

Abstract
Duchenne muscular dystrophy (DMD) is a deadly and common childhood disease caused by mutations that disrupt dystrophin protein expression. Several miniaturized dystrophin/utrophin constructs are utilized for gene therapy, and while these constructs have shown promise in mouse models, the functional integrity of these proteins is not well described. Here, we compare the biophysical properties of full-length dystrophin and utrophin with therapeutically relevant miniaturized constructs using an insect cell expression system. Full-length utrophin, like dystrophin, displayed a highly cooperative melting transition well above 37°C. Utrophin constructs involving N-terminal, C-terminal or internal deletions were remarkably stable, showing cooperative melting transitions identical to full-length utrophin. In contrast, large dystrophin deletions from either the N- or C-terminus exhibited variable stability, as evidenced by melting transitions that differed by 20°C. Most importantly, deletions in the large central rod domain of dystrophin resulted in a loss of cooperative unfolding with increased propensity for aggregation. Our results suggest that the functionality of dystrophin therapeutics based on mini- or micro-constructs may be compromised by the presence of non-native protein junctions that result in protein misfolding, instability and aggregation.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a common (1 of 3500 live male births) and deadly childhood disease that is characterized by a fragile muscle cell membrane and progressive rounds of muscle cell death and regeneration (1–3). To date, many potential therapies have shown promise in animal models but no cure or effective treatment for human patients currently exists (4).
DMD is primarily caused by the loss of dystrophin protein expression due to gene deletions, duplications or mutations (5). Dystrophin is a large, multi-domain protein that functions intracellularly in muscle to stabilize cell membranes (6,7). Dystrophin binds actin filaments through a tandem calponin homology (CH) domain at its N-terminus and through an electrostatic interaction between basic spectrin repeats 11–17 and acidic actin (8–11). In addition, a C-terminal domain of dystrophin interacts with the transmembrane protein β-dystroglycan, a component of the dystrophin glycoprotein complex that in turn is anchored to the extracellular matrix (ECM) (12–14). Therefore, dystrophin forms a structural link between the intracellular cytoskeleton and the ECM that protects muscle from contraction-induced injury (7).
Multiple strategies have been explored to rescue dystrophin expression in animal models of DMD, including exon skipping to restore a functional reading frame, stop codon suppression and viral gene therapy. One of the most promising approaches has been the expression of miniaturized dystrophin genes through adeno-associated viral (AAV) delivery (4,15). Since the packaging capacity of AAV is significantly smaller than the large dystrophin cDNA (16), a portion of the dystrophin sequence was internally deleted to decrease its size while retaining critical ligand binding domains located at the N- and C-termini (15). Recently, a viral vector encoding such a miniaturized dystrophin construct was tested in a phase I clinical trial to determine efficacy and safety in DMD patients with large deletions of the dystrophin gene (17). Unfortunately, very few muscle fibers were found to express dystrophin long-term while, at the same time, an immune response specific for peptides unique to the internally deleted dystrophin protein was reported (17).
To circumvent possible immune reactions to exogenously delivered dystrophin, multiple studies have explored whether the closely related protein utrophin could serve as a surrogate in dystrophin-deficient muscle (18). Utrophin is an autosomal homolog of dystrophin that is upregulated in dystrophin-deficient muscle and has been shown to functionally substitute for dystrophin when transgenically overexpressed in dystrophic mouse models (19–23). Utrophin is expressed at high levels in muscle perinatally, but is restricted to the myotendinous and neuromuscular junctions in adult muscle (21). Additionally, utrophin has been shown to bind actin filaments and β-dystroglycan (24,25), but through modes of contact distinct from those employed by dystrophin (11). Multiple gene therapy studies have used internally deleted utrophin-based constructs to rescue the dystrophic phenotype in mdx mice (18,26). Importantly, utrophin is expressed in adult muscle of DMD patients and is therefore less likely to stimulate an immune response. However, expression of utrophin may not substitute for all of dystrophin's functions, such as anchoring neuronal nitric oxide synthase (nNOS) to the sarcolemma (27).
A common theme of several proposed DMD treatments is the generation of proteins encoding non-native junctions between spectrin-like repeats in dystrophin (4). Two studies of small recombinant dystrophin fragments examined the stability of non-native junctions expected from exon skipping approaches for DMD (28,29). Ruszczak et al. (28) found that hybrid repeats generated from skipping of exons 43 and 44 were significantly less stable, while skipping exons 44 and 45 had almost no effect on protein stability. Carag-Kreiger et al. (29) measured forced unfolding for non-native repeat junctions generated by exon skipping, or resulting from in-frame deletions of the dystrophin gene found in patients with Becker muscular dystrophy. The authors reported variable mechanical stability as well as aggregation in the proteins that were tested (29). These studies highlight the potential for decreased stability or protein aggregation when non-native repeat junctions are created in the dystrophin protein. Similar to exon skipping strategies, the success of a gene therapy approach is also dependent on the stability of internally deleted dystrophin or utrophin mini-genes with non-native spectrin-like repeat junctions. A study by Banks et al. (30) reported that viral delivery of an internally truncated dystrophin micro-gene led to skeletal muscle abnormalities. A number of human patient studies have documented missense mutations or small deletions that lead to a loss of dystrophin protein levels causing disease (31–36). Our previous work (37), recently confirmed and expanded by Singh et al. (38), demonstrated that disease-causing missense mutations in dystrophin can lead to marked loss of protein stability and increased aggregation. In order to optimize therapeutic constructs for the treatment of dystrophinopathies in humans, it is imperative to characterize the biophysical consequences of internal deletions within dystrophin and utrophin. Here, we report that full-length utrophin exhibits a lower intrinsic thermal stability compared with dystrophin and displays a uniform thermal stability from N- to C-terminus. Additionally, internal deletion of spectrin-like repeats in utrophin is highly tolerated. In sharp contrast, we show that the thermal stability of dystrophin varies from N- to C-terminus, and internal deletion of hinge 2 through repeat 19 causes significant loss of protein stability.
RESULTS
Biophysical analysis of utrophin and internally deleted utrophin proteins
In the first biophysical comparison of dystrophin and utrophin, we performed circular dichroism (CD) spectroscopy on full-length utrophin and two large fragments encoding the N-terminus through spectrin-like repeat 10 and repeat 11 through the C-terminus (Fig. 1A). All three proteins exhibited similar alpha helical content (Fig. 2A) as previously published for full-length dystrophin (37). Analysis of thermal denaturation revealed that all three utrophin proteins exhibited highly cooperative unfolding transitions at nearly identical melting points (Tm ≈ 49°C), which were 10° lower than the Tm of full-length dystrophin (Fig. 2B and Table 1).
Figure 1.

Domain structure of proteins analyzed. (A and B) Tilted rectangles represent hinge regions, ovals represent spectrin-like repeats, lighter shaded ovals represent spectrin-like repeats that participate in actin binding and dashed lines represent deleted region. (A) Utrophin proteins analyzed. (B) Dystrophin proteins analyzed.
Figure 2.
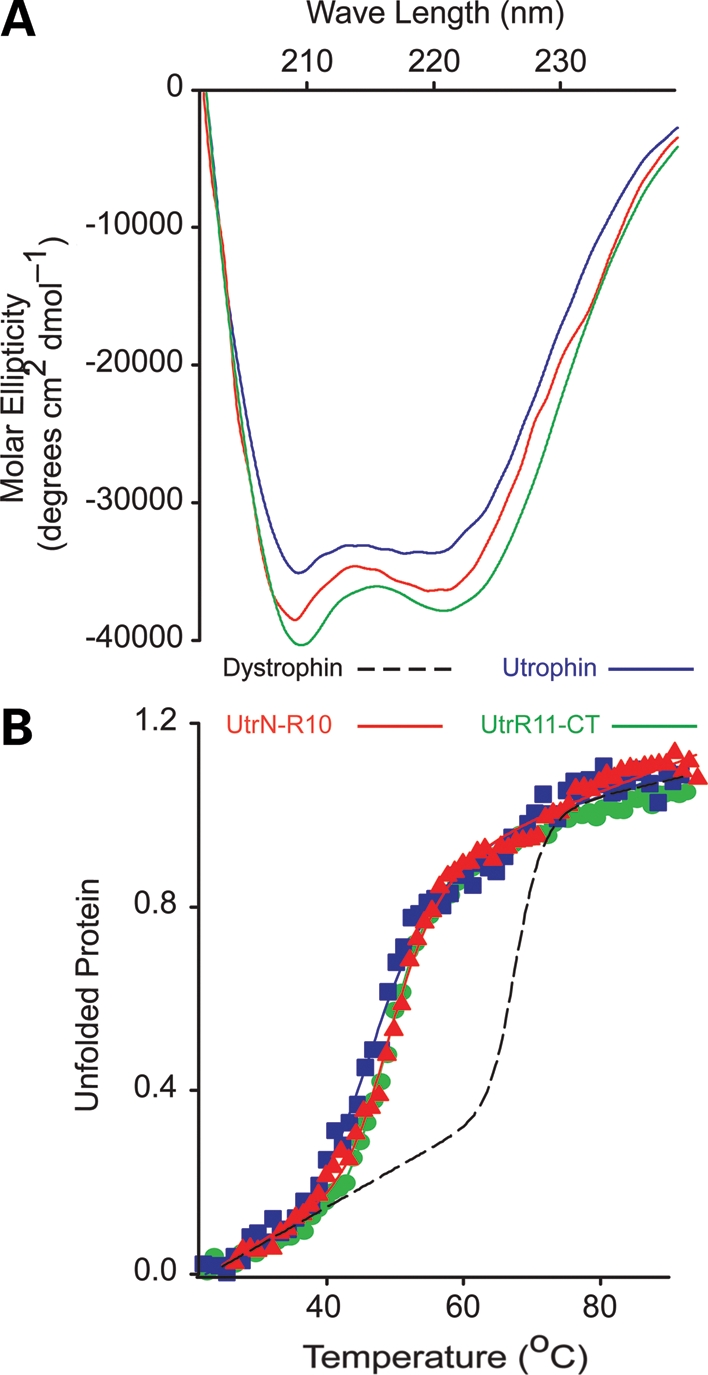
CD analysis of utrophin. (A) Far UV spectra of full-length utrophin (blue line), Utr-N-R10 (red line) and Utr-R11-CT (green line). (B) Thermal unfolding of utrophin, Utr-NT-R10 and Utr-R11-CT from 20 to 90°C. Dashed line represents previously published full-length dystrophin thermal unfolding provided for reference.
Table 1.
Melting point and PK50 values for each protein assayed
| Tm (°C) | PK50 (ng) | |
|---|---|---|
| Utrophin | 48.7 ± 0.64 | 7.0 ± 0.08 |
| Utr-N-R10 | 48.2 ± 0.40 | 51.1 ± 8.4 |
| Utr-R11-CT | 48.2 ± 0.40 | 22.8 ± 7.7 |
| TAT-Utrophin | 49.1 ± 0.19 | 43.4 ± 11.6 |
| TAT-Utr▵R11-22 | 48.6 ± 0.28 | 12.8 ± 1.49 |
| TAT-Utr▵R7-22 | 47.6 ± 0.18 | 18.9 ± 2.38 |
| TAT-Utr▵R4-21 | 49.1 ± 0.12 | 27.7 ± 0.92 |
| Dystrophin | 59.6 ± 0.29 (37) | 25.1 ± 2.09 |
| Dys-N-R10 | 46.1 ± 1.13 | Nd |
| Dys-N-R17 | 50.9 ± 1.37 | 0.19 ± 0.10 |
| DysR18-CT | 65.5 ± 0.76 | 29.5 ± 4.9 |
| DP260 | 50.2 ± 1.63, 69.6 ± 2.73 | 15.1 ± 3.39 |
| Dys▵H2-R19 | Na | 89.4 ± 22.7 |
| hDys▵H2-R19▵CT | Na | 25.25 ± 0.79 |
We also performed CD analysis on four different utrophin constructs that we are investigating as a potential protein replacement therapy for DMD (39), each tagged with an 11 amino acid protein transduction domain of the HIV-1 TAT protein (TAT). All four proteins yielded highly alpha helical spectra similar to full-length utrophin (Fig. 3). Full-length TAT-utrophin yielded a spectrum nearly identical to non-TAT-utrophin, suggesting that the TAT tag did not grossly affect protein structure. Despite differences in large internal deletions, all of the TAT-utrophin constructs displayed highly cooperative unfolding at very similar melting temperatures (Fig. 3 and Table 1), which indicates that utrophin folding and thermal stability are uniform and unaffected by internal deletion.
Figure 3.

CD analysis of TAT-utrophin proteins. (Top) Far UV spectra of TAT-utrophin and internally deleted TAT-utrophin proteins. (Bottom) Thermal unfolding of TAT-utrophin and internally deleted TAT-utrophin proteins.
In addition to thermal stability, we examined how the presence of non-native spectrin-like repeat junctions affected sensitivity to protease digestion. We subjected each protein, at a constant concentration, to increasing concentrations of the non-specific protease, proteinase K (PK) (28,40). Proteins with well-structured and compact folding are typically degraded at a slower rate than proteins with compromised folding or unstructured regions (41,42). We suspected that either the large deletions or non-native junctions would lead to a less stable protein and cause an increased rate of degradation compared with the native protein as previously reported by Ruszczak et al. (28) for small recombinant fragments of dystrophin. In contrast, we observed that each of the internally deleted utrophin proteins showed similar sensitivities to protease digestion compared with native protein (Fig. 4, Supplementary Material, Fig. S1 and Table 1), which again suggests that internal deletions and resulting non-native junctions had little effect on utrophin stability. We conclude that utrophin exhibits a lower intrinsic thermal stability compared with dystrophin, but is uniformly stable from N- to C-terminus and is highly tolerant to internal deletion.
Figure 4.

Protease sensitivity analysis of TAT-utrophin proteins. (A) Exponential decay plots representing the fraction (ng) of TAT-utrophin protein remaining over a range of PK concentrations. Data were plotted using a log scale and fit to a first-order exponential decay function using regression analysis. Error bars represent standard error of the mean (SEM). (B) Representative Coomassie stained SDS polyacrylamide gels.
Biophysical analysis of truncated dystrophin proteins
In stark contrast to the invariant biophysical properties of all analyzed utrophin constructs (Figs 2–4, Supplementary Material, Fig. S1 Table S1), truncated dystrophin proteins (Fig. 1B) showed more marked differences in biophysical properties. Two recombinant fragments encoding the N-terminus through the first 10 or 17 spectrin-like repeats (Dys-N-R10, Dys-N-R17) exhibited significantly lower melting transitions compared with full-length dystrophin, while the C-terminal retinal isoform DP260 and a construct encoding spectrin repeat 18 through the C-terminus (Dys-R18-CT) melted at 10°C higher than the full-length protein (Fig. 5B and Table 1). Consistent with the results of Mirza et al. (41), we observed a double transition in DP260 at 50 and 70°C (Fig. 5C and Table 1), which is also apparent upon closer inspection of data previously reported for full-length dystrophin (37). Our data demonstrate that the thermal stabilities of dystrophin fragments are context-dependent. Note that despite differences in Tm between N- and C-terminal fragments of dystrophin, all displayed highly cooperative denaturation transitions (Fig. 5B) indicative of well-folded proteins.
Figure 5.
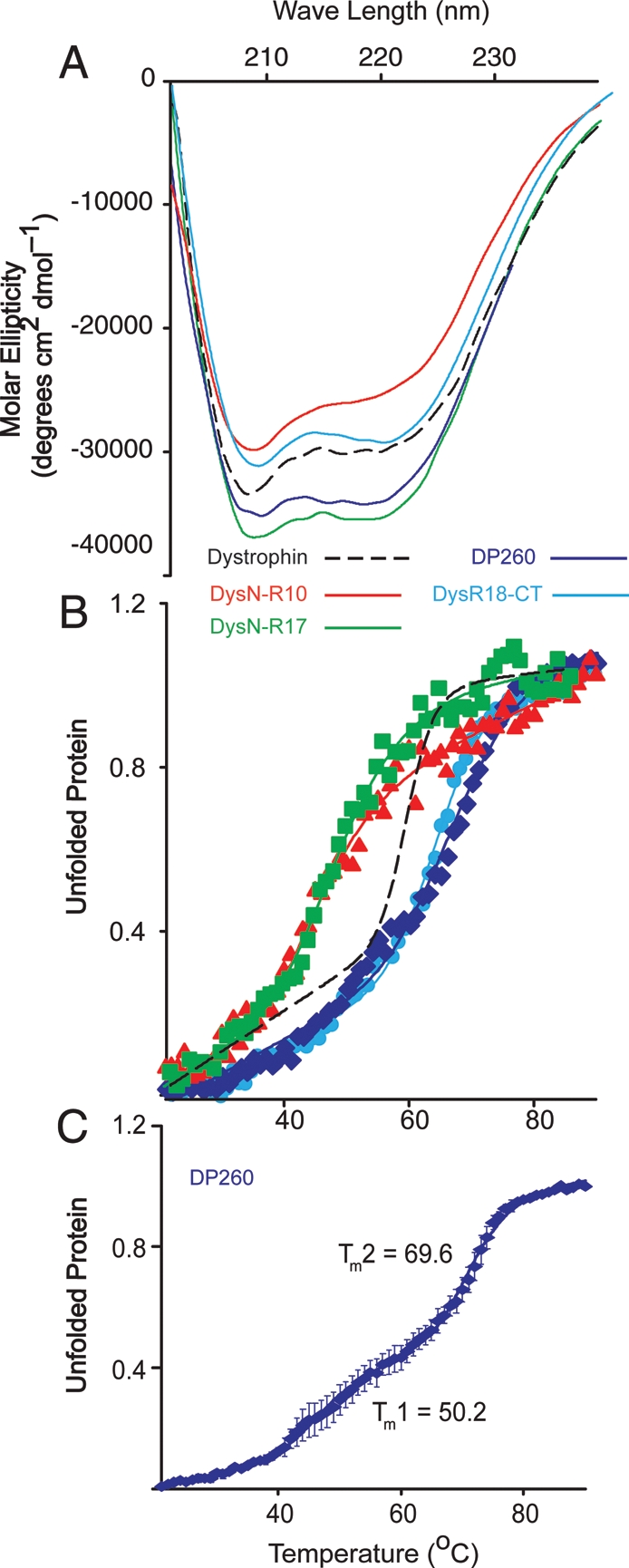
CD analysis of dystrophin proteins. (A) Far UV spectra of Dys-N-R10 (red), Dys-N-R17 (green), DP260 (blue) and DYS-R18-CT (light blue). (B) Thermal unfolding analysis of Dys-N-R10, Dys-N-R17, DP260 and Dys-R18-CT. (A and B) Dashed line represents previously published data for full-length dystrophin for reference. (C) Detailed thermal unfolding of DP260 showing two transitions states. Error bars represent SEM from three independent purifications and experiments.
In order to better understand how the differential thermal stabilities of the N- and C-terminal regions affected the structural stability of the protein, we compared the PK resistance of full-length dystrophin protein with that of N- and C-terminally deleted fragments. We found that the protease sensitivities of the C-terminal fragments of dystrophin, DP260 and Dys-R18-CT were similar to the stability of the full-length protein. In contrast, we found that a construct containing the N-terminus and the first 17 spectrin-like repeats was significantly (P = < 0.01) more susceptible to PK degradation than either the full-length protein, DP260 or Dys-R18-CT, suggesting that the folding in the N-terminal region is less compact (Fig. 6 and Table 1) than in the C-terminal region of the protein. Since the protease stability of full-length dystrophin aligned closely with Dys-R18-CT and DP260 (Fig. 6 and Table 1) but has a lower thermal stability compared with the same C-terminal fragments (Fig. 5 and Table 1), we conclude that the correlation between protease sensitivity and thermal stability for full-length protein and large protein fragments is not as robust as previously observed for smaller dystrophin fragments (28,40).
Figure 6.

Protease sensitivity analysis of dystrophin proteins. (A) Exponential decay plots representing the fraction (ng) of dystrophin protein remaining over a range of PK concentrations. Data were plotted using a log scale and fit to a first-order exponential decay function using regression analysis. Error bars represent SEM. (B) Coomassie stained SDS polyacrylamide gels of a representative protease sensitivity assay.
Analysis of internally deleted dystrophin proteins
Finally, we characterized two internally truncated mini-dystrophin constructs containing non-native junctions (Fig. 1B). hDys-ΔH2-R19ΔCT is the cDNA that demonstrated the greatest efficacy in rescuing the dystrophic phenotype when transgenically overexpressed in mdx mice (15). mDysΔH2-R19 is the corresponding mouse construct generated for the current study, which differs from hDys-ΔH2-R19ΔCT in that it encodes an intact C-terminus. CD analysis revealed that the mouse construct with the intact C-terminus was highly alpha helical while the human construct lacking a portion of the C-terminus showed a stronger minimum at 209 nm (Fig. 7A), suggesting that an alternative fold had been adopted. Most surprisingly, we failed to detect a cooperative melting transition for either of the mini-dystrophin proteins (Fig. 7B). In fact, the unfolding isotherms (Fig. 7B) were most similar to those exhibited by full-length dystrophin mutants bearing a variety of disease-causing missense mutations in the tandem CH domain (37). Despite the loss of cooperative unfolding with thermal denaturation (Fig. 7B), both hDys-ΔH2-R19ΔCT and mDys-ΔH2-R19 displayed wild-type sensitivities to PK digestion, suggesting they were as stable as full-length dystrophin (Fig. 7C and D and Table 1).
Figure 7.

Analysis of mini-dystrophin proteins. (A) Far UV spectra of mouse and human mini-dystrophin proteins. (B) Thermal unfolding of mouse and human mini-dystrophin. No cooperative transition is observed for either protein. (C) Protease sensitivity decay isotherm for human and mouse mini-dystrophin proteins. Error bars represent SEM. (D) SDS polyacrylamide gels representing typical protease sensitivity assays for human and mouse mini-dystrophin.
To assess whether the protease stability of hDys-ΔH2-R19ΔCT and mDys-ΔH2-R19 was caused by aggregation, we measured the fraction of hDys-ΔH2-R19ΔCT and mDys-ΔH2-R19 pelleted by high-speed sedimentation compared with several well-folded, highly soluble controls (Fig. 8A). We found that hDys-ΔH2-R19ΔCT but not mDys-ΔH2-R19 exhibited a significantly higher percentage of aggregated protein after purification compared with all other proteins tested (Fig. 8B), which may help to explain the robust stability to protease challenge (Fig. 7C and D). In addition to the solubility of proteins after purification, we examined the propensity of hDys-ΔH2-R19ΔCT and mDys-ΔH2-R19 to aggregate immediately after lysis of infected insect cells. Previously, we demonstrated that disease-causing missense mutations in ABD1 cause a large percentage of the mutant protein to be redistributed to the insoluble fraction of insect cells (37). We performed a similar analysis with hDys-ΔH2-R19ΔCT and mDys-ΔH2-R19 compared with full-length utrophin and internally deleted TAT-micro-utrophin ▵R4-21 (Fig. 8C). Both utrophin and TAT-Utr-▵R4-21 were found predominantly in the soluble fraction while both mini-dystrophin proteins were equally distributed between the insoluble and soluble fractions (Fig. 8C). Thus, while prior studies clearly demonstrate that hDys-ΔH2-R19ΔCT can fully rescue the dystrophic phenotype of mdx mice when overexpressed (15), our in vitro biophysical analyses suggest that its thermal stability and solubility are compromised in comparison with full-length dystrophin, utrophin and numerous truncated constructs. Furthermore, because N- and C-terminal fragments of dystrophin were all highly soluble (Fig. 8A and B) and displayed highly cooperative unfolding transitions (Fig. 5B), we suggest that the compromised stability/solubility of mini-dystrophins may be due to the presence of non-native protein junctions.
Figure 8.

Aggregation and solubility of mini-dystrophins. (A) High-speed sedimentation assessment of purified protein aggregation levels. Supernatant and pellet fractions for each protein tested. (B) Densitometry analysis of SDS gels in A. Analysis was performed on three to five independent experiments. Error bars represent SEM. (C) Anti-Flag immunoblots of the soluble and insoluble fractions of Sf21 insect cells expressing utrophin, TAT-Utr▵R4-21, mDys▵H2-R19 or hDys▵H2-R19▵CT.
DISCUSSION
Dystrophin and utrophin are related proteins that share multiple common structural domains (7,21). Not surprisingly, the highest level of sequence identity occurs in the N-terminal actin binding and the C-terminal dystroglycan binding domains. The central rod regions of dystrophin and utrophin display the lowest sequence identity with one another. Multiple studies have investigated the biophysical properties of dystrophin rod domain fragments and demonstrated marked heterogeneity in the stability of individual spectrin-like repeats (28,40). Despite these intrinsic differences between isolated modules, we recently demonstrated that full-length dystrophin unfolds in a highly cooperative, two-state manner (37). To assess whether the low level of sequence identity between dystrophin and utrophin translates into differences in protein stability, we performed the first biophysical characterization of full-length utrophin as well as an array of N-terminal, C-terminal and internal truncated dystrophin and utrophin proteins. We report that like dystrophin, utrophin unfolds in a highly cooperatively manner, but with a lower melting temperature. Although utrophin exhibits lower thermal stability compared with dystrophin, it must be noted that utrophin is nonetheless a highly stable protein as it only unfolds at a temperature well above 37°C.
We recently reported that dystrophin displays a melting point of ∼60°C (37), but here we show that this value is an amalgamation of the differing thermal stabilities of the N- and C-terminal portions (Fig. 5) of the full-length protein. The N-terminal half of dystrophin exhibits a cooperative transition at ∼50°C while the C-terminal half exhibits a cooperative transition at >70°C. In contrast, utrophin showed little change in stability from N- to C-terminus and we observed nearly identical unfolding transitions for several internally deleted utrophin constructs. Most notable from the perspective of therapeutic development, we observed no cooperative transition during thermal denaturation and significant protein aggregation for two internally deleted dystrophin mini-gene constructs (Figs 7 and 8). One of the mini-dystrophin constructs analyzed here has previously been shown to effectively prevent dystrophy in the mdx mouse, albeit at expression levels that greatly exceeded dystrophin expression in normal skeletal muscle (15). Because our analyses relied on proteins expressed in insect cells, it remains possible that mini-dystrophin folding may be more efficient in mammalian cells, or muscle cells in particular. Equally notable, however, our findings with mini-dystrophins expressed in insect cells were analyzed in parallel with an array of 11 dystrophin and utrophin constructs that displayed full solubility and highly cooperative unfolding transitions. Therefore, we believe that our biophysical analyses of recombinant dystrophin and utrophin constructs provide the basis for a rapid and highly sensitive assay of protein stability that may be useful in optimizing mini- or micro-gene constructs prior to extensive testing in animals.
Our results establish the importance of protein stability as a new factor to consider in the design of therapeutic vectors for the treatment of DMD. While our results on a limited number of internally deleted dystrophin and utrophin constructs immediately suggest that utrophin is more amenable to the sequence deletions necessary for AAV-mediated gene therapy approaches, we believe they also provide the basis for a functional assay to engineer more stable miniaturized dystrophin constructs as well. Finally, further analysis of junctions within internally truncated dystrophins modeling exonic deletions in Becker muscular dystrophy patients may provide a basis for the variability in protein expression and phenotype seen in these patients (43,44).
MATERIALS AND METHODS
Cloning
TAT-Flag-utrophin protein coding sequences were assembled using PCR as described in (39). The construction and characterization of DP260 have been previously described by Prins et al. (45). Flag DysR18-CT was also assembled using PCR and cloned into pFastBac Dual. The human mini-dystrophin cDNA was obtained from the Chamberlain laboratory and cloned into pFastBac using PCR and restriction digest. Mouse mini-dystrophin was assembled by PCR from full-length cDNA and cloned into pFastBac using restriction digest.
Expression and purification
All proteins for this study were expressed by Kinnakeet Biosciences in Sf9 insect cells using the Bac-to-Bac expression system (Invitrogen). Expressed proteins were purified using Flag affinity chromatography (Sigma-Aldrich) and dialyzed in two changes of phosphate buffered saline (PBS) at pH 7.5. After purification, proteins were concentrated using either 50 or 100 kDa cut-off centrifuge-based concentrators (Millipore). Protein concentration was determined by Dc protein assay (Bio-Rad) with a bovine serum albumin standard curve.
Circular dichroism
CD spectra and melt curves were acquired as previously described (37). Briefly, measurements were acquired on a Jasco J-815 spectropolarimeter. Temperature was varied from 20 to 90°C using an attached Peltier thermal regulator. Each protein was assayed with at least two different purifications and at least two different concentrations between 0.15 and 0.5 mg/ml. CD spectra were collected between 200 and 260 nm at 20°C at pH 7.5 in PBS. Melting point analysis was assayed by collecting spectra between 200 and 240 nm for every degree of temperature change and displayed by plotting the change at 222 nm. Data were fit using regression analysis in Sigma Plot (Systat Software) using a two-state unfolding model to obtain a melting point (46).
Protease sensitivity assay
Each protein was assayed at a constant concentration of 0.15 mg/ml for sensitivity to a non-specific protease, PK, similar to Ruszczak et al. (28). Samples were subjected to no PK and a range of PK from 1.5 to 0.003 ng. Every other sample represents a 1 : 10 dilution. Each series was incubated at 37°C for 30 min and then subjected to SDS-PAGE. Poly-acrylamide gels were stained with Coomassie blue and the remaining full-length protein was quantitated using software from UVP Bioimaging Systems. Values were fit using first-order exponential decay regression analysis in Sigma Plot to obtain a value that represents the PK concentration (ng) where half of the full-length protein is still present (PK50).
Analysis of aggregates and insoluble protein
For sedimentation analysis of purified proteins, each protein was subjected to ultracentrifugation at 100 000g to separate large molecular weight aggregates from soluble protein. The resulting supernatant and pellet fractions were separated by SDS-PAGE and the Coomassie stained gels were quantitated using densitometry. Analysis of soluble and insoluble proteins was performed in Sf21 insect cells by baculovirus expression. An identical viral load to large-scale expressions was used to standardize expression levels. Cells were infected and incubated for 72 h. The resulting cell pellets were lysed and solubilized identically to large-scale purifications as before (37). Each sample was centrifuged at 14 000g for 20 min to separate the soluble and insoluble fraction of the lysed insect cells. The pellet was resuspended in 6 Mm urea in PBS and incubated at 50°C to solubilize aggregated protein. 5× SDS protein sample buffer was then used to solubilize any remaining protein. The soluble and insoluble fractions were then separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes. Membranes were then immunoblotted using a monoclonal anti-Flag® antibody (Sigma) as recommended by the manufacturer. Goat anti-mouse Li-cor secondary antibody was used to detect primary antibodies and resulting membranes were imaged on a Li-cor Odyssey infrared scanner.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the NIH Training Program in Muscle Research (AR007612), National Institute of Arthritis, Musculoskeletal and Skin Diseases grant AR042423, the Muscular Dystrophy Association, the Nash Avery Foundation and Charley's Fund.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr David D. Thomas for access to his CD spectrometer.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Weller B., Karpati G., Carpenter S. Dystrophin-deficient mdx muscle fibers are preferentially vulnerable to necrosis induced by experimental lengthening contractions. J. Neurol. Sci. 1990;100:9–13. doi: 10.1016/0022-510x(90)90005-8. [DOI] [PubMed] [Google Scholar]
- 2.Menke A., Jockusch H. Decreased osmotic stability of dystrophin-less muscle cells from the mdx mouse. Nature. 1991;349:69–71. doi: 10.1038/349069a0. [DOI] [PubMed] [Google Scholar]
- 3.Matsuda R., Nishikawa A., Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with evans blue: evidence of apoptosis in dystrophin- deficient muscle. J. Biochem. (Tokyo) 1995;118:959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- 4.Muir L.A., Chamberlain J.S. Emerging strategies for cell and gene therapy of the muscular dystrophies. Expert. Rev. Mol. Med. 2009;11:e18. doi: 10.1017/S1462399409001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffman E.P., Brown R.H., Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 6.Koenig M., Monaco A.P., Kunkel L.M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–228. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- 7.Ervasti J.M., Sonnemann K.J. Biology of the striated muscle dystrophin-glycoprotein complex. Int. Rev. Cytol. 2008;265:191–225. doi: 10.1016/S0074-7696(07)65005-0. [DOI] [PubMed] [Google Scholar]
- 8.Rybakova I.N., Amann K.J., Ervasti J.M. A new model for the interaction of dystrophin with F-actin. J. Cell Biol. 1996;135:661–672. doi: 10.1083/jcb.135.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amann K.J., Renley B.A., Ervasti J.M. A cluster of basic repeats in the dystrophin rod domain binds F- actin through an electrostatic interaction. J. Biol. Chem. 1998;273:28419–28423. doi: 10.1074/jbc.273.43.28419. [DOI] [PubMed] [Google Scholar]
- 10.Hanft L.M., Rybakova I.N., Patel J.R., Rafael-Fortney J.A., Ervasti J.M. Cytoplasmic γ-actin contributes to a compensatory remodeling response in dystrophin-deficient muscle. Proc. Natl Acad. Sci. USA. 2006;103:5385–5390. doi: 10.1073/pnas.0600980103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rybakova I.N., Humston J.M., Sonnemann K.J., Ervasti J.M. Dystrophin and utrophin bind actin filaments through distinct modes of contact. J. Biol. Chem. 2006;281:9996–10001. doi: 10.1074/jbc.M513121200. [DOI] [PubMed] [Google Scholar]
- 12.Huang X., Poy F., Zhang R., Joachimiak A., Sudol M., Eck M.J. Structure of a WW domain containing fragment of dystrophin in complex with ß-dystroglycan. Nat. Struct. Biol. 2000;7:634–638. doi: 10.1038/77923. [DOI] [PubMed] [Google Scholar]
- 13.Ibraghimov-Beskrovnaya O., Ervasti J.M., Leveille C.J., Slaughter C.A., Sernett S.W., Campbell K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 14.Ervasti J.M., Campbell K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harper S.Q., Hauser M.A., DelloRusso C., Duan D., Crawford R.W., Phelps S.F., Harper H.A., Robinson A.S., Engelhardt J.F., Brooks S.V., Chamberlain J.S. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 16.Coura R.S., Nardi N.B. The state of the art of adeno-associated virus-based vectors in gene therapy. Virol. J. 2007;4:99. doi: 10.1186/1743-422X-4-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendell J.R., Campbell K., Rodino-Klapac L., Sahenk Z., Shilling C., Lewis S., Bowles D., Gray S., Li C., Galloway G., et al. Dystrophin immunity in Duchenne's muscular dystrophy. N. Engl. J. Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Odom G.L., Gregorevic P., Allen J.M., Finn E., Chamberlain J.S. Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol. Ther. 2008;16:1539–1545. doi: 10.1038/mt.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tinsley J.M., Blake D.J., Roche A., Byth B.C., Knight A.E., Kendrick-Jones J., Suthers G.K., Love D.R., Edwards Y.H., Davies K.E. Primary structure of dystrophin-related protein. Nature. 1992;360:591–593. doi: 10.1038/360591a0. [DOI] [PubMed] [Google Scholar]
- 20.Tinsley J.M., Potter A.C., Phelps S.R., Fisher R., Trickett J.I., Davies K.E. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384:349–353. doi: 10.1038/384349a0. [DOI] [PubMed] [Google Scholar]
- 21.Blake D.J., Weir A., Newey S.E., Davies K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 22.Rybakova I.N., Patel J.R., Davies K.E., Yurchenco P.D., Ervasti J.M. Utrophin binds laterally along actin filaments and can couple costameric actin with the sarcolemma when overexpressed in dystrophin-deficient muscle. Mol. Biol. Cell. 2002;13:1512–1521. doi: 10.1091/mbc.01-09-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tinsley J., Deconinck N., Fisher R., Kahn D., Phelps S., Gillis J.M., Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med. 1998;4:1441–1444. doi: 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- 24.Rybakova I.N., Ervasti J.M. Identification of spectrin-like repeats required for high affinity utrophin–actin interaction. J. Biol. Chem. 2005;280:23018–23023. doi: 10.1074/jbc.M502530200. [DOI] [PubMed] [Google Scholar]
- 25.James M., Nuttall A., Ilsley J.L., Ottersbach K., Tinsley J.M., Winder S.J. Adhesion-dependent tyrosine phosphorylation of ß-dystroglycan regulates its interaction with utrophin. J. Cell Sci. 2000;113:1717–1726. doi: 10.1242/jcs.113.10.1717. [DOI] [PubMed] [Google Scholar]
- 26.Wakefield P.M., Tinsley J.M., Wood M.J., Gilbert R., Karpati G., Davies K.E. Prevention of the dystrophic phenotype in dystrophin/utrophin-deficient muscle following adenovirus-mediated transfer of a utrophin minigene. Gene Ther. 2000;7:201–204. doi: 10.1038/sj.gt.3301066. [DOI] [PubMed] [Google Scholar]
- 27.Li D., Bareja A., Judge L., Yue Y., Lai Y., Fairclough R., Davies K.E., Chamberlain J.S., Duan D. Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J. Cell Sci. 2010;123:2008–2013. doi: 10.1242/jcs.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruszczak C., Mirza A., Menhart N. Differential stabilities of alternative exon-skipped rod motifs of dystrophin. Biochim. Biophys. Acta. 2009;1794:921–928. doi: 10.1016/j.bbapap.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krieger C.C., Bhasin N., Tewari M., Brown A.E., Safer D., Sweeney H.L., Discher D.E. Exon-skipped dystrophins for treatment of Duchenne muscular dystrophy: mass spectrometry mapping of most exons and cooperative domain designs based on single molecule mechanics. Cytoskeleton (Hoboken.) 2010;67:796–807. doi: 10.1002/cm.20489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banks G.B., Judge L.M., Allen J.M., Chamberlain J.S. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS. Genet. 2010;6:e1000958. doi: 10.1371/journal.pgen.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamed S., Sutherland-Smith A., Gorospe J., Kendrick-Jones J., Hoffman E. DNA sequence analysis for structure/function and mutation studies in Becker muscular dystrophy. Clin. Genet. 2005;68:69–79. doi: 10.1111/j.1399-0004.2005.00455.x. [DOI] [PubMed] [Google Scholar]
- 32.Hamed S.A., Hoffman E.P. Automated sequence screening of the entire dystrophin cDNA in Duchenne dystrophy: point mutation detection. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006;141B:44–50. doi: 10.1002/ajmg.b.30234. [DOI] [PubMed] [Google Scholar]
- 33.Tuffery-Giraud S., Beroud C., Leturcq F., Yaou R.B., Hamroun D., Michel-Calemard L., Moizard M.P., Bernard R., Cossee M., Boisseau P., et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum. Mutat. 2009;30:934–945. doi: 10.1002/humu.20976. [DOI] [PubMed] [Google Scholar]
- 34.Becker K., Robb S.A., Hatton Z., Yau S.C., Abbs S., Roberts R.G. Loss of a single amino acid from dystrophin resulting in Duchenne muscular dystrophy with retention of dystrophin protein. Hum. Mutat. 2003;21:651. doi: 10.1002/humu.9143. [DOI] [PubMed] [Google Scholar]
- 35.Lenk U., Oexle K., Voit T., Ancker U., Hellner K.A., Speer A., Hübner C. A cysteine 3340 substitution in the dystroglycan-binding domain of dystrophin associated with Duchenne muscular dystrophy, mental retardation and absence of the ERG b-wave. Hum. Mol. Genet. 1996;5:973–975. doi: 10.1093/hmg/5.7.973. [DOI] [PubMed] [Google Scholar]
- 36.Goldberg L.R., Hausmanowa-Petrusewicz I., Fidzianska A., Duggan D.J., Steinberg L.S., Hoffman E.P. A dystrophin missense mutation showing persistence of dystrophin and dystrophin-associated proteins yet a severe phenotype. Ann. Neurol. 1998;44:971–976. doi: 10.1002/ana.410440619. [DOI] [PubMed] [Google Scholar]
- 37.Henderson D.M., Lee A., Ervasti J.M. Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc. Natl Acad. Sci. USA. 2010;107:9632–9637. doi: 10.1073/pnas.1001517107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh S.M., Kongari N., Cabello-Villegas J., Mallela K.M. Missense mutations in dystrophin that trigger muscular dystrophy decrease protein stability and lead to cross-beta aggregates. Proc. Natl Acad. Sci. USA. 2010;107:15069–15074. doi: 10.1073/pnas.1008818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sonnemann K.J., Heun-Johnson H., Turner A.J., Baltgalvis K.A., Lowe D.A., Ervasti J.M. Functional substitution by TAT-utrophin in dystrophin-deficient mice. PLoS. Med. 2009;6:e1000083. doi: 10.1371/journal.pmed.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mirza A., Sagathevan M., Sahni N., Choi L., Menhart N. A biophysical map of the dystrophin rod. Biochim. Biophys. Acta. 2010;1804:1796–1809. doi: 10.1016/j.bbapap.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 41.Spolaore B., Bermejo R., Zambonin M., Fontana A. Protein interactions leading to conformational changes monitored by limited proteolysis: apo form and fragments of horse cytochrome c. Biochemistry. 2001;40:9460–9468. doi: 10.1021/bi010582c. [DOI] [PubMed] [Google Scholar]
- 42.Fontana A., de Laureto P.P., Spolaore B., Frare E., Picotti P., Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim. Pol. 2004;51:299–321. [PubMed] [Google Scholar]
- 43.Kesari A., Pirra L.N., Bremadesam L., McIntyre O., Gordon E., Dubrovsky A.L., Viswanathan V., Hoffman E.P. Integrated DNA, cDNA, and protein studies in Becker muscular dystrophy show high exception to the reading frame rule. Hum. Mutat. 2008;29:728–737. doi: 10.1002/humu.20722. [DOI] [PubMed] [Google Scholar]
- 44.Kaspar R.W., Allen H.D., Ray W.C., Alvarez C.E., Kissel J.T., Pestronk A., Weiss R.B., Flanigan K.M., Mendell J.R., Montanaro F. Analysis of dystrophin deletion mutations predicts age of cardiomyopathy onset in becker muscular dystrophy. Circ. Cardiovasc. Genet. 2009;2:544–551. doi: 10.1161/CIRCGENETICS.109.867242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prins K.W., Humston J.L., Mehta A., Tate V., Ralston E., Ervasti J.M. Dystrophin is a microtubule-associated protein. J. Cell Biol. 2009;186:363–369. doi: 10.1083/jcb.200905048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Legardinier S., Legrand B., Raguenes-Nicol C., Bondon A., Hardy S., Tascon C., Le R.E., Hubert J.F. A Two-amino Acid Mutation Encountered in Duchenne Muscular Dystrophy Decreases Stability of the Rod Domain 23 (R23) Spectrin-like Repeat of Dystrophin. J. Biol. Chem. 2009;284:8822–8832. doi: 10.1074/jbc.M805846200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.