

Abstract
A 16-year-old boy affected by Sotos syndrome was referred to our clinic for cardiac evaluation in order to play noncompetitive sport. Physical examination was negative for major cardiac abnormalities and rest electrocardiogram detected only minor repolarization anomalies. Transthoracic echocardiography showed left ventricular wall thickening and apical trabeculations with deep intertrabecular recesses, fulfilling criteria for isolated left ventricular noncompaction (ILVNC). Some sporadic forms of ILVNC are reported to be caused by a mutation on CSX gene, mapping on chromosome 5q35. To our knowledge, this is the first report of a patient affected simultaneously by Sotos syndrome and ILVNC.
1. Introduction
Sotos syndrome [1] is an autosomal dominant condition, mainly occurring as a neomutation, characterized by a distinctive facial appearance, learning disability, and overgrowth resulting in tall stature and macrocephaly. This syndrome is known to be associated in 25% of cases with renal and cardiac abnormalities, such as patent ductus arteriosus, ventricular and/or atrial septal defects, seizures and/or scoliosis, and less frequently with a broad variety of additional features (hearing loss, abnormal vision, and thyroid disorders). In 2002, Sotos syndrome was shown to be caused by mutations and deletions of NSD1 gene, which encodes a histone methyltransferase involved in chromatin regulation mechanism. Furthermore, NSD1, mapping in 5q35 region, has multiple functional domains and may play different roles in regulating transcriptional processes, thus playing a negative or positive role according to cellular environment. In most of cases, abnormalities occurring in NSD1 gene are missense mutations in functional domains, partial deletions, and 5q35 variable-size microdeletions [2] encompassing it. We report a case of a patient with Sotos syndrome, who met echocardiographic criteria for ILVNC.
2. Case Report
A 16-year-old boy affected by Sotos syndrome was referred to our outpatient cardiology clinic for fitness to noncompetitive sport. Diagnosis of Sotos syndrome had been established at birth and genetically confirmed. At physical examination, he presented with evident macrocephaly, height at the 96 percentile, normal psychomotoric development, and no hearing problems or thyroid disorders were detected. Rest electrocardiogram showed sinusal arrhythmia and an early repolarization pattern. Blood pressure was 120/70 mmHg, heart sounds were normal, no murmurs were present.
The 2D echocardiogram demonstrated mild thickening of the left ventricular wall in the inferior, posterior, and anterolateral segments of the apex, with a two-layered structure: a compacted epicardial layer and a non-compacted endocardial layer consisting of prominent trabeculations separated by deep intertrabecular recesses communicating, on color Doppler analysis, with the cavity (Figures 1 and 2). Furthermore, the non-compacted/compacted myocardium ratio, measured in short axis view at the end of systole, was >2, according to quantitative criteria proposed by Jenni et al. [3]. Left ventricular ejection fraction was normal and wall motion abnormalities were not detected. Diagnosis of ILVNC was considered. For confirmation of diagnosis, and in order to obtain perfusion imaging analysis, Cardiac Magnetic Resonance Imaging was performed in the affiliated Hospital Institution (Forlanini) and diagnosis of ILVNC was confirmed, with evidence of non-compaction also in the middle segments. In addition, research of areas of consistent fibrosis (by mean of delayed enhancement marker) resulted negative. Holter monitoring and stress electrocardiogram were substantially normal. Relatives of the patient were considered for screening, and none of them was positive for ILVNC detection. This reported cooccurrence of a rare genetical disease and an even rarer cardiac abnormality raises some considerations.
Figure 1.
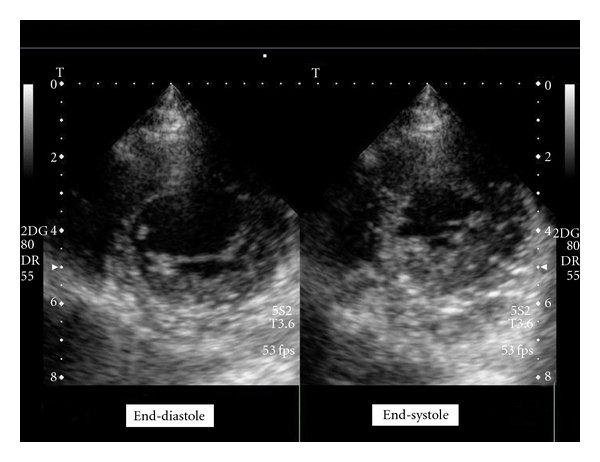
Echocardiographic short axis view showing apical segments of left ventricle during end-systole and end-diastole.
Figure 2.
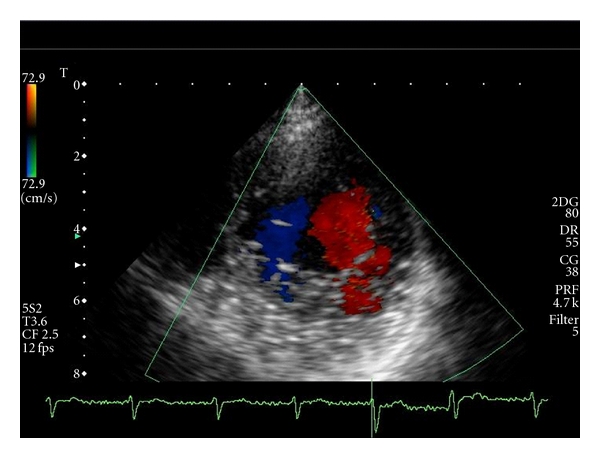
Transthoracic Doppler echocardiography (short axis view): the intertrabecular spaces are filled by direct blood flow from the ventricular cavity.
3. Discussion
ILVNC is a rare cardiomyopathy, characterized by the persistence of embryonic myocardial morphology, thought to be caused by an arrest of normal embryogenesis of the endocardium and myocardium, in the absence of other structural heart diseases which could explain the abnormal myocardial fibers development [4]. Clinical manifestations are highly variable, ranging from no symptoms to disabling congestive heart failure, arrhythmias, and systemic thromboembolism. Echocardiography represents the first-step procedure for diagnosis and shows prominent trabeculations with deep intertrabecular recesses in the left ventricle. ILVNC has shown to be associated with an heterogeneous genetic background [5], and autosomal dominant/recessive and X-linked familial recurrent forms have been implicated as causative. Most ILVNC are found in eumorphic individuals, but some complex genetic syndromes characterized by facial dismorphisms, skull malformation, and mild to severe delay in psychomotor development may include ILVNC as cardiac feature [6]. The loss of cardiac specific gene CSX, encoding for transcription factors with highly cardiac restricted expression, has been implicated in the development of some cases of ILVNC and deletion of distal chromosome 5q [7].
In this reported case, patient relatives were free from ILVNC, thus deposing for a sporadic form of the disease affecting the patient. If we consider that CSX gene maps at a distance of about 4 Mb from NSD1 gene [8] (the one involved in Sotos syndrome development), in chromosome 5q35, a possible wide microdeletion [9], although rare in Sotos syndrome, may encompass both of them, thus explaining the concomitance of these two pathologies. Another consideration relates to the possible, independent, specific role of gene NSD1, regardless of its defect, in genomic transcription and eventually in cardiac embryogenesis, as confirmed by the occurrence of congenital cardiac abnormalities, in about 20% of Sotos patients.
At present, we cannot establish whether in our patient the combination of ILVNC and Sotos syndrome is incidental or due to a common underlying genetic etiology, but the found association will help in considering further investigation on this topic.
In the meantime, we suggest cardiologists to consider also ILVNC together with the Sotos-associated cardiac anomalies when they examine patients affected by the syndrome.
Acknowledgment
The authors wish to thank Mr. Alessio Loreti for his contribution to scientific research.
References
- 1.Tatton-Brown K, Rahman N. Sotos syndrome. European Journal of Human Genetics. 2007;15(3):264–271. doi: 10.1038/sj.ejhg.5201686. [DOI] [PubMed] [Google Scholar]
- 2.Tatton-Brown K, Douglas J, Coleman K, et al. Multiple mechanisms are implicated in the generation of 5q35 microdeletions in Sotos syndrome. Journal of Medical Genetics. 2005;42(4):307–313. doi: 10.1136/jmg.2004.027755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenni R, Oechslin EN, Van Der Loo B. Isolated ventricular non-compaction of the myocardium in adults. Heart. 2007;93(1):11–15. doi: 10.1136/hrt.2005.082271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breckenridge RA, Anderson RH, Elliott PM. Isolated left ventricular non-compaction: the case for abnormal myocardial development. Cardiology in the Young. 2007;17(2):124–129. doi: 10.1017/S1047951107000273. [DOI] [PubMed] [Google Scholar]
- 5.Moric-Janiszewska E, Markiewicz-Łoskot G. Genetic heterogeneity of left ventricular noncompaction cardiomyopathy. Clinical Cardiology. 2008;31(5):201–204. doi: 10.1002/clc.20202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stöllberger C, Finsterer J, Blazek G. Left ventricular hypertrabeculation/noncompaction and association with additional cardiac abnormalities and neuromuscular disorders. American Journal of Cardiology. 2002;90(8):899–902. doi: 10.1016/s0002-9149(02)02723-6. [DOI] [PubMed] [Google Scholar]
- 7.Pauli RM, Scheib-Wixted S, Cripe L, Izumo S, Sekhon GS. Ventricular noncompaction and distal chromosome 5q deletion. American Journal of Medical Genetics. 1999;85(4):419–423. [PubMed] [Google Scholar]
- 8. NSD1 nuclear receptor binding SET domain protein 1 [Homo sapiens], Gene ID: 64324, updated on 21-May-2011, http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=DetailsSearch&Term=64324%5Buid%5D.
- 9.Saugier-Veber P, Bonnet C, Afenjar A, et al. Heterogeneity of NSD1 alterations in 116 patients with sotos syndrome. Human Mutation. 2007;28(11):1098–1107. doi: 10.1002/humu.20568. [DOI] [PubMed] [Google Scholar]