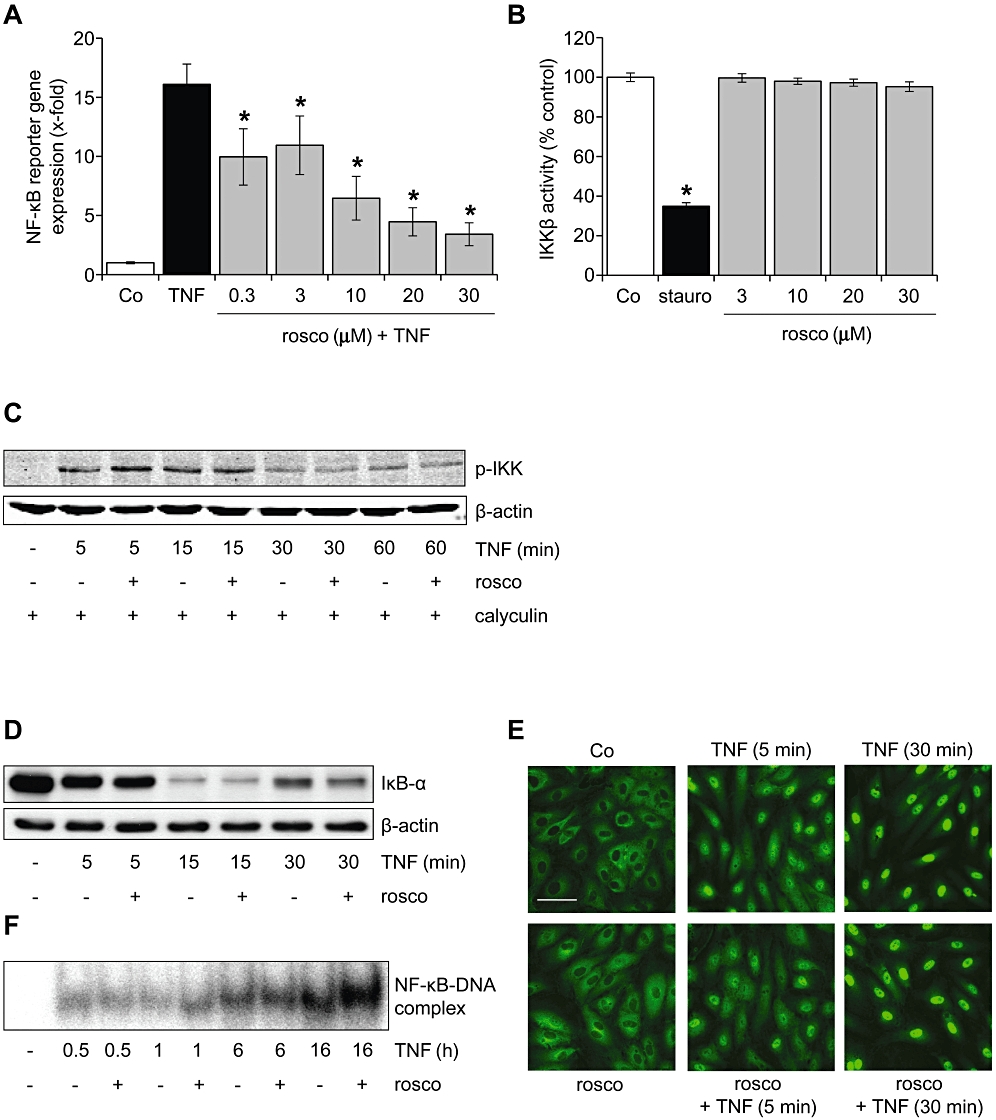
Roscovitine blocked NF-κB-dependent gene expression, but did not influence the NF-κB activation cascade. (A) HUVECs were either left untreated (Co) or pretreated with roscovitine (30 min) and subsequently treated with TNF-α (10 ng·mL−1) for 6 h. Levels of NF-κB-dependent gene expression were analysed by dual luciferase reporter gene expression. n = 3. *P < 0.05 versus TNF-α. (B) The influence of roscovitine on the activity of recombinant IKKβ was assessed by an in vitro kinase assay. Staurosporine (stauro, 100 µM) served as positive control. n = 3. *P < 0.05 versus Co. (C–F) HUVECs were either left untreated (Co) or pretreated with roscovitine (30 min, 10 µM) and subsequently treated with TNF-α (10 ng·mL−1). One representative image out of three independently performed experiments is shown for each assay. (C) Levels of phospho-IKKα/β and β-actin were assessed by Western blot analysis. HUVECs were additionally treated with the protein phosphatase inhibitor calyculin (1 nM) to stabilize the phosphorylation. (D) Levels of IκB-α and β-actin were assessed by Western blot analysis. (E) The NF-κB p65 subunit was visualized by immunocytochemistry and confocal microscopy. White bar = 20 µm. (F) NF-κB DNA-binding activity was determined by electrophoretic mobility shift assay (EMSA). HUVECs, human umbilical vein endothelial cells; IKK, IκB kinase; NF-κB, nuclear factor-κB; TNF-α, tumour necrosis factor-α.
Roscovitine blocked several protein kinases, but only the inhibition of CDK5 and CDK9 resulted in a decrease of endothelial activation
To investigate which signalling pathways were affected by roscovitine, a cellular kinome array was performed, i.e. the activity of kinases (phosphorylation of target peptides) in roscovitine-treated intact endothelial cells was determined. The kinases with the highest reduction of their activity in roscovitine- and TNF-α-treated human endothelial cells compared with only TNF-α-treated cells, as displayed in Table 1, are PKA, CDK5 and ribosomal S6 kinase (RS6K). Pharmacological inhibitors and a gene silencing approach were utilized to test if these kinases inhibited by roscovitine were crucial players in the pro-inflammatory action of TNF-α. ICAM-1 expression was used as a feasible read-out parameter to judge the role of kinase inhibition. As shown in Figure 4A, blockade of PKA activity by two different inhibitors did not change the ability of TNF-α to induce ICAM-1. Indirect inhibition of RS6K by rapamycin, a blocker of the RS6K upstream kinase mammalian target of rapamycin, evoked a decrease of ICAM-1 expression (Figure 4B), but this effect was not prominent enough to explain the strong attenuation observed by roscovitine. However, silencing of CDK5 in endothelial cells led to a pronounced reduction of ICAM-1 expression (Figure 4C).
Figure 4.
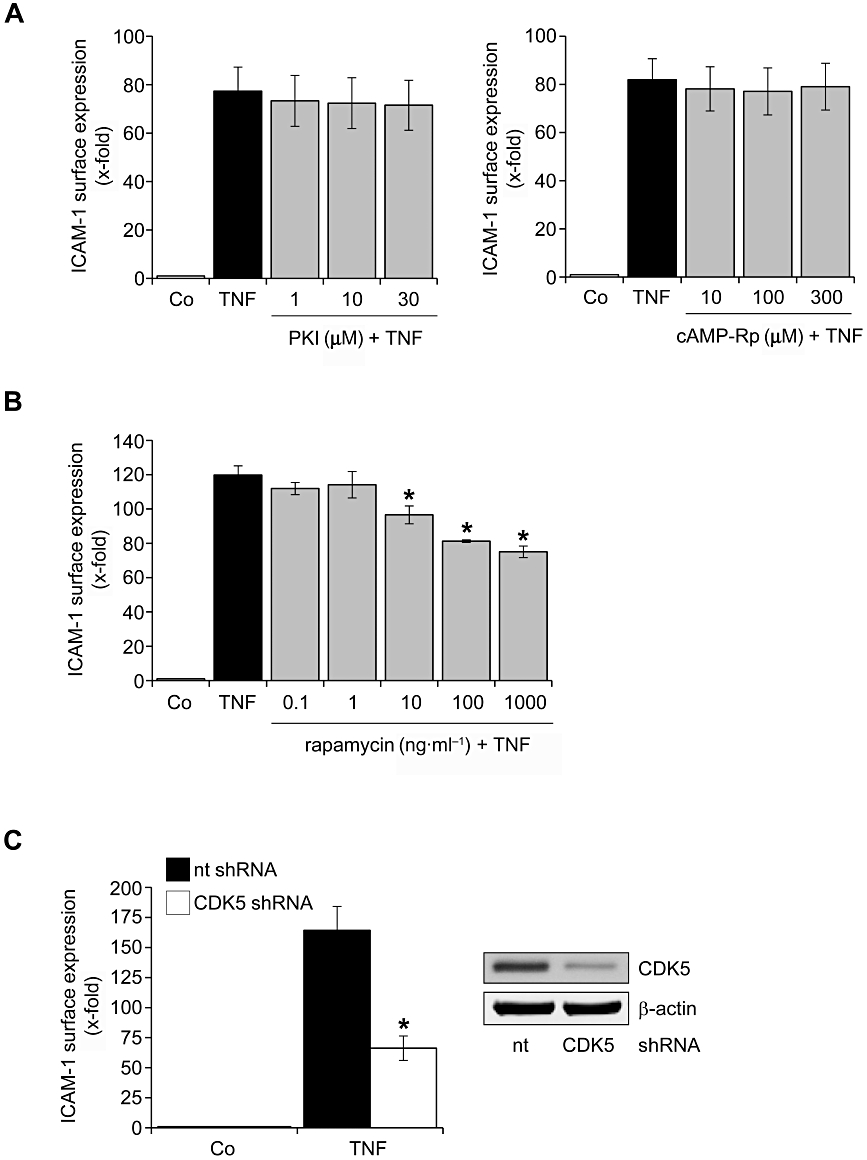
Silencing of CDK5 and inhibition of RS6K attenuates TNF-α-induced ICAM-1 expression, whereas inhibition of PKA has no influence. (A, B) HUVECs were either left untreated (Co) or pretreated for 30 min with the PKA inhibitors PKI (A, left panel) and cAMP-Rp (A, right panel) or the mTOR inhibitor rapamycin (B). TNF-α (10 ng·mL−1) was applied for 24 h. Surface protein expression of ICAM-1 was analysed by flow cytometry. n = 3, each. (C) In HUVECs, CDK5 gene expression was silenced via shRNA. Proof of successful gene silencing (Western blot) is depicted in the right panel. Seventy-two hours after transfection, cells were treated with TNF-α (10 ng·mL−1) for 24 h. Surface protein expression of ICAM-1 was analysed by flow cytometry. n = 3. *P < 0.05 versus TNF-α. CDK, cyclin-dependent kinase; HUVECs, human umbilical vein endothelial cells; ICAM, intercellular cell adhesion molecule; mTOR, mammalian target of rapamycin; PKA, protein kinase A; RS6K, ribosomal S6 kinase; TNF-α, tumour necrosis factor-α.
As the kinome array only covered a very limited number of different CDKs, we additionally tested the inhibitory action of roscovitine on recombinant CDKs in an in vitro kinase assay (Table 2). Due to the low IC50 value of roscovitine on adhesion molecule expression (1 µM), we infer from Table 2 that CDK2, 7 and 9 (IC50 on recombinant enzyme in the range of 1 µM) are likely to be targets underlying the action of roscovitine. Consequently, we silenced gene expression of these CDKs and found that CDK2 (Figure 5A) was not involved in the up-regulation of ICAM-1 by TNF-α. Despite the incomplete down-regulation of CDK7, our data also suggested that CDK7 did not play a major role in ICAM-1 up-regulation (Figure 5B), whereas CDK9 was of great importance for ICAM-1 expression (Figure 5C). Taken together, our findings suggest that the inhibition of CDK5 and CDK9 might be the basis for the action of roscovitine. As CDK9 is part of the positive transcription elongation factor P-TEFb, we tested whether the inhibition of CDK9 by roscovitine affected the transcriptional process. Thus, we measured the phosphorylation of Ser2/5 of the C-terminal domain (CTD) of the RNA polymerase (RNAP) II subunit Rpb1 and found that roscovitine was able to suppress RNAP II activity within 1 h, at concentrations >10 µM (Figure 5D).
Table 2.
In vitro CDK panel
| CDK | IC50 (µM) |
|---|---|
| CDK1 | 4.5 |
| CDK2 | 0.4 |
| CDK4 | 9.2 |
| CDK5 | 1.4 |
| CDK7 | 0.9 |
| CDK9 | 1.4 |
The table indicates the IC50 values of roscovitine on the respective recombinant CDK.
CDK, cyclin-dependent kinase.
Figure 5.
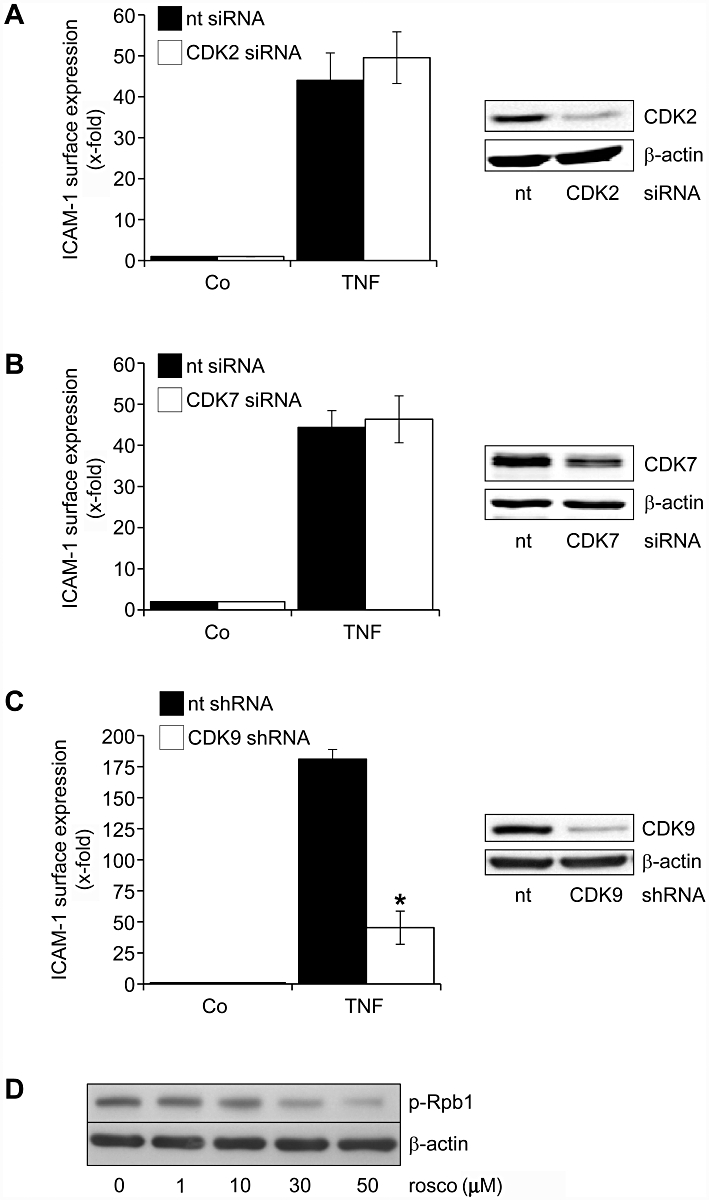
Silencing of CDK9, but not of CDK2 and CDK7, prevents the up-regulation of ICAM-1 by TNF-α. (A–C) In HUVECs, CDK2, CDK7 and CDK9 gene expression were silenced by siRNA (A, B) or shRNA (C). Proof of successful gene silencing (Western blot) is depicted in the right panel, each. Twenty-four hours (A, B) and 72 h (C) after transfection, cells were treated with TNF-α (10 ng·mL−1, 24 h). Surface protein expression of ICAM-1 was analysed by flow cytometry. n = 3, each. *P < 0.05 versus TNF-α. (D) HUVECs were either left untreated (Co) or treated with roscovitine for 1 h. Levels of phospho-Ser2/5-Rpb1 CTD and β-actin were analysed by Western blotting. n = 3, one representative image is shown for each assay. CDK, cyclin-dependent kinase; HUVECs, human umbilical vein endothelial cells; ICAM, intercellular cell adhesion molecule; TNF-α, tumour necrosis factor-α.
Discussion
Beyond its anti-cancer action, roscovitine has, in the last few years, been increasingly recognized to possess profound anti-inflammatory properties in vivo. Roscovitine protects against acute graft-versus-host disease by blocking the clonal expansion of T cells (Li et al., 2009) as well as against glomerulonephritis by inhibition of the proliferative response of T and B cells (Zoja et al., 2007). Moreover, roscovitine is effective in mouse models of neutrophil-dominant inflammation, such as carrageenan-induced pleurisy, bleomycin lung injury and arthritis by augmenting neutrophil apoptosis (Rossi et al., 2006). Mechanistically, roscovitine was found to promote neutrophil apoptosis in a caspase-dependent manner and via down-regulation of the cell survival protein Mcl-1 (Leitch et al., 2010). Thus, the anti-inflammatory effect of roscovitine has been ascribed to its inhibition of proliferation and induction of cell death in immune cells and, consequently, recent research focused on these actions in the context of resolution of inflammation (Leitch et al., 2009).
Surprisingly, the influence of roscovitine on the onset of inflammation – beyond its cell death-inducing action – was investigated to a much lesser extent: roscovitine was found to decrease the inflammatory activation of macrophages, such as NO generation or cytokine production (Du et al., 2009; Jhou et al., 2009). Liu et al. showed that roscovitine attenuated the spontaneous adhesion of Jurkat T cells and peripheral blood mononuclear cells to the extracellular matrix (Liu et al., 2008). Nevertheless, effects of roscovitine on the activation of vascular endothelial cells and the interaction of leukocytes with the activated endothelium – two steps of critical importance in the onset of inflammation – have not been evaluated so far. Thus, our work provides for the first time evidence that roscovitine effectively protects against leukocyte recruitment to inflamed tissue by inhibition of expression of adhesion molecule on endothelial cells.
In vitro, roscovitine effectively diminished the expression of E-selectin, which is necessary for leukocyte rolling, as well as that of ICAM-1 and VCAM-1, which are responsible for leukocyte firm adhesion. Interestingly, in vivo, leukocyte rolling was not affected by roscovitine and the leukocyte rolling velocity was only slightly increased, whereas firm adherence and transmigration were clearly inhibited. One can speculate that roscovitine might in vivo primarily act on ICAM-1 and VCAM-1 expression, but only marginally on E-selectin expression.
Importantly, an increased leukocyte apoptosis triggered by roscovitine could be ruled out in our in vivo and in vitro models. A damping action on leukocyte activation might play a role for the observed in vivo effect; however, the data suggest that roscovitine unexpectedly targets endothelial cells to a much higher degree. Reports of effects of roscovitine on endothelial cells are rare and have, as yet, only been concerned with a potential anti-angiogenic role of the compound (Maggiorella et al., 2009).
Regarding the dosage of roscovitine, we applied a bolus injection of 3.5 µg in order to reach an initial calculated plasma concentration of approximately 10 µM. This in vivo concentration was chosen as it causes an effective inhibition of endothelial activation in vitro. In other animal inflammation models, roscovitine was applied i.p. at 10–200 mg·kg−1 (Rossi et al., 2006; Zoja et al., 2007). Data about plasma concentrations in these animals are not available. In humans (phase I and II clinical trials) roscovitine was given orally over a very broad dose range, from 200–3200 mg·day−1 (Aldoss et al., 2009). In a study with patients suffering from nasopharyngeal tumours, 400–800 mg roscovitine were administered twice daily, which resulted in plasma levels of 1.3–6.6 µg·mL−1, i.e. 3.7–18.6 µM (Hsieh et al., 2009). These studies suggest that the doses used in our work are tolerable and can be easily attained in humans.
The profound suppression of endothelial activation by roscovitine can be easily explained by the strong inhibition of NF-κB-dependent gene expression, because NF-κB is a major pro-inflammatory transcription factor in the endothelium (De Martin et al., 2000). Inhibition of NF-κB activation by roscovitine has also been reported in macrophages (Du et al., 2009; Jhou et al., 2009) and different cancer cell lines (Dey et al., 2008). In these studies, roscovitine was clearly demonstrated to affect IKK activity, IκBα degradation or p65 nuclear translocation. Intriguingly, none of these key players of the canonical NF-κB activation cascade was influenced by roscovitine in endothelial cells. One can speculate that this discrepancy is due to cell type-dependent CDK expression patterns or to a different interaction of CDKs with the NF-κB pathway. However, information about these issues is lacking and further research is required to clarify this discrepancy.
Given the fact that roscovitine did also not affect NF-κB DNA-binding activity, but decreased expression of NF-κB-dependent mRNA (ICAM-1) and protein (ICAM-1 and luciferase reporter gene), it can be assumed that roscovitine interferes with gene transcription processes, such as transcription initiation, elongation, etc. By applying a cellular kinome array and a CDK kinase panel, we studied the action of roscovitine on kinases that might play a role in these events. Data about an influence of roscovitine treatment on the activity of a broad spectrum of kinases in intact endothelial cells are as yet lacking, whereas information on the influence of roscovitine on recombinant kinases are already available (Karaman et al., 2008). We revealed that the inhibition of CDK9 was highly relevant to the action of roscovitine. CDK9, which is not involved in the cell cycle, is part of the positive transcription elongation factor b (P-TEFb), which regulates the elongation phase of RNA polymerase (RNAP) II-dependent gene transcription and acts by phosphorylation of negative elongations factors as well as of RNAPII (Wang and Fischer, 2008). Studies on multiple myeloma cells demonstrated that roscovitine can in fact evoke a dephosphorylation of RNAPII (MacCallum et al., 2005). We could demonstrate that roscovitine leads to a dephosphorylation of RNAPII in endothelial cells, suggesting that the CDK9-mediated inhibition of RNAPII participates in the action of roscovitine.
Recently, a selective modulation of gene expression by CDK9 has been reported and CDK9 inhibition was found to not only decrease, but also to strongly up-regulate gene expression (Garriga et al., 2010). Moreover, P-TEFb has been recently discovered to play an important role in TNF-α-induced NF-κB activation by forming a protein complex with the NF-κB p65 subunit (Barboric et al., 2001) and to be of importance for the activation of a subset of NF-κB-dependent target genes (Nowak et al., 2008). Thus, CDK9 inhibition by roscovitine does not exert detrimental effects on global transcription, which would result in strong cytotoxicity, but seems to specifically interfere with the transcription of genes triggering endothelial activation, such as ICAM-1.
Besides CDK9, we found that inhibition of CDK5 was also linked to the anti-inflammatory action of roscovitine, whereas the cell cycle-associated CDK2 and the transcription-regulating CDK7 did not play a crucial role. CDK5 is known as an important player in the regulation of neuronal architecture and function. Although extra-neuronal effects of CDK5 have been increasingly recognized in the last years (Rosales and Lee, 2006), studies describing a role for CDK5 in inflammatory processes are still rare: Utreras et al. reported that TNF-α increases the activity of CDK5 in PC12 cells (Utreras et al., 2009). Utilizing N4-(6-aminopyrimidin-4-yl)-sulphanilamide as a CDK2/5 inhibitor, Du et al. suggested that CDK5 was involved in the LPS-induced activation of macrophages (Du et al., 2009). Moreover, CDK5 regulated the transcriptional activity of STAT3 (Fu et al., 2004), an important mediator of the IL-6-induced pro-inflammatory gene expression. Data on the effects of CDK5 on the NF-κB pathway and on endothelial activation in general are lacking. Our ongoing research is intended to clarify this issue.
In summary, our study shows that roscovitine exhibits anti-inflammatory properties not only by inducing leukocyte apoptosis and thus enhancing the resolution of inflammation, but also by inhibiting vital steps in the onset of inflammation, i.e. the activation of endothelial cells and the interaction of leukocytes with the endothelium. These novel insights might lead to the clinical evaluation of roscovitine as a promising anti-inflammatory compound. Moreover, this work also highlights CDK5 and CDK9 as interesting targets for the treatment of inflammation-associated diseases.
Acknowledgments
This study was supported in part by a grant from the European Community (FP6-2002-Life Science & Health, Prokinase Research Project, project no. LSHB-CT-2004-503467) and by the German Research Foundation (DFG, VO 376/11-1/2). We thank Bianca Hager and Jana Peliskova for excellent technical assistance.
Glossary
Abbreviations
- CDK
cyclin-dependent kinase
- ICAM
intercellular cell adhesion molecule
- IKK
IκB kinase
- IκB
inhibitor of κB
- NF-κB
nuclear factor-κB
- PKA
protein kinase A
- RS6K
ribosomal S6 kinase
- VCAM
vascular cell adhesion molecule
Conflicts of interest
None.
Supporting Information
Teaching Materials; Figs 1–5 as PowerPoint slide.
References
- Aldoss IT, Tashi T, Ganti AK. Seliciclib in malignancies. Expert Opin Investig Drugs. 2009;18:1957–1965. doi: 10.1517/13543780903418445. [DOI] [PubMed] [Google Scholar]
- van Baal JW, Diks SH, Wanders RJ, Rygiel AM, Milano F, Joore J, et al. Comparison of kinome profiles of Barrett's esophagus with normal squamous esophagus and normal gastric cardia. Cancer Res. 2006;66:11605–11612. doi: 10.1158/0008-5472.CAN-06-1370. [DOI] [PubMed] [Google Scholar]
- Baez S. An open cremaster muscle preparation for the study of blood vessels by in vivo microscopy. Microvasc Res. 1973;5:384–394. doi: 10.1016/0026-2862(73)90054-x. [DOI] [PubMed] [Google Scholar]
- Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell. 2001;8:327–337. doi: 10.1016/s1097-2765(01)00314-8. [DOI] [PubMed] [Google Scholar]
- Blagden S, de Bono J. Drugging cell cycle kinases in cancer therapy. Curr Drug Targets. 2005;6:325–335. doi: 10.2174/1389450053765824. [DOI] [PubMed] [Google Scholar]
- De Martin R, Hoeth M, Hofer-Warbinek R, Schmid JA. The transcription factor NF-kappa B and the regulation of vascular cell function. Arterioscler Thromb Vasc Biol. 2000;20:E83–E88. doi: 10.1161/01.atv.20.11.e83. [DOI] [PubMed] [Google Scholar]
- Dey A, Wong ET, Cheok CF, Tergaonkar V, Lane DP. R-Roscovitine simultaneously targets both the p53 and NF-kappaB pathways and causes potentiation of apoptosis: implications in cancer therapy. Cell Death Differ. 2008;15:263–273. doi: 10.1038/sj.cdd.4402257. [DOI] [PubMed] [Google Scholar]
- Du J, Wei N, Guan T, Xu H, An J, Pritchard KA, et al. Inhibition of CDKS by roscovitine suppressed LPS-induced *NO production through inhibiting NFkappaB activation and BH4 biosynthesis in macrophages. Am J Physiol Cell Physiol. 2009;297:C742–C749. doi: 10.1152/ajpcell.00138.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu AK, Fu WY, Ng AK, Chien WW, Ng YP, Wang JH, et al. Cyclin-dependent kinase 5 phosphorylates signal transducer and activator of transcription 3 and regulates its transcriptional activity. Proc Natl Acad Sci U S A. 2004;101:6728–6733. doi: 10.1073/pnas.0307606100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriga J, Xie H, Obradovic Z, Grana X. Selective control of gene expression by CDK9 in human cells. J Cell Physiol. 2010;222:200–208. doi: 10.1002/jcp.21938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh WS, Soo R, Peh BK, Loh T, Dong D, Soh D, et al. Pharmacodynamic effects of seliciclib, an orally administered cell cycle modulator, in undifferentiated nasopharyngeal cancer. Clin Cancer Res. 2009;15:1435–1442. doi: 10.1158/1078-0432.CCR-08-1748. [DOI] [PubMed] [Google Scholar]
- Jhou RS, Sun KH, Sun GH, Wang HH, Chang CI, Huang HC, et al. Inhibition of cyclin-dependent kinases by olomoucine and roscovitine reduces lipopolysaccharide-induced inflammatory responses via down-regulation of nuclear factor kappaB. Cell Prolif. 2009;42:141–149. doi: 10.1111/j.1365-2184.2009.00584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Leitch AE, Haslett C, Rossi AG. Cyclin-dependent kinase inhibitor drugs as potential novel anti-inflammatory and pro-resolution agents. Br J Pharmacol. 2009;158:1004–1016. doi: 10.1111/j.1476-5381.2009.00402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch AE, Riley NA, Sheldrake TA, Festa M, Fox S, Duffin R, et al. The cyclin-dependent kinase inhibitor R-roscovitine down-regulates Mcl-1 to override pro-inflammatory signalling and drive neutrophil apoptosis. Eur J Immunol. 2010;40:1127–1138. doi: 10.1002/eji.200939664. [DOI] [PubMed] [Google Scholar]
- Li L, Wang H, Kim J, Pihan G, Boussiotis V. The cyclin dependent kinase inhibitor (R)-roscovitine prevents alloreactive T cell clonal expansion and protects against acute GvHD. Cell Cycle. 2009;8:1794–1802. doi: 10.4161/cc.8.11.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Schwartz B, Tsubota Y, Raines E, Kiyokawa H, Yonekawa K, et al. Cyclin-dependent kinase inhibitors block leukocyte adhesion and migration. J Immunol. 2008;180:1808–1817. doi: 10.4049/jimmunol.180.3.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, et al. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005;65:5399–5407. doi: 10.1158/0008-5472.CAN-05-0233. [DOI] [PubMed] [Google Scholar]
- Maggiorella L, Aubel C, Haton C, Milliat F, Connault E, Opolon P, et al. Cooperative effect of roscovitine and irradiation targets angiogenesis and induces vascular destabilization in human breast carcinoma. Cell Prolif. 2009;42:38–48. doi: 10.1111/j.1365-2184.2008.00570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mempel TR, Moser C, Hutter J, Kuebler WM, Krombach F. Visualization of leukocyte transendothelial and interstitial migration using reflected light oblique transillumination in intravital video microscopy. J Vasc Res. 2003;40:435–441. doi: 10.1159/000073902. [DOI] [PubMed] [Google Scholar]
- Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- Nowak DE, Tian B, Jamaluddin M, Boldogh I, Vergara LA, Choudhary S, et al. RelA Ser276 phosphorylation is required for activation of a subset of NF-kappaB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol Cell Biol. 2008;28:3623–3638. doi: 10.1128/MCB.01152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales JL, Lee KY. Extraneuronal roles of cyclin-dependent kinase 5. Bioessays. 2006;28:1023–1034. doi: 10.1002/bies.20473. [DOI] [PubMed] [Google Scholar]
- Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12:1056–1064. doi: 10.1038/nm1468. [DOI] [PubMed] [Google Scholar]
- Utreras E, Futatsugi A, Rudrabhatla P, Keller J, Iadarola MJ, Pant HC, et al. Tumor necrosis factor-alpha regulates cyclin-dependent kinase 5 activity during pain signaling through transcriptional activation of p35. J Biol Chem. 2009;284:2275–2284. doi: 10.1074/jbc.M805052200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Fischer PM. Cyclin-dependent kinase 9: a key transcriptional regulator and potential drug target in oncology, virology and cardiology. Trends Pharmacol Sci. 2008;29:302–313. doi: 10.1016/j.tips.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Zoja C, Casiraghi F, Conti S, Corna D, Rottoli D, Cavinato RA, et al. Cyclin-dependent kinase inhibition limits glomerulonephritis and extends lifespan of mice with systemic lupus. Arthritis Rheum. 2007;56:1629–1637. doi: 10.1002/art.22593. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.