

Abstract
Nickel-catalysed reductive coupling reactions of alkynes have emerged as powerful synthetic tools for the selective preparation of functionalized alkenes. One of the greatest challenges associated with these transformations is control of regioselectivity. Recent work from our laboratory has provided an improved understanding of several of the factors governing regioselectivity in these reactions, and related studies have revealed that the reaction mechanism can differ substantially depending on the ligand employed. A discussion of stereoselective transformations and novel applications of nickel catalysis in coupling reactions of alkynes is also included.
Introduction
1.1 Addition reactions of alkynes
Alkynes are very useful functional groups in synthetic chemistry. They are stable to many common nucleophiles and electrophiles and generally resistant to mild acids, bases, and oxidants. Thus, the selective functionalization of alkynes via hydromet alation has proven to be a versatile tool in organic 1 synthesis. However, many of the common hydrometalations of alkynes require stoichiometric use of a metal reagent, and there has been growing interest in reactions of alkynes that are catalysed by a transition metal.2 Nickel has been associated with the catalytic reactions of alkynes s ince the seminal work of Reppe and Wilke3 and has since been shown to catalyse many such alkyne functionalization transformations4,5 including the addition of alkynes to enones (eq 1).6,7 The appearance in recent years of several excellent reviews on nickel-catalysed transformations reflects the growing interest in nickel catalysis.8 This account will focus on advances since 2004 in the area of nickel-catalysed carbon-carbon bond-forming reactions of alkynes, with an emphasis on improvements made to control selectivity, increase reactivity, and expand the substrate scope of certain alkyne addition reactions.
1.2 Nickel-catalysed reductive and alkylative coupling reactions of alkynes to form allylic alcohols
Allylic alcohols are a found in a variety of natural products (Figure 1).9 While numerous methods for the synthesis of allylic alcohols have been reported,10 routes that offer improved convergence and functional group compatibility continue to attract significant interest. For example, coupling reactions of alkynes and aldehydes that employ a stoichiometric reducing agent and a catalytic nickel source allow for single-step generation of this versatile functional group array.
Fig. 1.

1.2a) Intramolecular nickel-catalysed reductive and alkylative cyclization of α,ω-alkynals
In 1997, Montgomery and coworkers reported the first example of a nickel-catalysed reductive cyclization of an alkynal. In this transformation, diethylzinc served as the stoichiometric reducing agent, while the catalyst was composed of bis(1,5-cyclooctadiene)nickel(0) (Ni(cod)2) and tributylphosphine (eq 2).11 The authors observed that, in the absence of a catalytic phosphine additive, the alkylative cyclization product (i.e., transfer of Et instead of H from diethylzinc) is observed (eq 3).11 These cyclization strategies have been further developed and found several applications in total synthesis.12
1.2b) Intermolecular nickel-catalysed reductive and alkylative coupling of alkynes
The first report of an intermolecular nickel-catalysed coupling reaction of alkynes and aldehydes also employed diethylzinc as the stoichiometric reducing agent, and afforded the alkylative coupling product (i.e., transfer of Et from diethylzinc) (eq 4).11 To achieve intermolecular reductive coupling, our group examined a variety of reducing agents and catalytic ligand additives, eventually determining that use of triethylborane in the presence of catalytic Ni(cod)2 and tributylphosphine provided the E-trisubstituted allylic alcohol with excellent yield and selectivity (eq 5).13,14 We later found that a similar transformation was also possible using epoxides as coupling partners, which afforded homoallylic alcohol products (eq 6).15,16 An intermolecular, nickel-catalysed three-component coupling of alkynes, imines, and organoborane reagents (either boronic acids or trialkylboranes) was also developed (eq 7).17 These reactions all proceed with exclusive syn-addition across the alkyne, and often in excellent regioselectivity, making them attractive candidates for use in total synthesis. As such, nickel-catalysed coupling reactions of alkynes have been used in both fragment coupling reactions and macrocyclizations by ourselves18 and others.19
2 Regioselectivity in nickel-catalysed reductive coupling reactions
Several classes of alkynes have previously been shown to afford excellent regioselectivity in nickel-catalyzed coupling reactions, including aryl-substituted alkynes (Ar–C≡C–alkyl), alkynyl silanes (R–C≡C–SiR3), and terminal alkynes (R–C≡C–H) (Table 1).14,18,20 However, alkynes substituted with two sterically and electronically similar groups, such as dialkyl alkynes (i.e., alkyl–C≡C–alkyl’) typically afford poor regioselectivity (Table 1, entry 4).
Table 1.
Nickel-catalysed reductive coupling of alkynes and aldehydes
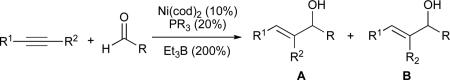 | |||
|---|---|---|---|
| entry | R1 | R2 | A:Ba |
| 1 | Ph | Me | 92:8 |
| 2 | n-hex | H | 96:4 |
| 3 | n-Bu | SiMe3 | >98:2 |
| 4 | n-hex | Et | 50:50 |
Determined by 1H NMR.
2.1 1,3-Enynes
To address this deficiency and based on a hypothesis that the high regioselectivity observed with aryl-substituted alkynes is likely due to an electronic differentiation between the alkyland the aryl- substituents, we considered the possibility that another conjugating group would provide similar regiocontrol (Figure 2). Of possible choices for such groups, a simple olefin seemed most intriguing as it might then be possible to convert this directing group into the corresponding saturated alkyl chain after the nickel-catalysed reductive coupling via site-selective hydrogenation.
Fig. 2.

1,3-Enynes as equivalents of aryl alkynes.
We found that coupling reactions of 1,3-enynes are highly regioselective when trialkylphosphines are used as ligands (Scheme 1).21 A variety of substitution patterns on the enyne are tolerated and both aldehydes and terminal epoxides can be employed as coupling partners. As terminal epoxides are readily available in highly enantiomerically-enriched form,22 this method represents a convenient synthesis of enantiomerically-enriched homoallylic dienes.
Scheme 1.

1,3-Enynes as highly regioselective substrates in nickel-catalysed reductive coupling reactions.
We initially hypothesized that the high regioselectivity observed with 1,3-enynes was simply due to an electronic distinction between the two alkyne substituents; however, several results dispute this assertion. First, complete regioselectivity was observed in a reaction of 1-phenyl-3-butenyne, suggesting that a vinyl group is a significantly more potent directing group than a phenyl ring (Scheme 2). Second, coupling reactions of 5,5-dimethyl-hexenyne not only proceed efficiently, but in excellent regioselectivity to favour C-C bond formation at the more hindered alkyne carbon. The latter result is in stark contrast to other tert-alkyl-substituted alkynes, which either do not react at all under these conditions, or favour exclusive formation of the opposite, less-hindered regioisomer. Thus, the alkene substituent appears to strongly direct regioselectivity and also significantly increase reactivity. We believe that this unique effect is a result of the ability of the olefin to form a favourable bonding interaction with the nickel in a high-energy intermediate such as 1, which serves to lower the energy of the transition state and thus influence regioselectivity.23 A site-selective, rhodium-catalysed reduction of the less substituted olefin in the dienyl alcohols products obtained provides access to alkyl-substituted allylic and homoallylic alcohols not otherwise accessible in a regioselective fashion using nickel-catalysed reductive coupling chemistry (eq 8).
Scheme 2.

Highly regioselective Ni-catalysed reductive coupling reactions of 1,3-enynes and aldehydes
2.2 1,6-Enynes
In order to determine whether the directing effect of the olefin observed with 1,3-enynes could be extended to non-conjugated systems, a series of enynes was synthesized and evaluated in nickel-catalysed reductive couplings with isobutyraldehyde (Table 2).24 Remarkably, in the absence of a phosphine additive, a marked difference in reactivity and selectivity was observed when the alkyne and alkene were separated by three methylene units (entry 4, Table 2). As it is very unlikely that enyne 4 is significantly different in a steric or electronic sense to alkynes 2, 3, or 5, it seems that direct involvement of the olefin in the reaction occurs uniquely in the case of the 1,6-enyne.
Table 2.
Directing effects of tethered alkenesa
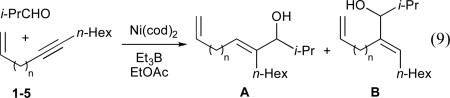 | ||||
|---|---|---|---|---|
| entry | alkyne | n | yield (%) | regioselectivity (A : B)b |
| 1 | 1 | 0 | <5 | n.d. |
| 2 | 2 | 1 | <5 | n.d. |
| 3 | 3 | 2 | <5 | n.d. |
| 4 | 4 | 3 | 53 | >95 : 5 |
| 5 | 5 | 4 | <5 | n.d. |
| 6 | n-pentyl–C≡C-n-hexyl | n.a. | 28 | 50 : 50 |
Ni(cod)2 (10 mol %), Et3B (200 mol %) in EtOAc (0.5 mL), 15 h at room temperature.
Determined by 1H NMR and/or GC.
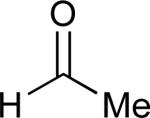
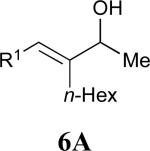
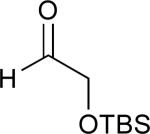
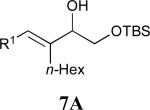
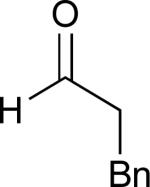
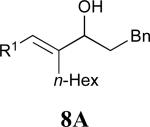
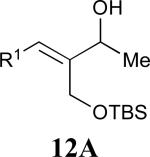
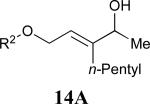
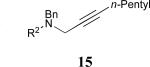
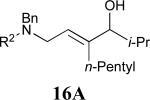
Considering that almost all other nickel-catalysed reductive coupling reactions previously reported required the addition of an external ligand,25 the coupling reaction of 1,6-enynes and aldehydes proved remarkably general (Table 3). The presence of an olefin tether was sufficient to overcome an inherent steric preference for the B regioisomer (entry 4, Table 3).26 Heteroatoms were also well tolerated and augment the versatility of this directed transformation (entries 6-8, Table 3).
Table 3.
Catalytic reductive coupling reactions directed by a remote alkenea
| enyne | aldehyde | product | yield | |
|---|---|---|---|---|
| 1 |

|
|
|
69% |
| 2 | 4 |
|
|
58% |
| 3 | 4 |
|
|
60% |
| 4 |
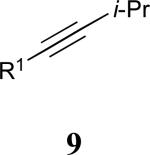
|
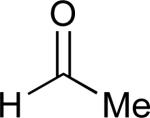
|
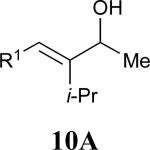
|
64% |
| 5 |
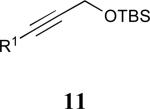
|
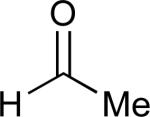
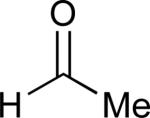
|
|
62% |
| 6 |
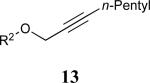
|
|
|
60% |
| 7 |
|
|
|
62% |
| 8 |
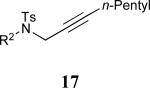
|
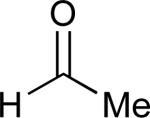
|
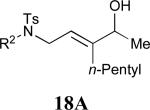
|
68% |
See eq 9, Table 2 for representative reaction. R1 = (CH2)3CH=CH2. R2 = CH2CH=CH2. Regioselectivity > 95:5 in all cases, determined by 1H NMR and/or GC.
When conducted in the presence of an organophosphine additive (PCyp3), the regioselectivity of the transformation showed a complete reversal, favouring regioisomer B (Scheme 3). The switch in regioselectivity upon the addition of a catalytic amount of an additive is highly unusual amoung tethered olefin-directed metal-mediated reactions.27 The effect of the ligand additive seemed dependant upon the size of the phosphine, as smaller phosphines such as PBu3 provided a mixture of regioisomers.20
Scheme 3.

Additive effects on regioselectivity.
We hypothesize that this pronounced ligand effect could be explained by considering a planar, 3-coordinate nickel complex28 that undergoes stereospecific ligand substitution with retention of stereochemistry (Scheme 4). 29,30 The olefin tether must bind ‘cis’ to carbon b to give 19. A series of stereospecific ligand substitutions then conserve this initial bias, thus controlling regioselectivity even if the C–C bond is formed when the olefin is not coordinated to the nickel.31
Scheme 4.

Origin of regioselectivity in nickel-catalysed coupling reactions of 1,6-enynes and aldehydes. See eq 6, table 6.
In the absence of a phosphine, the olefin remains bound to the nickel and the aldehyde displaces a weakly bound ligand L to afford complex 20. A phosphine ligand (e.g., PCyp3) binds strongly to the nickel center to give 21. The olefin tether is displaced preferentially by the aldehyde to afford 22, which undergoes C–C bond formation to give regioisomer B. Phosphines with smaller cone angles (e.g., PBu3) likely form 2:1 complexes with nickel, and thus will displace both L and the olefin. In this case, the aldehyde must displace either of two identical phosphine ligands, and a mixture of regioisomers results (24 and 25).
By incorporating a chiral stereocenter into the tether, we sought to probe the likelihood that the olefin remained bound, and hence imparted greater diastereoinduction, in the type I pathway, but not the type II and type III pathways (Table 4).32 In the absence of a phosphine ligand, a single regioisomer was observed (as expected), and in excellent diastereoselectivity (95:5) (entry 1). This result is consistent with the olefin being bound to the nickel center during the C-C bond forming step. In the presence of PCyp3, the opposite regioisomer is formed (>95:5) as approximately a 1:1 mixture of diastereomers (entry 2), suggesting that in this case the reaction proceeds through selective formation of complex 22. With PBu3, a mixture of regioisomers is observed, each of which is a mixture of diastereomers (entry 3), a result that is consistent with formation of a mixture of complexes 24 and 25. The observation of stereochemical induction in the presence of a chiral phosphine ligand (entries 4 and 5) offers further support for the proposal that the phosphine is bound in the type III and, by extrapolation, the type II pathways.
Table 4.
Coupling reactions of a chiral 1,6-enyne
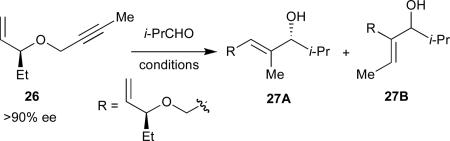 | ||||
|---|---|---|---|---|
| entry | reaction conditionsa | A : Bb | dr A (S:R)c | dr Bc |
| 1 | I | >95:5 | 95:5 | -- |
| 2 | II | <5:95 | -- | 45:55 |
| 3 | III | 55:45 | 50:50 | 45:55 |
| 4 | IV | 48:52 | 30:70 | 28:72 |
| 5 | V | 55:45 | 66:34 | 68:32 |
I: Ni(cod)2 (10 mol %), Et3B (200 mol %). II: Reaction conditions I + PCyp3 (20 mol %). III: Reaction conditions I + PBu3 (20 mol %). IV: Reaction conditions I + (R)-FcP(o̱-i-Pr)Ph (20 mol %). V: Reaction conditions I + (S)-FcP(o̱-i-Pr)Ph (20 mol %)
Based on isolated yields.
Determined by 1H NMR.
All of these results are best explained via stereospecific ligand substitution on a trisubstituted planar nickel complex. Therefore, for nickel-catalysed reductive coupling reactions of alkynes and aldehydes using triethylborane as the reducing agent and organophosphine ligands we propose that the reaction proceeds through complex 22.
2.3 Effect of N-heterocyclic carbene ligands
N-Heterocyclic carbenes (NHC's) are sterically hindered, electron-rich ligands that have proven useful in a wide variety of metal-catalysed transformations (Figure 3).33 Recent applications in nickel-catalysed reductive coupling reactions have further illustrated the influence of the ligand on both reaction mechanism and regioselectivity.34,35 Montgomery examined the macrocyclization of 28 and found that when R1 is phenyl, both phosphine and NHC ligands afford product 30; however, when R1 is methyl, different regioselectivities were observed (Scheme 5).33 Use of triethylborane as the reducing agent and PMe3 as the ligand favoured formation of endocyclic product 29 (9:2 ratio), whereas a combination of IPr and triethylsilane led to predominant formation of the exocyclic double bond (5:1). The use of a larger phosphine (PBu3) resulted in a reduced preference (3:1) for the endocyclic product and, similarly, a smaller NHC ligand (IMes) resulted in a 1:1 mixture of 29b:30b. A study which compared the ratio of crossover products for the two sets of conditions revealed an inherent difference in their mechanisms (Table 5).34 Very little crossover was observed when using the NHC ligand (entries 2/3:1/4, >96:4), suggesting that addition of hydride and silane occur simultaneously. However, in the presence of PBu3 significant amounts of crossover products are formed, indicating that, in this case, the hydride and silane are added in separate steps (entries 2/3:1/4, 57:43). This same study revealed that, when NHC's are employed as ligands, it is possible to use trialkylsilanes as reducing agents in intermolecular, nickel-catalysed reductive coupling of alkynes and aldehydes (Table 6).34,36 This method generates silyl-protected allylic alcohols in excellent yield and regioselectivity when aryl-substituted alkynes (Ar–C≡C–R), terminal alkynes (R–C≡C–H), and 1,3-enynes are used (Table 6, entries 1-4).
Fig. 3.

Representative NHC ligands.
Scheme 5.

Nickel-catalyzed reductive cyclizations.
Table 5.
Ligand dependence in the observation of crossover products
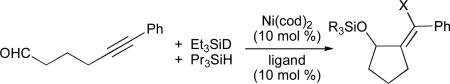
Crossover product.
Table 6.
Nickel-catalysed intermolecular reductive coupling reactions
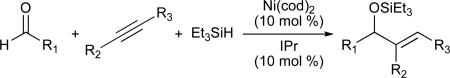 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | R3 | yield (regioselectivity) |
| 1 | Ph | CH3 | Ph | 84% (>98:2) |
| 2 | C6H13 | CH3 | Ph | 82% (>98:2) |
| 3 | Ph | H | C6H13 | 71% (>98:2) |
| 4 | Ph | Ph | C(CH3)=CH2 | 84% (>98:2) |
3 Stereoselective nickel-catalysed reductive coupling reactions
Despite the success of nickel-catalysed coupling reactions of alkynes and aldehydes, until recently very few techniques existed for controlling the configuration of the allylic alcohol stereocenter generated during the reaction. There were only two, excluding our own work, examples of stereoselective intermolecular reductive coupling reactions of alkynes with carbonyls in the 2004 review on nickel-catalysed reductive coupling reactions by Montgomery.12b In the past few years, many methods for the control of stereochemistry in intermolecular nickel-catalysed coupling reactions have been developed.
3.1 Enantioselective reactions
In 2003, we reported the first examples of catalytic, enantioselective reductive couplings of alkynes and aldehydes, using two distinct classes of chiral organophosphines as ligands (Figure 4). Superior results for alkynes containing one aromatic substituent (aryl–C≡C–alkyl) were achieved using neomenthyldiphenylphosphine ((+)-NMDPP) as a chiral ligand (Scheme 6).37 Alkynes substituted with two distinct alkyl groups (alkyl–C≡C–alkyl’) afforded reductive coupling products with modest enantioselectivities when P-chiral ferrocenyl phosphines 45/46 were employed.38
Fig. 4.

Scheme 6.

Asymmetric induction
A ligand-controlled, stereoselective nickel-catalysed reductive coupling was used in the synthesis of terpestacin Scheme 7).39 Fragments 33 and 34 were coupled to give the E-trisubstituted allylic alcohols 36 and 37 with good regioselectivity. The configuration of the allylic alcohol stereocenter was controlled by the configuration of the chiral phosphine. In comparison, the use of tributylphosphine gave a 1:1 mixture of 36:37 allowing for the synthesis of both terpestacin and epi-C11-terpestacin. It was originally believed that epi-C11-terpestacin was its own natural product, siccanol. However, when we obtained a sample of synthetic epi-C11-terpestacin it was determined that its spectra did not match that of isolated siccanol but rather that of terpestacin itself.40 Macrocyclization via an intramolecular allylation, followed by methylation and α-oxidation led to the completed synthesis of terpestacin and epi-C11-terpestacin.
Scheme 7.

Ligand control in the total synthesis of terpestacin
The P-chiral ferrocenyl phosphines developed in our laboratory have found use as ligands in several other asymmetric nickel-catalysed coupling reactions of alkynes. 1,3-Enynes underwent reductive coupling with a series of aromatic aldehydes in modest enantioselectivities in the presence of a ferrocenyl phosphine 32 (Scheme 8).41 Although higher enantiomeric excesses are available in the coupling of aryl-alkynes and aliphatic aldehydes (Scheme 9), the enantiomerically enriched dienols afforded via 1,3-enyne couplings offer significant flexibility for further modification.
Scheme 8.

Asymmetric induction with Ni-catalysed reductive coupling reactions of 1,3-enynes and aldehydes
Scheme 9.

Asymmetric induction with Ni-catalysed reductive coupling reactions of 1,3-enynes with ketones
The enhanced reactivity of 1,3-enynes observed in nickel-catalyzed reductive coupling reactions also allowed for the use of ketones as electrophiles. Such couplings promoted by 32 afforded 1,3-dienes with an adjacent quaternary carbinol stereocenter in excellent regioselectivity and up to 70% ee (Scheme 9).42 Site-selective reduction of the less-hindered olefin provides access to enantiomerically enriched tertiary allylic alcohols, and ozonolysis affords α-hydroxy ketones.
Enantioselective alkylative coupling reactions of alkynes and imines have been achieved using 32 to afford allylic amines in up to 89% ee (Scheme 10).43 The use of a removable (trialkylsilyloxy)ethyl protecting group allows for facile generation of primary allylic amines, which can then be recrystallized to optical purity as their maleic acid salts.
Scheme 10.

Asymmetric induction in the Ni-catalysed alkylative coupling of alkynes and imines.
3.2 Diastereoselective reactions
We recently reported the diastereoselective addition of aryl-alkynes (Ar–C≡C–alkyl) to α-hydroxy aldehydes (Scheme 11).44,45 High anti-selectivity is obtained regardless of protecting group on the α-hydroxy substituent. This is in sharp contrast to the syn-product typically observed when nucleophiles are added to aldehydes that possess an α-hydroxy group capable of coordination.46
Scheme 11.

Diastereoselective reductive coupling reactions of aryl-alkynes
In a related report by Montgomery, high anti-selectivity (>98:2 for R1 = n-pentyl or CH2Bn) was again observed (Scheme 12).47 In this case, a variety of silyl-substituted alkynes were employed (R3Si–C≡C–R), including terminal and non-aromatic variants, and it was demonstrated that global deprotection of all three silyl groups was possible using TBAF. With respect to the aldehyde, the reaction was most effective when the R1 substituent was unbranched; this was complimentary to our own work in which improved diastereoselectivities were obtained when R1 was 3°.
Scheme 12.

Diastereoselective reductive coupling reactions of silyl-alkynes
The modes of diastereoinduction in these systems are unknown. Although the results are consistent with a Felkin model in the absence of chelation, the mechanistic considerations involved in this system are significantly more complex than a classical metalated nucleophile. That being said, the observation that identical senses of induction are obtained both in the absence and presence of a possible chelating group suggests that the α-alkoxy group does not interact with nickel during the C-C bond-forming step.
Diastereoselectivity can also be influenced by substitution on the alkyne, as was observed in our work on the reductive coupling of 1,6-enynes and aldehydes. Very high levels of diastereomeric induction could be obtained by placing a chiral centre within the enyne tether, even when the substituent was quite small (Scheme 13). Although the scope of the diastereoselective nickel-catalysed reductive coupling reactions of 1,6-enynes and aldehydes have not been fully explored, the high selectivities observed would suggest it as a viable strategy for the stereoselective formation of allylic alcohols.
Scheme 13.

Diastereoselective phosphine-free nickel-catalysed coupling reaction.
By using the allylic alcohols formed in stereoselective nickel-catalysed reductive macrocyclizations as masked α-hydroxy ketones, we have completed the total syntheses of amphidinolides T1 and T4 (Scheme 14).48 In both natural products, the macrocyclization proceeds with excellent regioselectivity and diastereoselectivity. The regioselectivity is likely controlled by the phenyl substituent, while the diastereoselectivity may be a function of the cyclization since intermolecular variants were not as highly diastereoselective. Protection of the allylic alcohol followed by ozonolysis, selective methylenation and HF deprotection afforded the respective amphidinolides.
Scheme 14.

Diastereoselective nickel-catalysed reductive cyclization.
Conclusions
This review discusses recent developments in the area of nickel-catalysed coupling reactions of alkynes.49,50 In the nickel-catalysed reductive coupling of alkynes and aldehydes, significant advances have been made in improving substrate scope, controlling regioselectivity, and understanding operative reaction mechanisms. Development of novel ligand systems has allowed for additional control of regio-, diastereo-, and enantioselectivities. The use of both imines and weakly electrophilic ketones as coupling partners has been realized, and good to excellent enantioselectivities can be achieved in many cases.
It is clear that numerous challenges in the field remain, including, for example, better control of regioselectivity in couplings involving alkynes containing two distinct alkyl substituents (alkyl–C≡C–alkyl’). However, as understanding of these catalytic systems increases, enhancements in selectivity and generality may be obtained, thus supplementing the versatility of these selective transformations.
 |
(1) |
 |
 |
 |
(8) |
Notes and references
- 1.For a review discussing hydroboration, hydrosilylation, and hydrostannation of terminal and internal alkynes, see: Trost BM, Bell ZT. Synthesis. 2005:853.Hart DW, Blackburn TF, Schwartz J. J. Am. Chem. Soc. 1974;96:679. Hydrozirconation.Ashby EC, Noding SA. J. Org. Chem. 1980;45:1035. Hydroalumination.
- 2.For examples of regioselective catalytic additions to alkynes, see: Larock RC, Yum EK. J. Am. Chem. Soc. 1991;113:6689.Molander GA, Retsch WH. Organometallics. 1995;14:4570.Trost BM, Ball ZT. J. Am. Chem. Soc. 2001;123:12726. doi: 10.1021/ja0121033.
- 3.For reviews describing the early role of nickel in catalysis, see: Reppe W, Kutepow N. v., Magin A. Angew. Chem. Int. Ed. 1969;8:727.Wilke G. Angew. Chem. Int. Ed. 1988;27:185.
- 4.Tsuda T, Kiyoi T, Saegusa T. J. Org. Chem. 1990;55:2554. [Google Scholar]
- 5.Lautens M, Klute W, Tam W. Chem. Rev. 1996;96:49. doi: 10.1021/cr950016l. [DOI] [PubMed] [Google Scholar]
- 6.Montgomery J, Savchenko AV. J. Am. Chem. Soc. 1996;118:2099. [Google Scholar]
- 7.Ikeda S.-i., Yamamoto H, Kondo K, Sato Y. Organometallics. 1995;14:5015. [Google Scholar]
- 8.An entire issue was recently devoted to nickel-catalysis. Jamison TF, editor. Tetrahedron. 2006;62:7499–7610.
- 9.a Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Cancer Res. 1995;55:2325. Epothilones. [PubMed] [Google Scholar]; b Oka M, Iimura S, Tenmyo O, Sawada Y, Sugawara M, Ohkusa N, Yamamoto H, Kawano K, Hu S-L, Fukagawa Y, Oki T. J. Antibiotics. 1993;46:367. doi: 10.7164/antibiotics.46.367. (–)-Terpestacin. [DOI] [PubMed] [Google Scholar]; c Barchi JJ, Jr., Moore, R. E. RE, Patterson GML. J. Am. Chem. Soc. 1984;106:8193. (+)-Acutiphycin. [Google Scholar]
- 10.Nozaki-Hiyama-Kishi: Review: Fürstner A. Chem. Rev. 1999;99:991. doi: 10.1021/cr9703360. Enantioselective variant: Choi HW, Nakajima K, Demeke D, Kang FA, Jun HS, Wan ZK, Kishi Y. Org. Lett. 2002;4:4435. doi: 10.1021/ol026981x. Hydrometalation followed by transmetalation to vinyl zinc: Oppolzer W, Radinov RN. Helv. Chim. Acta. 1992;75:170.Wipf P, Kendall C. Chem. Eur. J. 2002;8:1778. doi: 10.1002/1521-3765(20020415)8:8<1778::aid-chem1778>3.0.co;2-h.
- 11.Oblinger E, Montgomery J. J. Am. Chem. Soc. 1997;119:9065. [Google Scholar]
- 12.For an extensive review of this topic, see: Montgomery J. Acc. Chem. Res. 2000;33:467. doi: 10.1021/ar990095d.Montgomery J. Angew. Chem. Int. Ed. 2004;43:3890. doi: 10.1002/anie.200300634.
- 13.Use of triethylborane as a stoichiometric reducing agent in the nickel-catalysed addition of 1,3-dienes to aldehydes: Kimura M, Ezoe A, Shibata K, Tamaru Y. J. Am. Chem. Soc. 1998;120:4033.
- 14.a Huang W-S, Chan J, Jamison TF. Org. Lett. 2000;2:4221. doi: 10.1021/ol006781q. [DOI] [PubMed] [Google Scholar]; b Miller KM, Huang W-S, Jamison TF. J. Am. Chem. Soc. 2003;125:3442. doi: 10.1021/ja034366y. [DOI] [PubMed] [Google Scholar]
- 15.Molinaro C, Jamison TF. J. Am. Chem. Soc. 2003;125:8076. doi: 10.1021/ja0361401. [DOI] [PubMed] [Google Scholar]
- 16.For a review, see: Miller KM, Molinaro C, Jamison TF. Tetrahedron: Asymmetry. 2003;14:3619.
- 17.Patel SJ, Jamison TF. Angew. Chem., Int. Ed. 2003;42:1364. doi: 10.1002/anie.200390349. [DOI] [PubMed] [Google Scholar]
- 18.a Chan J, Jamison TF. J. Am. Chem. Soc. 2003;125:11514. doi: 10.1021/ja0373925. [DOI] [PubMed] [Google Scholar]; b Colby EA, O'Brien KC, Jamison TF. J. Am. Chem. Soc. 2004;126:998. doi: 10.1021/ja039716v. [DOI] [PubMed] [Google Scholar]
- 19.See ref 12.
- 20.Miller KM. Ph.D. Thesis. Massachusetts Institute of Technology; Cambridge, MA: Jun, 2005. Selective, Nickel-Catalyzed Carbon–Carbon Bond-Forming Reactions of Alkynes. [Google Scholar]
- 21.Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J. Am. Chem. Soc. 2004;126:4130. doi: 10.1021/ja0491735. [DOI] [PubMed] [Google Scholar]
- 22.a Tokunaga M, Larrow JF, Kakuchi F, Jacobsen EN. Science. 1997;277:936. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]; b Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J. Am. Chem. Soc. 2002;124:1307. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 23.Crystallographic evidence for a similar interaction in a related Pt-complex: Benyunes SA, Fries A, Green M, Mahon MF, Papworth TM. J. Chem. Soc., Dalton Trans. 1993:3785.
- 24.a Miller KM, Jamison TF. J. Am. Chem. Soc. 2004;126:15342. doi: 10.1021/ja0446799. [DOI] [PubMed] [Google Scholar]; b Moslin RM, Jamison TF. Org. Lett. 2006;8:455. doi: 10.1021/ol052719n. [DOI] [PubMed] [Google Scholar]; c Moslin RM, Miller KM, Jamison TF. Tetrahedron. 2006;62:7598. [Google Scholar]
- 25.For example, see ref 12-18.
- 26.Colby EA, Jamison TF. J. Org. Chem. 2003;68:156. doi: 10.1021/jo0264123. [DOI] [PubMed] [Google Scholar]
- 27.Trost reported a Pd-catalysed enyne isomerization that proceeded with >15:1 regioselectivity in the absence of an additive and 1:2.5 in the presence of one. Trost BM, Tanoury GJ, Lautens M, Chan C, MacPherson DT. J. Am. Chem. Soc. 1994;116:4255.
- 28.Pörshke reports X-ray crystal structures of 3-coordinate Ni-diene and Ni-diyne complexes, the third ligand being an organophosphine: (diene) Proft B, Pörschke K-R, Lutz F, Krüger C, Proft B, Pörschke K-R, Lutz F, Krüger C. Chem. Ber. Chem. Ber. 1991;1994;124127:2667, 653. (diyne)
- 29.For discussions of stereospecific ligand substitution in square planar metal complexes see: Cattalini L. Prog. Inorg. Chem. 1970;13:263–327.Cross RJ. Chem. Soc. Rev. 1985;14:197–223.Basolo F, Pearson RG. Mechanisms of Inorganic Reactions. John Wiley and Sons; New York: 1967. Chapter 5.Collman JP, Hegedus LS, Norton JR, Finke RG. Principles and Applications of Organotransition Metal Chemistry. University Science Books; Mill Valley, California: 1987. Chapter 4.
- 30.Drawn as a Ni(II) complex for illustrative purposes. The oxidation state of the nickel prior to formation of the C–C bond is unknown.
- 31.Based on the work of Montgomery (ref 10), we describe the reaction as proceeding through an oxanickelacyclopentene. Hydride transfer from triethylborane, as evidenced by the evolution of ethene, and reductive elimination are believed to complete the catalytic cycle (ref 20).
- 32.The pathways were defined as follows: type I: a pathway which forms exclusively regioisomer A. type II: a pathway which forms exclusively regioisomer B. type III: any pathway that forms a mixture of regioisomers A and B.
- 33.For a review, see: Herrmann WA. Angew. Chem. Int. Ed. 2002;41:1290. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y.
- 34.Knapp-Reed B, Mahandru GM, Montgomery J. J. Am. Chem. Soc. 2005;127:13156. doi: 10.1021/ja054590i. [DOI] [PubMed] [Google Scholar]
- 35.Mahandru GM, Liu G, Montgomery J. J. Am. Chem. Soc. 2004;126:3698. doi: 10.1021/ja049644n. [DOI] [PubMed] [Google Scholar]
- 36.While intermolecular reductive coupling reactions of alkynes and aldehydes to afford allylic alcohols had been previously reported (ref 11), this is the first example of the use of a silane as a reducing agent, and affords a silyl-protected allylic alcohol.
- 37.See ref 14b.
- 38.See ref 26.
- 39.Chan J, Jamison TF. J. Am. Chem. Soc. 2004;126:10682. doi: 10.1021/ja0470968. [DOI] [PubMed] [Google Scholar]
- 40.Nihashi Y, Lim C-H, Tanaka C, Miyagawa H, Ueno T. Biosci. Biotechnol. Biochem. 2002;66:685. doi: 10.1271/bbb.66.685. [DOI] [PubMed] [Google Scholar]
- 41.Miller KM, Colby EA, Woodin KS, Jamison TF. Adv. Synth. Catal. 2005;347:1533. [Google Scholar]
- 42.Miller KM, Jamison TF. Org. Lett. 2005;7:3077. doi: 10.1021/ol051075g. [DOI] [PubMed] [Google Scholar]
- 43.Patel SJ, Jamison TF. Angew. Chem., Int. Ed. 2004;43:3941. doi: 10.1002/anie.200460044. [DOI] [PubMed] [Google Scholar]
- 44.Luanphaisarnnont T, Ndubaku CO, Jamison TF. Org. Lett. 2005;7:2937. doi: 10.1021/ol050881k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.(–)-NMDPP provided slightly diminished, but similar, dr. No achiral phosphine provided comparable yields.
- 46.a Cram DJ, Abd Elhafez RA. J. Am. Chem. Soc. 1952;74:5828. [Google Scholar]; b Gawley RE, Aube J. Principles of Asymmetric Synthesis. Vol. 14. Pergamon Press; Elsevier, Oxford: 1996. pp. 121–160. Tetrahedron Organic Chemistry Series. [Google Scholar]
- 47.Sa-ei K, Montgomery J. Org. Lett. 2006;8:4441. doi: 10.1021/ol061579u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.a Colby EA, O'Brien KC, Jamison TF. J. Am. Chem. Soc. 2005;127:4297. doi: 10.1021/ja042733f. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Colby EA, Jamison TF. Org. Biomol. Chem. 2005;3:2675. doi: 10.1039/b507315b. [DOI] [PubMed] [Google Scholar]
- 49.Recently, the nickel-catalysed coupling of olefins and aldehydes has been reported: Ng S-S, Jamison TF. J. Am. Chem. Soc. 2005;127:14194. doi: 10.1021/ja055363j.Ho C-Y, Ng S-S, Jamison TF. J. Am. Chem. Soc. 2006;128:5362. doi: 10.1021/ja061471+.Ng S-S, Ho C-Y, Jamison TF. J. Am. Chem. Soc. 2006;128:11513. doi: 10.1021/ja062866w.
- 50.For a more comprehensive overview of recent developments in the field of nickel-catalysed coupling reactions, see ref 8.