

Abstract
Summary: Acute Trypanosoma cruzi infections can be asymptomatic, but chronically infected individuals can die of Chagas' disease. The transfer of the parasite mitochondrial kinetoplast DNA (kDNA) minicircle to the genome of chagasic patients can explain the pathogenesis of the disease; in cases of Chagas' disease with evident cardiomyopathy, the kDNA minicircles integrate mainly into retrotransposons at several chromosomes, but the minicircles are also detected in coding regions of genes that regulate cell growth, differentiation, and immune responses. An accurate evaluation of the role played by the genotype alterations in the autoimmune rejection of self-tissues in Chagas' disease is achieved with the cross-kingdom chicken model system, which is refractory to T. cruzi infections. The inoculation of T. cruzi into embryonated eggs prior to incubation generates parasite-free chicks, which retain the kDNA minicircle sequence mainly in the macrochromosome coding genes. Crossbreeding transfers the kDNA mutations to the chicken progeny. The kDNA-mutated chickens develop severe cardiomyopathy in adult life and die of heart failure. The phenotyping of the lesions revealed that cytotoxic CD45, CD8+ γδ, and CD8α+ T lymphocytes carry out the rejection of the chicken heart. These results suggest that the inflammatory cardiomyopathy of Chagas' disease is a genetically driven autoimmune disease.
INTRODUCTION
Chagas' disease is the most lethal endemic infectious ailment in the Western Hemisphere, with a devastating effect upon populations in rural areas of Latin America. Chagas' heart disease typically kills people in the age range of 30 to 50 years. The disease is considered incurable, and its high mortality rates translate to hundreds of thousands of deaths per year (407). Perhaps the most important problem in Chagas' disease is the determination of its pathogenesis (293). (Reference 293 appeared in a July 2004 issue of Cell, and it was retracted [September 2005] after an editor-instigated inquisition regarding the authenticity of the kinetoplast DNA [kDNA] integration site, which was considered “open to alternative interpretations,” and the article can no longer be obtained from the Cell website as a legible transcript. Primary data on the integration of parasite DNA into vertebrate genomes have taken on a controversial nature [259a], with justifications that are not reasonable to many in the scientific community. The original work has been supported subsequently by additional published studies [173, 366, 402], of humans, rabbits, and chickens, from the authors' laboratory. After six years, the editor did not show experimental data to refute the original observations of kDNA integration. Now, unadulterated copies of the Cell article can be obtained from Google-cited independent websites.) Various theories have been proposed, beginning with the mechanical action of the parasite encysted in host cells, followed by the subsequent degradation of the affected tissue by inflammation, which was challenged by the neurogenic theory, stemming from a hypothetical parasite-released neurotoxin, and displaced by the current autoimmune theory, in which host tissues are self-rejected by immune system effector lymphocytes (173, 293 [see comments regarding this retracted article at first citation], 344, 398, 402, 408).
Here we review basic parasitologic, immunologic, molecular biology, genetic, clinical, and pathology aspects required to approach questions related to the pathogenesis of Chagas' disease. Life-long, cryptic Trypanosoma cruzi infections provide the grounds for the transfer of parasite mitochondrial minicircle sequences that accumulate and spread DNA insertions throughout the human host genome over time (173, 293 [see comments regarding this retracted article at first citation], 402). Genotype modifications of the host's cells are associated with the pathogenesis of autoimmune Chagas' disease in the cross-kingdom, parasite-free chicken model system.
THE PROTOZOA
The eukaryotic protozoa may have originated from drastic biochemical alterations in an ancestor prokaryotic eubacterium that modified membrane sterol synthesis by replacing murein peptidoglycans by N-linked glycoproteins to form a flexible surface coat (58–61). This membrane flexibility enabled the prokaryote-derived eukaryote to develop key properties, including phagotrophy; an internal membrane system with peroxisomes, a cytoskeleton, and a nucleus; cell division; and sex (59–61). The resulting eukaryotic plasticity was central to symbiogenesis and, ultimately, to mitochondrial formation from an internalized alphaproteobacterium. Thus, a bifurcation between prokaryotes and eukaryotes was resolved circa 1 billion years ago. In that epoch, major suites of evolution occurred, including the origin of cellular structures and organelles with all the novelties included in the compendium of eukaryotic biology. With the evolution of phagotrophy (118, 368), contributions by lateral DNA transfer (LDT) allowed significant improvements made by the acquisition of genes for internal membrane compartmentation, while retroelements and reverse transcriptase (RT) further shaped the eukaryote genome.
Quantum Revolution and Acquisitions
The controversy over whether eukaryogenesis resulted from the acquisition of an entire genome, according to the symbiogenesis theory, or from quantum evolution is an important issue discussed elsewhere (58, 260). Currently, 13 protozoan phyla are recognized, among which is the subkingdom Excavata (Euglenozoa) (58). These early protozoa, using either pseudopods or flagella for locomotion, are in the class Zoomastigophorea, subclass Mastigophora. Among the latter are flagellate protozoa containing a large amount of mitochondrial DNA, which comprise the order Kinetoplastida (438).
The protozoa in the order Kinetoplastida form a group that contains free-living commensals and parasitic flagellates. All members of this order have variable amounts of DNA (kDNA) localized in a mitochondrial subcompartment. The variable total amount of kDNA in members of different orders is remarkable (248), possibly resulting from the protists having different modes of organelle acquisition (58). For example, either haploid alphaproteobacterium mitochondrial DNA was acquired in a single event, or members of an order, family, or genus made a complete acquisition of the mitochondrial DNA from a dividing alphaproteobacterium, possibly by quantum acquisitions on several occasions.
The ancestors of protozoa were likely similar to free-living bacterivorous flagellates called jakobids (367). Ultrastructural studies have associated kinetoplastids with the diplonemids, forming a group of facultative parasites of invertebrates (258), but analyses of small-subunit (SSU) rRNA suggested that the organisms most closely related to the kinetoplastids are the euglenids (32, 220, 441). According to their morphology, eukaryotes of the order Kinetoplastida are divided into two main suborders: the uniflagellate Trypanosomatida and the biflagellate Bodonina (437). Phylogenetic analyses based on SSU rRNA and heat shock proteins (Hsp90) supported the placement of the root for the trypanosomatids next to the free-living bodonids, with Bode saltans as the closest extant relative (367). B. saltans is a nonparasitic species included among the bacterivorous kinetoplastid flagellate protozoans especially abundant in organically enriched water. B. saltans diverged early on from the evolutionary line of kinetoplastids, and this species of bodonid is more closely related to the trypanosomatids (115) and is a predecessor of the single-flagellum trypanosomatids that lost the second flagellum in the course of their evolution (59).
The family Trypanosomatidae is comprised of genera including Trypanosoma and Leishmania (213). The genus Trypanosoma can be divided in subgroups, including a rodent clade, an avian clade, and aquatic clade, in addition to the African salivarian trypanosomes (Trypanosoma brucei and relatives) and the American stercorarian groups of Trypanosoma cruzi and related species (367, 466).
In the family Trypanosomatidae there are parasites of plants and animals, including trypanosome and leishmanial parasites of medical and veterinary importance (277). These parasites likely invaded insects during the acquisition of hematophagy, followed by their transmission to mammals and other vertebrates (167, 379, 380). An alternative view posited that the genus evolved from primary parasites of vertebrates and incorporated invertebrate vectors secondarily (444). Stercorarian T. cruzi organisms that are passed in insects' feces and the salivarian T. brucei group that contaminates prey via insect saliva inoculation are among the blood trypanosomes of medical and veterinary importance. This review will focus on T. cruzi, which is limited to the American continent. T. cruzi infection is estimated to have initiated as a limited enzootic infection about 90 million years ago (mya), probably requiring stepwise adaptation to invertebrate and vertebrate hosts before it became a widespread zoonosis in the American continent (408).
ULTRASTRUCTURE, GENOME ORGANIZATION, AND FUNCTION OF TRYPANOSOMA CRUZI
Ultrastructural studies on the developmental forms of T. cruzi are performed largely with epimastigotes, which grow luxuriously in vitro, the closest approximation of the parasite found in the gut of the invertebrate host (312). A detailed description of the structures and functions can be found in specialized texts (103, 437, 438). Blood trypomastigotes and intracellular amastigotes are the T. cruzi forms encountered in mammalian hosts (Fig. 1). The parasite membrane interacts with the extracellular medium, including host blood, the intercellular space, and the lumen of the insect gut. The membrane contains a coat of glycosylphosphatidylinositol (GPI)-anchored proteins, glycoproteins, and the carbohydrate portion of glycolipids. These well-characterized surface-associated macromolecules play a fundamental role in parasite biology. Mucins, trans-sialidase, and the Tc85 family of glycoproteins are included in the array of constituents (5, 12, 240, 390). The glycoconjugate-rich rugous surface of the cytostome region plays a role in the preferential binding of macromolecules and their intake by endocytosis.
Fig. 1.

Trypanosoma cruzi parasitic forms present in human tissues. (Left) Schematic representation of a trypomastigote form of Trypanosoma cruzi. (Reprinted from reference 63 with permission of the publisher.) (Right) Ultrastructure of a T. cruzi amastigote free in the cytoplasm of a muscle cell. N, nucleus; K, kinetoplast; F, flagellum.
A complete description of morphological and functional properties of specialized structures and organelles of the T. cruzi epimastigote and trypomastigote stages has been reviewed elsewhere (103). Following cell invasion, the trypomastigotes are enclosed in a vacuole of lysosomal origin. The trypomastigotes escape from these acidic environments and transform into amastigotes, the mammalian replicative forms of the parasite, in the cytosol. The in vitro exposure of trypomastigotes to an acidic pH (pH 5.0 for 2 h) induces their transformation into rounded amastigote forms (422) that are ultrastructurally and biochemically indistinguishable from mammalian-tissue-derived amastigotes. Extracellular transformation reveals the stage-specific surface antigen of the acidic pH-transformed amastigotes (Ssp-4) and the concurrent loss of a stage-specific trypomastigote antigen (Ssp-3) while retaining the characteristic replicative property of intracellular amastigotes. Electron microscopic analyses revealed the kinetoplast structure, with general morphological features of the intracellularly derived amastigotes (422).
Nuclear DNA
T. cruzi stores genomic DNA (nuclear DNA [nDNA]) in a nucleus enveloped by the pore-containing membranes typical of eukaryotes (52). During cell division the nuclear membrane keeps its typical structures, but the chromatin becomes dispersed, and intranuclear microtubules become apparent in association with dense plates (105). The dense plates correspond to chromosomes. The DNA is condensed in the chromatin-dispersed nucleoplasm, with a nucleolus found only in the epimastigote stage (124, 371). The total DNA content ranges from 125 to 280 fg/cell (79, 122, 123, 233).
The T. cruzi genome sequence of 60.3 Mbp has been assembled into 4,008 contigs that can be reduced to 784 scaffold assemblies (125, 446). Mostly due to difficulties arising from two haplotype sequences, the assembly of the scaffold to generate complete chromosome sequences has been hindered by the large numbers of genes in repetitive gene families and by large amounts of repetitive noncoding DNA. This might explain why completely assembled sequences for all of the T. cruzi chromosomes have not yet been achieved. Protein-encoding genes are organized into directional gene clusters (DGCs), similar to the organization of bacterial operons (53). Adjacent DGCs are usually found on opposite strands of the DNA, either in a diverging or in a converging orientation. The order of genes on a particular chromosome is conserved in large blocks (45, 125, 148), and the regions between DGCs, which are named strand switch regions (SSRs), may represent islands of transcription initiation and termination (267, 268). The T. cruzi genome sequences and predicted protein sequences can be accessed through public databases, including GeneDB (177), TcruziDB (9), and TriTrypDB (http://tritrypdb.org/tritrypd/). Studies of T. cruzi heterozygous hybrids can be greatly enhanced by an available database (3) of single-nucleotide polymorphisms (SNPs).
The T. cruzi 93-kb contig in chromosome 3 illustrates the primary DNA sequence of an SSR between divergent DGCs (14), showing strand asymmetry with respect to base com- position (292). The GC-rich SSRs of chromosomes 1 and 3 comprise degenerate retrotransposon transposable elements termed L1Tc and VIPER/SIRE (295), and the GC-rich regions cleaved by etoposide/topoisomerase II bear a centromere function associated with the origin of DNA replication (295). The SSR constitutes transcriptional promoters in which the L1Tc retrotransposon sequences activate gene transcription (46, 175).
The T. cruzi genome encodes approximately 22,570 distinct proteins, including 12,570 allelic pairs (27). The observation that 18% of the protein-encoding genes may be present at ≥14 copies suggests that the number of variant proteins may be greater than 20,000 (29). Two-dimensional (2D) gel and mass spectrophotometric analyses (300, 301) have identified 2,784 proteins, accounting for approximately 10% of the predicted proteome (29).
Kinetoplast DNA
The T. cruzi kinetoplast lamellar, almost spiral-like structure is retained in a membrane lining a vacuole (378). The remarkable molecular configuration of the kDNA network (279, 326, 360) continues directly into a typical mitochondrion canal system, as seen for any tissue cell (309). The kDNA is located within the mitochondrial matrix, and the position of the kinetoplast relative to the nucleus changes during the cell life cycle. However, the kinetoplast is always located close to the basal body, to which it is physically linked by a set of filaments. This connection is crucial during the segregation of the trypanosome mitochondrial genome (297).
The kDNA comprises 15% to 30% of the total cellular DNA and differs from nuclear DNA in its buoyant density, base ratio, and degree of renaturation (103, 104). Different from any other known DNAs, the kDNA of trypanosomatids is composed of circular molecules that are topologically relaxed and interlocked to form a single network. Two types of DNA rings are present in the kinetoplast: minicircles and maxicircles. There are approximately 15,000 T. cruzi minicircles, averaging 1.4 kb in size, and a few dozen maxicircles, varying from 20 to 40 kb in length (382). The minicircle is composed of four interspersed conserved and hypervariable regions. The conserved region carries a CA-rich sequence block (CArsb), which can be the origin of replication, transcription, recombination, and a specific site mediating kDNA transfer to the host genome (173, 178, 382, 383).
The minicircles encode guide RNAs (gRNAs), which modify the maxicircle transcripts by extensive uridine insertion or deletion in a process known as RNA editing (257). Information for this process is given by small gRNA molecules encoded primarily on the kDNA minicircles. The sequence heterogeneity of thousands of minicircles in each cell represents an enormous potential for this kDNA component to bring forth additional genetic diversity. The maxicircles are structurally and functionally analogous to mitochondrial DNA from higher eukaryotes, encoding rRNAs and subunits of respiratory complexes (382).
The unusual organization of kinetoplastid genes in DGCs requires equally unorthodox mechanisms to generate functional eukaryotic mRNA (53, 163). Most, if not all, protein-encoding genes are transcribed as polycistronic units (191, 369). The expression of kinetoplastid genes is not controlled at the level of transcription initiation. Sequence-specific initiation by kinetoplastid RNA polymerase II has been demonstrated for the spliced-leader (SL) RNA gene (53, 92). A directed initiation of transcription has been shown for some kinetoplastid protein-encoding genes (92, 163, 187, 225, 268).
Biochemical and molecular evidence confirmed the presence of basic proteins in the kinetoplast, and histone H1 participates in kDNA condensation (307, 370). Fluorescence microscopy revealed bromodeoxyuridine (BrdU)-labeled free gaped minicircles (343), which replicate in approximate synchrony with the nuclear S phase. This mechanism requires a repertoire of molecules, including type II topoisomerases; DNA polymerases; universal minicircle sequence binding proteins, primases, and ribonucleases (158); and the p166 protein localized between the kDNA disk and the flagellar body (300).
TRIATOMINE VECTOR (HEMIPTERA: REDUVIID) OF TRYPANOSOMA CRUZI
The emergence of Hemiptera insects during the Paleozoic and Silurian periods, over 400 mya, occurred when drastic changes in the mixture of atmospheric gases and increasing oxygen diffusion led to changes in environmental temperatures. The plants grew vascular systems for conducting the phloem sucked in by arthropods. Among the insects, a complete adaptation to plant sucking resulted from the development of mouth parts with a pump connected to a proboscis (112). The Hemiptera that inhabited the earth (146) during the Devonian period (360 mya) were second-stage vehicles for the delivery of macromolecules widely exchanged between species. The insects' newly acquired machineries could be used for the subsequent transportation and delivery of microorganisms to newcomers.
The reduviids of the subfamily Triatominae, which are obligatorily hematophagous during their entire life cycle, establish a strict relationship with their food sources, mainly birds and mammals and rarely other animals such as reptiles and amphibians, which decisively influences the insects' biology and behavior. Gradually, the triatomines developed hematophagy, initially a biochemical requirement for insect growth, which has contributed to approximate bugs and vertebrate animals. The triatomines do not disseminate infections by the inoculation of parasites with saliva. Instead, the insect infects the host usually after the elimination of excreta with flagellates, with the ensuing distension of its abdominal cavity during a successful blood meal. Multiple insect bites induce an allergic reaction that stimulates vasodilation, which facilitates blood sucking. Additionally, the allergy provokes the scratching of the skin and inoculation of the T. cruzi infective forms at the site of the insect bite (174).
Lately, 136 triatomine species have been found to be widespread in the American continent (139, 316). A majority of the triatomine species are present exclusively in wildlife, but some species that colonize houses are of primary importance for the transmission of T. cruzi. The main species of triatomine vectors of T. cruzi for the human population are Triatoma infestans (Fig. 2), Panstrongylus megistus, Rhodnius prolixus, Triatoma pseudomaculata, Triatoma brasiliensis, and Triatoma sordida (112). Additionally, the triatomines are intermediate hosts and transmitters of protozoan infections to omnivorous mammals (112). The oral route of acquisition of the protozoan infection is considered particularly frequent among skunks, armadillos, and anteaters. The triatomines living in wildlife or adapted to human dwellings generate the great endemic zoonantroponosis named American trypanosomiasis (407, 408).
Fig. 2.

Triatoma infestans (Hemiptera: reduviid), the main transmitter of Trypanosoma cruzi to humans. The adult kissing bug inserts its stylet into the skin for blood feeding from the forearm.
TRYPANOSOMA CRUZI INFECTIONS OF COLD-BLOODED ANIMALS
Early reports showed that T. cruzi can replicate and differentiate in cells from amphibians and reptiles (120, 247). The T. cruzi developmental stages that grow at 26°C to 37°C can infect exothermic and homoeothermic animals (43). The infection is insect vector transmitted to lizards, or they become infected upon the ingestion of a triatomine contaminated with T. cruzi (337). Cold-blooded animals retain cryptic infections (106, 319). Mice inoculated with blood from an infected lizard can become infected with T. cruzi (429).
REFRACTORINESS OF AVES TO TRYPANOSOMA CRUZI INFECTIONS
Birds are notoriously refractory to T. cruzi infections (107). The refractoriness of chickens to T. cruzi is dependent on innate immune factors (281, 293 [see comments regarding this retracted article at first citation]) and does not require a natural antibody; bursectomized chicks are refractory to T. cruzi infections (291). The lytic effect of the chicken complement system alternate pathway upon infection by T. cruzi trypomastigotes cannot be totally ruled out (203, 204). The refractoriness of chicks is present at hatching (293 [see comments regarding this retracted article at first citation], 402), but the infection can be installed by the inoculation of infective T. cruzi trypomastigotes into the air chamber of fertile chicken eggs; dividing parasite amastigotes can be seen in embryo cells until the eighth day of growth (293 [see comments regarding this retracted article at first citation], 407). The innate immune mechanism eliminates infection, and hatched chicks are parasite free (164, 293 [see comments regarding this retracted article at first citation], 402). The chicken blood in the insect gut does not inhibit T. cruzi growth (399). The triatomine feeding upon birds does not represent a barrier against the insect transmission of T. cruzi infections to humans (407). The sympatry of birds and triatomines favors American trypanosomiasis endemicity.
THE BEGINNING OF A GREAT ENZOOTY
Orders of Mammals Infected by Trypanosoma cruzi
The origin of a new parasitic infection is usually recognized when the epidemiological triad (microorganism, vectors, and hosts) is fulfilled. Accordingly, the kinetoplastid protozoan of the genus Trypanosoma emerged by the epoch when the insect vector developed hematophagy and transmitted T. cruzi infection to marsupials (456). The mammal hosts that eliminated the parasite in urine and anal gland secretions then contaminated other wild mammals (99). American trypanosomiasis was set in motion in the ecosystem, where there is a sympatry of triatomine vectors and mammal reservoirs of T. cruzi infections. Hematophagy guided the spread of the triatomines, which propagated infections in the American continent (410).
T. cruzi infections have been detected in hundreds of species of mammals belonging to eight different orders widely distributed in all regions of the phytogeographic Neotropics: the Artiodactyla, Carnivora, Chiroptera, Didelphimorphia, Perissodactyla, primates, Rodentia, and Xenarthra (76, 101, 259, 456). The armadillo species Tatusia novencincta (Edentata: Dasypodide) is constantly seen to have T. cruzi infection. The interactions between the Xenarthra and T. cruzi remain poorly characterized (456). Marsupials of the genus Didelphis (Marsupialia: Didelphidae) are considered the earliest hosts of T. cruzi in South America. Moreover, as Didelphis species have a broad distribution in sylvatic and domestic habitats, marsupials are considered an important T. cruzi reservoir. Opossums (Didelphis sp.) have a peculiar interaction with the parasite: they are able to maintain amastigotes in the tissues and epimastigotes in anal gland luminal secretions, from where the parasitic forms contaminate the environment. It has been postulated that, in addition to serving as a reservoir host, skunks can participate in a nonvectorial route of T. cruzi transmission (452). Wild and peridomestic marsupials serve as food to people in rural areas of the Great Amazon, and therefore, the oral route of contamination by the eating of improperly cooked meat cannot be discarded at present (418). Currently, the epidemiologic importance of this alternative route of transmission remains unknown. Also, the drinking of milk from an infected lactating host is considered an alternative route for T. cruzi infections (33, 55).
Rodents (Rodentia: Echimyidae; Rodentia: Cricetidae; Rodentia: Muridae) play a major role in the dissemination of T. cruzi infections. Various species of rodents with a broad ecological distribution can be infected by T. cruzi. The prevalence (30%) of infections in Rattus rattus correlate with sustained high rates of infections in humans (143). In this respect, it is important that 43% of all South America mammals are rodents (145). Actually, caviomorph rodents (i.e., guinea pigs and relatives) are particularly important because they have an ancient coevolutionary history with T. cruzi, widely spread in the American continent (138). The use of murine species has been fundamental for studies of T. cruzi infections aiming at the assessment of therapeutic regimes (89, 364, 410).
In the order Carnivora, dogs (family Canidae) and cats (family Felidae) show high prevalences of natural T. cruzi infections. They are the main domestic animals participating in the peridomicile and domicile transmission of this parasite infection. The triatomines feed better upon dogs than upon chickens or cats (408). In the southern United States, wild carnivores are exposed to T. cruzi infections, which have been detected in raccoons (Procyon lotor), coyotes (Canis latrans), gray foxes (Urocyon cinereoargenteus), and bobcats (Lynx rufus) (48). In Brazil, infected carnivores (mainly Nasua nasua) have been reported for different biomes: the Amazon Forest, Pantanal, Caatinga, and the Atlantic Rainforest (176, 239, 454).
In the Amazon Basin, T. cruzi infections have been detected in wild primates from different families: the Callitrichidae (Saguinus midas, Saguinus fuscicollis, Saguinus labiatus, and Saguinus ustus), Aotidae (Aotus sp.), and Cebidae (Cebuella pygmaea, Saimiri sciureus, and Cebus albifrons) (259). Epidemiological studies of neotropical species of primates (Leontopithecus rosalia and Leontopithecus chrysomelas) revealed that about 70% of tamarins sampled had T. cruzi infections (283). At the Southwest National Primate Research Center (San Antonio, TX) 182 baboons in an open-pan colony tested seropositive for T. cruzi, but the primary route of infection was not established (450). The demonstrated high prevalence rates indicate the importance of primates in the maintenance of the T. cruzi cycle in the surroundings of modern cities.
T. cruzi infections in a majority of wild and domestic animal hosts can run asymptomatic. In these hosts cryptic infections may produce minimal harm that does not endanger the animal's life, and T. cruzi infections can remain dormant for years or decades. However, T. cruzi-infected mammals may have clinical and pathological manifestations similar to those described for human Chagas' disease (408). Naturally T. cruzi-infected baboons, rabbits, dogs, cats, rats, and guinea pigs develop heart insufficiency, cyanosis, edema, hydrothorax, and hydropericardium. Also, electrocardiographic alterations such as bradycardia, arrhythmias, branch blocks, and other ventricular conduction defects in the course of experimental infections have been described (397, 401, 404, 411, 415, 417). The heart becomes enlarged, and the individual dies either suddenly or by heart failure. Fatal acute and chronic manifestations of Chagas' disease have been reported for chimpanzees, dogs, cats, rabbits, guinea pigs, rats, and mice, and every T. cruzi-infected mammal may end up with a gamut of clinical and pathological manifestations that characterize Chagas' disease (408). In summary, the clinical and pathological manifestations of Chagas' disease described for five different orders of mammals are similar to those described for humans (408).
INTERACTIONS OF TRYPANOSOMA CRUZI WITH VERTEBRATE HOSTS
T. cruzi is an obligate intracellular protozoan showing a complex cell invasion mechanism. Updates regarding the parasite-host interactions have been reviewed elsewhere (16, 127, 128). Infective metacyclic trypomastigotes that invade a host's body generally infect local macrophages, fibroblasts, and muscle tissue. Inside a host cell the trypomastigotes transform into amastigotes that replicate by binary fission every 15 to 18 h. Following several cycles of division, the daughter amastigotes differentiate back into trypomastigotes that disseminate the infection to the tissues. The host cell burst outs of amastigotes, and trypomastigote blood forms are likewise infective.
The protozoan interacts with specific molecules present in the host cells and the extracellular matrix (122, 181). The glycoproteins that share glycosylphosphatidylinositol (GPI), undergoing extensive sugar and side-chain modifications, fuse with the plasma membrane as extracellular membrane-associated proteins (47, 420, 424, 459) with adhesion, paracrine signaling, surface enzymes, and cell differentiation functions (458). Trypanosome GPI-anchored proteins serve as adhesion anchors (137), but they form a coat critical to immune evasion (134, 135). Additionally, GPI-anchored proteins may associate and define the lipid raft microdomain compartment (195). These surface molecules initiate parasite-host signaling events. The plasma membrane environment contains lipid microdomains and rafts, which regulate signaling events through the temporal-spatial organization of proteins (196). The lipid rafts on the parasite membrane appear to regulate membrane fluidity, and an impairment of the host-parasite signaling pathways favors the invasion of the host's body by the pathogen (34, 136).
Approximately 50% of the T. cruzi genome encodes a diversity of surface proteins, distributed into the gp63 proteases, the gp85/trans-sialidase superfamily (TS), the mucins, and the mucin-associated proteins (13, 125). The long-standing adaptation of T. cruzi to vertebrate hosts has been credited to the structural and functional polymorphism of cysteine protease isoforms of the parasite cruzipain genes showing different substrate preferences and susceptibility inhibitors (119, 237). The generation of kinins by cruzipain results in bradykinin receptor (B2R)-mediated signaling through phospholipase C (PLC) and inositol triphosphate (IP3)-kinase to release endoplasmic reticulum (ER)-bound calcium (128). Signaling through the bradykinin receptor (B1R) favors cell invasion (421). This pathway is opposed by the actions of the kininases with angiotensin-converting activities (345–348, 439, 440). The proteolytic generation of kinin in tissues of T. cruzi-infected mice depends on chemokine secretion by macrophages activated by Toll-like receptors (353). The naturally occurring protease inhibitors play a role in cellular invasion by T. cruzi (346). Oligopeptidase B acting upon its substrate generates an agonist for host cell calcium release through adenylate cyclase and phospholipase C (51). The mitogen-activated protein kinase (MAPK) implicated in macrophage activity through gp83 signaling (440) favors the parasite invasion of the host cell (346).
Infective T. cruzi trypomastigote plasma membrane ATP receptor and cyclic AMP (cAMP) levels decrease during parasite interactions with host cells in vitro (186, 442). T. cruzi trypomastigotes present a receptor-mediated ATP transport system regulated by tyrosine and serine/threonine phosphokinases (338). Its adhesion to trypomastigotes is made by ligands of muscarinic cholinergic and beta-adrenergic receptors, and the attachment to receptors on the monocyte membrane modulates the intracellular signal transduction pathways (41). The parasite in contact with the host cell triggers signaling pathway checkpoints critical for the invasion process (15, 16, 19). Parasite invasion occurs after the recruitment of vacuoles beneath the plasma membrane, which invaginates to increase the rate of fusion with lysosomes. Parasitophorous vacuole-lysosomal fusion triggered by parasite-induced stress at the plasma membrane level is essential for the retention of amastigotes inside the host cell (15, 16, 21, 49). In the course of parasite internalization, there are increasing intracellular Ca2+ levels prior to lysosome fusion. Increasing levels of cAMP play an important role in the internalization of the parasite, and adenylyl cyclase inhibitors reduce the rate of invasion. In contrast, increasing intracellular levels of cAMP and Ca2+ may induce exocytosis in many cell types (16).
Parasite invasion evokes plasma membrane vesicle transportation and the blockage of the actin cytoskeleton prior to fusion with lysosomes. Inside the cell, in the parasitophorous vacuole, parasite permanence is short. The trypanosome disrupts the lysosome-like vacuole, and the parasitic forms are set free for replication in the cytosol of the host cell (16). The escape of the parasites into the host cell cytoplasm may occur during the S phase, in which a stress-induced burst of oxygen leads to an increase in glucose consumption and energy production, thus triggering MAPK signaling pathways for parasite and host cell growth and differentiation. In this stage of infection, iron transport proteins are considered essential for parasite replication within macrophages and nonphagocytic cells.
Actually, parasitic overload with a great number of dividing amastigotes in the cytoplasm kills the host cell over a short period of time. In contrast, in vitro T. cruzi infections of monocytes, macrophages, and tissue histiocytes can be self-limiting in phagocytes, because the parasite may be killed in the acidic parasitophorous vacuole (122, 173, 214). In keeping with these observations, natural infections in humans and in laboratory animals show low-chronicity profiles. In the course of cryptic infections, T. cruzi amastigotes, in the absence of inflammatory reactions in their surroundings, undergo latency. The dormant amastigote (hypnomastigont) forms can be seen within nonphagocytic muscle cells (402), where they may persist for decades (Fig. 3) without any significant damage to the host (402, 408). The mechanism involved in the latency of a protozoan parasite inside a host cell has been demonstrated for Plasmodium malaria sporozoites (462). Parasite latency is an active process regulated by an initiation factor (α subunit of eukaryotic initiation factor 2 [eIF2α]) kinase (IK2), the cell cycle of which is downregulated by a phosphatase. When the eIF2α phosphatase removes PO4 from phosphorylated eIF2α (eIF2α-P), repressed translation then gives rise to latency (434). Thus, latency and long-lasting persistence, which are important requirements for the completion of parasite-host life cycles, may consist of concerted ability to exploit the stress response mechanism in eukaryotic cells.
Fig. 3.

Trypanosoma cruzi amastigote nest in the heart of a naturally infected baboon. (A) Parasite nest (arrow) seen in a healthy myocardium section. Shown is hematoxylin and eosin (HE) staining. Magnification, ×200. (B) T. cruzi amastigotes fluorescing with a fluorescein-conjugated streptavidin 188-nt probe, which was PCR amplified with nDNA primers. The amastigote parasites hibernate in the nest in the absence of an inflammatory reaction in their surroundings. (Reprinted from reference 408 with permission of the publisher.)
MECHANISMS OF RESISTANCE TO TRYPANOSOMA CRUZI INFECTIONS
Innate Immunity
Mammals of several orders are susceptible to T. cruzi infections, although different levels of natural resistance can be manifested (399). The important natural mechanism of resistance to T. cruzi infections is phagocytosis. Virulent metacyclic trypomastigotes replicate inside macrophages, and although many parasites are destroyed in the phagocytic vacuole, intracellular dividing amastigotes transform into trypomastigotes that escape into the blood to infect any other cell in the host's body. The resident nonactivated macrophage can control an infection at a 5:1 parasite-to-host cell ratio, but the macrophage is destroyed if heavily infected at a 10:1 ratio (214). Ultrastructural studies showed that the immediate survival of T. cruzi forms in the macrophage appears to be related to its ability to escape from the phagolysosomes, replicating freely in the cytoplasm. Although a lethal synergism between T. cruzi infections and macrophages activated by lipopolysaccharides has been described (303), the macrophage can propagate the virulent parasite transiently in a susceptible host.
An important role of innate immune factors is associated with the lysis of trypanosomes and natural resistance to the infection and its curtailment: cytokines, cationic proteins, transferrins, and other proteins in the complement system activated by the alternative pathway have some toxic activity against T. cruzi forms. Like some virus infections and other infections by parasitic protozoa, T. cruzi infections induce the upregulation of interferons and other cytokines by mononuclear phagocytes and natural killer (NK) cells of innate immunity (170). The levels of serum interferons correlate with the activities of splenic NK cells (189). Daily inoculations of interferons increase the resistance of mice to T. cruzi infections compared with mock-infected controls receiving a placebo. This finding suggests that the activity of alfa interferon-induced NK cells augments the resistance and survival of mice against T. cruzi infections (170, 171, 342). On the other hand, cytokines do not affect directly the viability, motility, infectivity, and virulence of the parasite. However, an increasing resistance is achieved with administrations of alfa and beta interferons, resulting from the stimulation of phagocytosis by macrophages (189, 201). An update of innate humoral factors that influence the course of viral and protozoan infections producing myocarditis, and of Leishmania parasites producing skin ulcers, is given in Table 1. On the one hand, the multiple positions of the arrows shown in Table 1 suggest intraspecific different results in terms of cytokine expression, and on the other hand, different infections may show similar cytokine expression profiles. Overall, innate-immunity-associated cytokine profiles are retained during late acquired immunity. In this regard, a word of caution is needed in order to interpret the data, because the studies have been conducted with mammalian hosts with different levels of susceptibility in the early acute, intermediate, and chronic phases of the infections.
Table 1.
Regulation of cytokine gene expression in the course of viral and protozoan chronic infections
| T cell response | Cytokine | Regulation of expressione |
|||||||
|---|---|---|---|---|---|---|---|---|---|
|
Trypanosoma cruzia |
Leishmania sp.b |
Toxoplasma gondiic |
Coxsackievirus B3d |
||||||
| Acute | Chronic | Acute | Chronic | Acute | Chronic | Acute | Chronic | ||
| Th1 | IFN-γ | ↑ | ↑ | ↑ ↓ | ↑ ↓ → | ↑ | ↓ ↑ | ↑ | ↑ |
| TNF-α | ↑ | ↑ → | ↑ | ↑ ↓ | ↑ → | ↑ ↓ → | ↑ | ↑ → | |
| IL-2 | → | ↓ → | ↑ | ↑ ↓ | ↓ ↑ | ↑ | ↓ ↑ | ↑ | |
| IL-12 | ↑ ↓ → | ↑ | ↑ | ↓ ↑→ | ↑ ↓ → | ↑ ↓ → | ↑ | ↑ | |
| Th2 | IL-4 | ↑ | ↓ ↑ → | ↑ ↓ | ↑ ↓ | ↑ | ↑ | ↑ ↓ | ↑ |
| IL-5 | ↑ | ↑ | ↓ | ↑ | ↑ | ↑ | ↓ | ↑ | |
| IL-6 | ↑ | ↑ | ↑ | ↑ → | ↓ ↑ | ↑ | ↑ | ↑ | |
| IL-10 | ↑ ↓ | ↓ ↑ → | ↑ ↓ → | ↑ → | ↑ ↓ | ↓ ↑ | ↑ ↓ | ↑ | |
| IL-13 | ↑ | ↑ → | ↑ | ↑ | NF | NF | ↑ | NF | |
| Th17 | IL-17 | ↑ | ↑ | ↑ | ↑ | NF | ↓ ↑ | ↑ | ↑ |
| Treg | TGF-β | ↑ | ↑ | ↑ → | ↑ → | ↑ | ↑ | ↑ | ↑ |
↑, superexpression; ↓, underexpression; →, no alteration; NF, not found. Multiple arrows mean that different results were reported in the literature.
Acquired Immunity
Direct evidence showing a role played by acquired immune factors in resistance to T. cruzi infections stems from the observation that human and laboratory animal survivors of acute infection do not undergo an onset with symptoms similar to those of the prime infection upon challenge with virulent trypomastigotes (264). The reinfection with virulent trypanosomes may run symptomless, and the low levels of parasitemias are usually of a short duration. The concept of acquired immunity in T. cruzi infections therefore addresses nonsterile immunity. Various mechanisms of acquired immunity afford some degree of protection against T. cruzi infections.
Humoral immunity.
There is some evidence in the literature showing that humoral factors play a role in the control of infections despite T. cruzi escape by a quick penetrance into a nonphagocytic cell, mainly muscle cells, where it remains inaccessible to serum lytic factors. The transient action of protective humoral antibodies against infection can be shown in mice. The pretreatment of mice with immune serum containing high titers of IgM, IgG, IgG1, or IgG2 plus IgG3 antibodies revealed that the protective antibodies are of the IgG2 subclass, particularly IgG2b. IgM and IgG1 afforded minimal or no protection at all (389). This finding suggests that the IgG response in the acute phase is important for the control of the infection. A role played by humoral immunity was shown with congenitally athymic mice deficient in the T cell compartment, which are hypersusceptible to T. cruzi (426). Athymic mice treated with immune serum had low-level parasitemia and prolonged survival compared to athymic control mice that received normal serum and that showed high levels of parasitemia and mortality in a short period of time. Also, a deficiency of B lymphocytes by the injection of anti-Ig antibodies in the neonatal period resulted in an augmented susceptibility to T. cruzi infections in mice with a selective depression of IgG and IgG2a (331).
Complement-mediated lysis plays a role in the control of T. cruzi infections. A specific antitrypomastigote antibody can be detected by complement-mediated lysis in the course of an active infection (157, 215, 276). The parasite surface antigen targeted by the lytic antibody is a 160-kDa glycoprotein (157, 215). Also, a monoclonal antibody of the IgM isotype with a high level of complement-mediated lytic activity against T. cruzi bloodstream trypomastigote targets a 72-kDa protein on the cell surface (157). Blood trypomastigotes can be made highly susceptible to complement-mediated lysis by treatment with trypsin and, to a lesser degree, with sialidase. These findings suggest that the escape of the trypomastigote from complement-mediated lysis is mediated by regulatory molecules susceptible to digestion by trypsin and sialidase on the parasite membrane (207, 208). Moreover, a complement activator factor released by T. cruzi culture forms inactivates the lytic factors in human serum. The administration of that factor to mice reduced the complement hemolytic activity against T. cruzi by 50% (86, 87).
Antibody-dependent anti-T. cruzi cytotoxicity against opsonized spleen cells is mediated by blood leukocytes (2, 56, 208, 244, 342). This cytotoxic effect is dependent on IgG binding to the Fc receptor on the effector cell surface coat (56). During the intracellular destruction of the parasitic form, there is an augmentation of oxygen consumption, increasing myeloperoxidase activity, and the generation of free radicals (242).
Cellular immunity.
A crucial observation showing the importance of cellular immune mechanisms in the control of T. cruzi infections stems from the dampening of the mononuclear phagocytic system with thorium dioxide or by the intravenous administration of silica particles in mice, before a challenge infection with virulent T. cruzi. In the test animals the parasitemias and the levels of mortality increased, contrasting with the low levels of T. cruzi parasitemias in the untreated control mice. The thymic dependence on the partially effective cellular immune responses was observed for groups of mice treated with cyclophosphamide, treated with total body irradiation, or receiving antithymocyte antiserum to suppress the T cell population. An exacerbation of T. cruzi infections was detected in the infected-treated group, contrasting with the low levels of parasitemias in the control, untreated group of mice (155, 217, 327).
The passive transfer of cellular immunity by lymphoid cells restores the resistance of nu/nu mice that are highly susceptible to T. cruzi infections (216, 328). Interestingly, it was shown that T-lymphocyte-rich spleen cells from mice that recover from an acute infection are highly protective, whereas a B-cell-rich population does not afford protection (322). Therefore, the main mechanism affording partial protection against T. cruzi infections is thymus-dependent cellular immunity. Additionally, cell-mediated immunity is particularly important for the control of the parasitic forms hidden intracellularly, which are considered inaccessible to the humoral factors.
The importance of thymus-dependent cellular immunity in the control of T. cruzi infections was further demonstrated by mice subjected to neonatal thymectomy, which underwent overwhelming infections much worse than those of control mice. Interestingly, the grafting of the neonatal thymus resulted in a restoration of thymic function and a recovery of resistance to the level seen for control mice (202). In the course of T. cruzi infections the subversion of host cell sialylation compromises the antigen-specific CD8+ T cell response (142). The protective immunity to T. cruzi infection appears to depend on the CD4+ and CD8+ T cell expressions of gamma interferon (IFN-γ) (98, 330). A central role played by CD8+ T cells in the control of T. cruzi infection in mice was suggested during the acute and chronic stages, when antigen-independent immunodominant memory T cells reach persistently high levels (102, 302). Regulatory CD4+ CD25+ T cells have a limited role in infection by T. cruzi (340). Additionally, the generation of parasite-specific CD8+ T cell immunity is unaffected by the absence of type I interferon signaling (38, 265). The importance of CD8+ T cells in T. cruzi-infected mice has been challenged by experiments with a restriction of its immunodominance, which avoids pathogen elimination and inversely correlates with the severity of lesions (426–428). However, the cryptic infection induced CD4+ T cells to senescence (11).
The patterns of cytokines that may influence the course of intracellular chronic microbial infections are given in Table 1. These patterns are concerned with the leishmanias, causing widespread intracellular parasitism in the mononuclear phagocytic system, and with coxsackievirus B3, Toxoplasma gondii, and T. cruzi, which cause cryptic chronic infections and myocarditis in humans. Regardless of the source of the infectious agent, there is an overexpression of several cytokines, which can be further upregulated by T helper 1 (Th1), Th2, Th17, and T regulatory (Treg) cell immune responses. Furthermore, it has been observed that some cytokines shifted the regulation patterns observed during the acute and chronic courses of the infections. The analyses suggested that the acute and chronic stages of the infections appear to be under different, yet undisclosed, polygenic regulations of complex interactions between the pathogenic organism and its mammal host; consequently, various patterns of immune system cell activation influence the activity of cytokines and the parasite-host equilibrium (159). During the infectious process the outcome of functional changes of differentiated T cell subsets seems to be influenced by the phase of the infection, and the decision to commit to a specific phenotype of T cells depends on the peculiar signaling pathways to which an effector T cell is exposed (39). CD4+ T cells differentiate a variety of patterns of interactions with the cognate antigen exposed by antigen-presenting cells, and an expected efficient host defense against invading pathogenic microorganisms depends on the coordination of complex intragenomic signaling networks that link the innate and adaptive immune systems (463).
CD4 T helper (Th1) cells play crucial roles in adaptive immune responses to T. cruzi infections, because they recruit and activate other immune cells, including macrophages, B cells, mast cells, neutrophils, basophils, eosinophils (343), and CD8 T cells. Th cells differentiate from naïve CD4 T cells and subdivide to in four major lineages, Th1, Th2, Th17, and T regulatory (Treg) cells, which differ in their functions, patterns of cytokine secretion, and expressions of specific transcription factors. However, a great heterogeneity among Th cell subsets and lineages influences the release of different effector cytokines, giving rise to cells with different functions in the immune host. On the other hand, immune cells secrete the cytokines interleukin-2 (IL-2), IL-9, and IL-10, depending on the environmental circumstances, possibly because immune cells belonging to different lineages, showing an enormous plasticity, may switch patterns of secretion (463). For example, upon stimulation, naive CD4+ T cells produce IL-2, a major cytokine that stimulates the growth of T cells and increases the production of other cytokines, such as IFN-γ and IL-4. Studies in vitro and in vivo with murine CD4+ T cells showed that IL-12 skews toward Th1 cells, IL-4 skews toward Th2 cells, transforming growth factor β (TGF-β) skews toward Treg cells, and IL-6 and TGF-β skew toward Th17 cells (8).
The Th1 cell subset may initiate and participate in cell-mediated immune reactions that trigger efficient macrophage responses to kill intracellular pathogens. Usually, Th1 cells produce IL-2, IFN-γ, and tumor necrosis factor alpha (TNF-α), whereas Th2 cells predominantly secrete IL-4, IL-5, IL-6, IL-10, and IL-13. These T cell subsets are reciprocally regulated by IL-4, IL-10, and IFN-γ. On the other hand, Th2 cell subsets are involved in humoral immune reactions against parasitic infection with an increased production of IgE, eosinophils, and mast cells, but they can suppress cell-mediated immune responses as well (75, 355). In this regard, it may not be possible to determine whether an illness is caused by a predominance of a Th1- or a Th2-induced cytokine response, in view of their interactions. The overlapping patterns of type 1 and type 2 cytokine-producing cells in both the acute and chronic stages of T. cruzi infection demonstrate that long-term infections do not necessarily lead to a dominance of either type of cytokine production (460). Moreover, in response to T cell receptor (TCR) stimulation in the presence of TGF-β, naive CD4+ T cells can differentiate into either Th17 or inducible Treg (iTreg) cells, depending on the cytokine environment. Many autoimmune diseases, which had been attributed to the activity of Th1 cells, are induced by Th17 cells, which produce many cytokines, including IL-17 and IL-22 (464).
Immunosuppression
The reactivation of T. cruzi infections in cases of acute or chronic leukemias requiring chemotherapy with cytostatics and antimetabolic drugs highlights the importance of cellular immune mechanisms in the control of cryptic infections (414). This aspect of the natural course of infections can be modified by coinfections. The important modifying factor is represented mainly by HIV and T. cruzi coinfections mutually affecting each other (168). HIV infection profoundly affects some T. cruzi-infected individuals, sometimes presenting a long silent clinical course and other times presenting an overwhelming recrudescence of the myocarditis and/or meningoencephalitis (88, 433). A growing concern is our understanding of these coinfections, which demand further investigations in regions of the world where this disease is both endemic and not endemic (410). In this respect, a comprehension of the pathogeneses of coinfections involved is crucial for the delivery of new therapeutic strategies, and chemokine receptors may become important therapeutic targets for immunosuppressed patients (263).
TRYPANOSOMA CRUZI INFECTIONS IN HUMAN HOSTS
Humans were introduced into the T. cruzi epidemiological chain possibly early upon their arrival to the American continent approximately 50,000 years ago (408). The earliest report of T. cruzi infection was made for Homo sapiens mummies found in Chinchorro, Chile (30), which date to ∼9,000 years ago. The observation suggests that by that epoch, T. cruzi infections and Chagas' disease affected the Amerindian people living in the Andean region between Chile and Peru. T. cruzi infections spread by human migrations throughout the American continent and possibly reached North America early in the year 1150 BC; parasite DNA was found in a male mummy showing chagasic megacolon (113, 114, 323). Among the factors that favored the transmission of T. cruzi infections to the Amerindians were the acquisition of sedentary habits, agriculture implementation, and domestication of animals that attracted triatomines to domiciles and peridomiciles (24). There were chroniclers that reported triatomine insects existing in human dwellings in the early 16th century (111, 162).
The true history of Chagas' disease began when Carlos Chagas (63) described a new human disease, determining its etiological agent, vectors, and reservoirs. Subsequently, Chagas (63–69) described T. cruzi infections (American trypanosomiasis) in wildlife invertebrate and vertebrate animals. Also, Chagas (67) reported clinical manifestations of the acute and chronic phases of the illness (Chagas' disease) in people with T. cruzi infections. The pathology of acute and chronic Chagas' disease was described in the following years (68, 69, 108, 110, 222, 423–425). After Chagas' discovery, several studies determined the endemicity of Chagas' disease in Latin America countries (109, 143, 147, 272–274, 350, 381, 452).
The World Health Organization estimates that there are approximately 18 million T. cruzi-infected people and 100 million people at risk of contracting infections in Latin America (453). Chagas' heart disease is considered incurable, and high mortality rates have been recorded for the chronic phase of the infection (221, 317). Currently, autochthonous American trypanosomiasis and Chagas' disease are recognized in a vast region between the parallels at 42° North in the State of California and 42° South in the Province of Chubut, Argentina (453). The few autochthonous cases of human Chagas' disease that have been reported in the United States (392) are considered here to be an underestimation. In addition to bug-transmitted T. cruzi infections, new cases resulting from contaminated blood transfusions and also congenital transmission from mother to offspring have been documented (37). As the population in Latin America has more than doubled in the last 3 decades, previous estimates of disease prevalence, mortality, and morbidity require updating.
Approximately one-third of those patients infected will develop chronic manifestations and will die of Chagas' disease. Chagas' disease is emerging (406) in the Amazon Basin, where an increasing number of people have acquired T. cruzi infections in recent years (77, 313), and novel outbreaks are occurring monthly. In Caracas, Venezuela, an outbreak of acute Chagas' disease affected a school community. An epidemiological investigation identified infected children, and the T. cruzi infections were acquired possibly by the ingestion of contaminated food (10). Chagas' disease has become cosmopolitan (354, 406). These reports show a small tip of the huge iceberg named Chagas' disease.
A novel chapter in the history of Chagas' disease has yet to be described, that is, as migration from Latin America to other parts of the world leads to a steady increase in the number of chronic cases of Chagas' disease in countries considered T. cruzi insect transmitter free. In those countries where the disease is not endemic, the suspected steady low rates of these infections are caused by transmission congenitally from mothers to offspring, via blood transfusion or organ transplantation, and accidentally in research laboratories and hospitals. Therefore, transplacental transmission may become an increasing risk for the spread of Chagas' disease in parts of Europe, the United States, Canada, Japan, Australia, and other countries favored by Latin American migrants. The emergence of Chagas' disease in countries where it is not endemic has made it a global health problem. Insights into the problem suggest caution, because T. cruzi infections are usually asymptomatic, although encrypted parasites may last life long in the human host (404, 408). This type of infection requires health systems to be prepared to deal with chronic Chagas' heart disease, digestive system syndromes, and other manifestations that require the delivery of specific medical care (406).
Clinical and Pathological Presentations of Chagas' Disease
What are the consequences for the health of millions of people affected by the American trypanosomiasis? The answer is that approximately 70% of individuals harboring cryptic T. cruzi infections will have a perfectly healthy life, as would any other person not having this protozoan infection. The remaining T. cruzi-infected cases will develop clinical manifestations of Chagas' disease (110). The triatomine-transmitted prime infections most frequently seen in children below 10 years of age are usually not perceived by the patient and by the physician in the absence of symptoms and signs of an acute illness. Approximately 99.5% of the T. cruzi-infected population enter an intermediate stage of chronic infection in the absence of clinically detected symptoms or signs of disease but show specific nucleic acid profiles and serum antibodies against T. cruzi antigens. Some clinically silent but chronically infected patients may yield positive hemoculture and/or xenodiagnosis results. Although unstoppable, T. cruzi infections are separated into acute, intermediate, and chronic stages (Table 2).
Table 2.
Clinical, parasitological, immunological, and pathological findings in Chagas' disease
| Clinical form of Chagas' disease | Symptom(s) | Result for T. cruzi in blooda | Serum antibody detectede | DNA markerb | Gross and/or microscopic finding(s) |
|---|---|---|---|---|---|
| Acute | Asymptomatic (95%) | Positive | IgM and IgG | nDNA | Amastigotes in tissue cells |
| Symptomatic (5%) | Myocarditis, meningitis, and encephalitis | ||||
| Intermediate | Asymptomatic | Negativec | IgG | nDNA | Minimal inflammatory lesion in the heart; parasites usually not seen in tissue sections |
| Chronic | |||||
| Heart | Arrhythmias, bradycardia, cardiomegaly | Negativec | IgG | nDNA | Heart enlargement; severe myocarditis in the absence of parasites in lesions; lysis of nonparasitized target cells by cytotoxic lymphocytes |
| Megasyndromes | Dilation of esophagus or dilation of colon | Negativec | IgG | nDNA | Thickening of the wall of esophagus and colon and lysis of neurons by cytotoxic lymphocytes |
| Neuroendocrine syndromes | Heart insufficiency, megasyndromes | Negativec | IgG | nDNA | Catecholamine cardiotoxicity, myocytolysis, and neuronolysis |
| Congenitald | |||||
| Absence of disease | Asymptomatic | Negativec | Pos/Negc | nDNA | Cryptic infection in the absence of clinical disease |
| Heart disease | Heart insufficiency | Negativec | Pos/Negc | nDNA | Heart enlargement; myocarditis |
| Digestive disease | Dilation of esophagus and/or colon | Negativec | Pos/Negc | nDNA | Megaesophagus and/or megacolon, lysis of parasympathetic and sympathetic neurons |
Usually identified in cases showing clinical symptoms or during routine examination of blood smears.
Nuclear DNA is indicative of a living infection. The kDNA can be integrated and vertically transferred to progeny in the absence of a living infection (173, 402).
Parasitemias may be detected in immunosuppressed patients.
Infections before the third month of gestation are tolerized and do not yield humoral factors.
Pos/Neg, positive/negative.
Acute Chagas' disease.
The triatomines popularly known as “cone-nosed kissing bugs” inoculate the protozoan through a skin injury produced by the insect proboscis (stylet or stinger) while it sucks the blood from the human prey. Acute infection in an immunocompetent host elicits a delayed-type hypersensitivity reaction at the site of entry of the T. cruzi trypomastigotes into the skin (399, 409, 413), but it is usually unnoticed in a host with acquired immunosuppression, as the patient may not complain about an acute illness (404, 408).
The incubation period of T. cruzi in the human body lasts 72 h, during which the parasite undergoes multiplication cycles within the host cell and the immune system triggers an inflammatory skin (chagoma) or conjunctiva (indurated unilateral periorbital injury known as Romanã's sign) reaction. The injury is present in a minority (<5%) of cases with clinical manifestations. In these cases there have been complaints of indisposition, fever, headache, joint and muscle pain, anorexia, vomiting, diarrhea, drowsiness, apathy, lymphadenopathy, hepatosplenomegaly, edema, and convulsion. Acute infection may be detected by a microscopic examination of blood smears stained by Giemsa stain, where the parasite can be seen directly usually during a period of 6 to 8 weeks. In this early phase, acute infections can be detected by specific IgM antibodies against T. cruzi antigens. Sinusal tachycardia, first-degree AV (atrium-ventricular) blockage, a low voltage of the QRS wave, and primary alterations of the T wave can be registered on an electrocardiogram (ECG). Chest X rays may show an enlargement of the cardiac silhouette (110). Among these very ill acute cases, the mortality rate is below 10%. The cause of death in acute Chagas' disease can be cardiac failure, meningitis, or encephalitis.
Typically, the heart of a patient who succumbs to acute Chagas' disease presents an increase in size, dilatation, softness, and congestion. The lymph nodes located between the aorta and the pulmonary arteries are ingurgitated. Microscopically, nests of dividing T. cruzi amastigote forms can be seen in muscle fibers and in histiocytes. Mononuclear cells, especially small and large lymphocytes with expanded cytoplasmic processes, infiltrate the myocardium and adhere to the membrane of cardiac fibers. In the inflammatory sites, other types of cells can be found in variable proportions, such as plasmocytes, neutrophils, eosinophils, and mastocytes (405). Parasitized myofibers can be found in the destructive inflammatory lesions (Fig. 4A). In the lesions, however, the noninfected cardiac cells are rejected by effector mononuclear cells from the immune system. The confluence of multiple rejection units generates the diffuse myocarditis of acute Chagas' disease. Figure 4B shows a typical rejection of the muscle fiber by mononuclear cells of the immune system. The inflammatory infiltration into the heart conduction system is associated with ECG alterations. The inflammatory cells invade the parasympathetic cardiac ganglia, where glia and Schwann cells may be parasitized, but the neurons are always spared (404). Interestingly, lymphocytes adhered to the neuron lead to its rejection and lysis. In addition, the lymphocytic infiltrate reaches sympathetic nerves in the epicardium and intramural structures in the heart. T. cruzi can be recovered from the cerebrospinal fluid in 72.7% of patients with acute infection (179), but pathological lesions (Fig. 4C) in the central nervous system are scarce.
Fig. 4.

Pathology of acute human Chagas' disease. (A) Heart section with a nest of T. cruzi amastigote forms (arrows) and mononuclear cell infiltrates associated with lysis of parasite-free target myofibers. (B) Skeletal muscle showing inflammatory mononuclear cell infiltrates and target cell destruction. (C) Nodular inflammatory lesion in the gray matter of the brain. (Reprinted from reference 408 with permission of the publisher.)
Intermediate phase.
Chronically infected individuals become an intermediate-phase reservoir of T. cruzi infection based on the following criteria: (i) a positive serological test with a specific IgG antibody or parasitological demonstration of the infectious agent; (ii) the absence of signs and symptoms of Chagas' disease; (iii) the absence of electrocardiographic abnormalities; or (iv) normal-sized heart, esophagus, and colon without alterations upon X-ray examinations. Over two-thirds of T. cruzi-infected individuals remain in the clinically intermediate phase throughout their life. Usually, the intermediate phase of T. cruzi infection is detected during job admission tests or during triage for blood donors. The life expectancy of these patients is similar to those recorded for noninfected individuals from the same region (250, 321).
The intermediate phase is detected by immunological and genetic markers of cryptic T. cruzi infection. There is no significant gross lesion in the organs (192, 243), but discrete inflammatory infiltrates in biopsy specimens from the heart (252) and target cellular lysis (Fig. 5) have been reported for chagasic patients (219, 363). In the digestive tube, inflammatory lesions result in the depopulation of parasympathetic neurons (314). The pathological lesions found in chronic chagasic patients classify the disease accordingly to the affected organ in the body.
Fig. 5.

Heart lesion in the chronic human intermediate form. The minimal rejection unit consists of the lysis of the target myofiber by immune system mononuclear cells. (Reprinted from reference 408 with permission of the publisher.)
Chronic Chagas' heart disease.
Clinical manifestations of chronic Chagas' disease are seen in fewer than one-third of those in the intermediate phase of the infection, usually 25 ± 7 years after the acquisition of the acute infection (318). Among chagasic patients with clinical manifestations, 94.5% of the cases are affected by heart trouble: 38.5% of these chagasic patients die suddenly, and 56% will succumb to heart failure. The remaining 5.5% develop digestive system syndromes, either megaesophagus or megacolon (318). The clinical manifestations of Chagas' disease include central and peripheral nervous system disturbances and endocrine dysfunctions (Fig. 6A). This scenario depicts the public health problem stemming from chronic Chagas' disease (95, 96).
Fig. 6.

Pathology of chronic human Chagas' heart disease. (A) Cardiomegaly in an adult with increased ventricles, bulging pulmonary artery conus, lymphatic vessel engorgement, and whitish soldier's patch. (B) Parasympathetic ganglion showing ganglionitis and neuronolysis. (C) Histopathological lesion consisting of strike mononuclear cell infiltrates and diffuse target heart cell lysis. A typical minimal rejection unit is encircled. (Reprinted from reference 408 with permission of the publisher.)
Sudden death can occur abruptly and unexpectedly without trauma or other evident causes (320). Sudden death that often occurs during exercise has been associated with cardiac arrhythmia and with heart rate turbulence (HRT) in patients with chronic chagasic infection (445). HRT is considered a vagal alteration of a baroreflex response to a low systolic volume, which may be abolished by atropine and is insensitive to beta-blockers. Abnormal HRT values have been reported for patients with chronic Chagas' heart disease (243). The pathological substrate of the functional alterations is minimal inflammatory injuries shown in biopsy specimens from the hearts of intermediate cases (81, 252). Intermediate-stage chagasic patients can die unexpectedly and show spared inflammatory injuries in the heart (243).
A random population study showed a high prevalence (18%) of Chagas' disease among street sweepers from Brasília City in Federal District, Brazil (224). Among 245 chagasic street sweepers, 2 were recognized as being in the acute phase of the disease because of an indurate skin lesion (chagoma sign). This finding showed consistently that for each known acute case, there are at least approximately 125 cases with no recording of any initial phase of infection. In a series of chagasic patients in the age group of 30 to 50 years, T. cruzi infections led to severe heart failure with ECG alterations. Premature ventricle contractions, blockage of the right branch of the His bundle, combined blockage of the branch, intraventricle conduction disturbance, and ventricle repolarization alterations are increasingly reported in proportion to the patient's age. Ominous signs of impending death are cardiomegaly, arrhythmias, and thromboembolism. Caution is very necessary because chagasic patients with a severely compromised heart may die during 24-h Holter monitoring electrocardiography (224).
Echocardiography reveals a hypokinesis of the ventricle wall and intraventricle thrombus. A gross finding frequently found for patients with chagasic cardiomyopathy is cerebral infarction due to a dislodged thrombus from the left ventricle (23). Chagasic cardiomyopathy-associated heart failure occurs in a mean period of 7 months to 2 years. The disease affects both genders equally for patients between 30 and 50 years of age. Parasite nDNA can be detected in the tissues of chagasic patients (42, 224).
The main microscopic finding for the heart of a chagasic patient who succumbs to Chagas' disease is an inflammatory infiltrate that palisades the target heart cells. The lymphocytic infiltrates destroy parasite-free neurons (Fig. 6B) and cardiac fibers (Fig. 6C). Parasite nests are seen for approximately 10% of cases, usually in healthy areas of the myocardium. In the lesions there are lymphocytes that adhere to the membrane of the noninfected heart cell and induce the lysis of the target unit. The confluence of several units of rejection leads to diffuse myocarditis. The self-destructive inflammatory process shifts from one region to another in the myocardium. Thus, some areas of the heart are actively destroyed by inflammation, while others are spared. At the ultrastructural level the myofibers shows mitochondrial swellings, hyaline degeneration, and necrosis (388). The lesion is repaired by the connective tissue, and the scar remains in the heart (334).
The lumina of the coronary vessels in a Chagas' disease-affected heart are wide, but the arterioles can be buried in the inflammatory scar in the myocardium. The small blood vessels may exhibit endothelial proliferation and basal membrane thickness associated with the inflammatory process (333, 424). However, an occlusive lesion in the main coronary arteries is not a common feature of Chagas' heart disease.
Digestive forms of Chagas' disease.
Megaesophagus and megacolon that stem from autonomous nervous system alterations can be detected early in chagasic patients (250), confirmed by the detection of specific anti-T. cruzi antibodies and DNA footprints (430, 431). The alterations in the esophageal motility affect young people and cause swallowing difficulties, regurgitation of ingested food, and epigastralgia. The X-ray films may show several esophageal disturbances: (i) normal diameter but difficulty in emptying the barium contrast ingested; (ii) moderate dilatation of diameter and barium contrast retention; (iii) great dilatation, hypotonia, and minimum contractility; or (iv) immense dilatation and elongation over the diaphragm. The disease evolves during periods of dysphagia followed by years with an absence of symptoms (37).
The conspicuous symptom of megacolon of a chagasic etiology is constipation. The indurate fecal matter leads to dilatation and the thickening of the sigmoid colon and rectum. The difficulty in defecation incites the dilatation of the colon, causing pain and discomfort. Megacolon is classified as follows, based on X-ray findings: stage I, with spontaneous elimination of fecal matter; stage II, without spontaneous elimination of fecal matter; or stage III, with complete obstruction and impossibility of elimination after pharmacological stimulus. Laxatives ulcerate the mucosa of the viscera, and peritonitis and septicemia can be fatal. Patients with megacolon (Fig. 7A) present long-term waves and hypercontraction of muscle fibers, and these alterations are not detected in control individuals; the sigmoid colons of chagasic patients show low mobility with higher wave frequency rates than those of nonchagasic individuals (278). These abnormalities are attributed to neuron loss (Fig. 7B). The innervation of the bowel is compromised by neuritis (Fig. 7C). The most frequent complications of megacolon are obstruction and rupture of the viscus (317, 329).
Fig. 7.

Pathology of the megacolon in a chronic chagasic baboon. (A) Huge dilation and thickening of the walls of a segment of sigmoid colon and rectum. (B) Parasympathetic ganglionitis and neuronitis and loss of neurons. (C) Section of a sympathetic nerve on the serosa surface of the colon showing peri- and intraneuritis. (Reprinted from reference 408 with permission of the publisher.)
The alteration of the mobility of the hollow viscera stems from inflammatory lesions in the smooth muscle fibers of the intestinal wall (6, 7) and in the intramural parasympathetic neurons. These lesions are distributed randomly in the esophagus, stomach, and small and large intestines, but the physiopathological implications are evident in the barrier-built terminal regions of the esophagus and of the colon. Inflammatory lesions and parasympathetic neuron depopulation in megaesophagus and in megacolon have been reported (166, 387). The clinical manifestations of the megacolon syndrome occur when the loss (depopulation) of neurons exceeds 55% of the overall intramural parasympathetic units (211, 212). The suggestion of a T. cruzi-secreted neurotoxin is refuted by data from experimental studies (17, 18, 88, 386). The death of the neuron is credited to lymphocyte-mediated lysis, exactly as described above for the minimal rejection unit in the heart of chagasic patients. In this respect, the minimal rejection unit (Fig. 5) is the common denominator of the pathology of human Chagas' heart disease, megacolon, and megaesophagus.
Systemic neuroendocrine manifestations.
Other clinical manifestations of the disease are frequently associated with lesions in the peripheral nervous system (278). Paresthesia, hypoesthesia, inadequate tendinous reflexes, postural and vibration sensibility losses, and muscle weakness have been reported for chagasic patients (304, 408). Also, an increase in the glycolytic function and a decrease in the oxidative capacity of peripheral muscles of chagasic patients have been reported, which were not observed for a control group of nonchagasic patients (269). Proportions of type II muscle fibers with a low activity of adenine nicotinamide dinucleotide diaphorase, a high proportion of fibers densely stained with alpha-glycerol phosphate, and low levels of citrate synthase in chagasic patients has been shown, and these findings were in different proportions for a healthy control group (140, 141).
Chagas' disease affects the sympathetic and parasympathetic ganglia (251). The sympathetic and parasympathetic synapses in the heart of chagasic patients show decreased activities of catecholamines and acetylcholinesterase (141). This finding correlates with the progressive autonomic enervation loss associated with Chagas' disease, and the intracardiac autonomic nerves are severely affected; the inflammatory infiltrates surrounding the neurons induce target cell lyses (298, 333). Therefore, neuronal cell loss is a typical pathological finding in Chagas' disease.
THEORIES ABOUT THE PATHOGENESIS OF CHAGAS' DISEASE
Several theories have tried to explain the lesions in the tissues of a chagasic patient: (i) parasite persistence, (ii) the unified neurogenic proposal, and (iii) autoimmunity. From a clinical point of view, the discrepancies among these theories can be justified, possibly, by the difficulty in determining pathogenicity after a long average time lapse between the acquisition of the T. cruzi infection and the development of severe lesions mainly in the heart, peripheral nervous system, and digestive tube in chronic Chagas' disease (318, 398, 399, 404, 407). Specific features in the course of natural T. cruzi infections and of Chagas' disease parallel some postvirus syndromes (210, 305). For example, the absence of the microbe in physical proximity to the destructive pathological lesions is considered a daunting gap (344). Moreover, the multifaceted clinical and pathological findings of Chagas' disease need to be emphasized in order to explain its unique pathogenesis. In this regard, a plethora of review articles discussed the various arguments in favor of or against any theory for explaining the pathogenesis of Chagas' disease (18, 36, 40, 82, 126, 150–153, 182, 188, 197–202, 227–230, 232, 262, 302, 347, 391–396). Actually, the pathogenesis of Chagas' disease concerns those life-threatening lesions seen in fewer than one-third of all those individuals chronically infected with T. cruzi.
To explain the pathogenesis of Chagas' disease, important questions are as follows: (i) why humans and other mammals belonging to several orders usually do not become sick in the course of acute T. cruzi infections; (ii) why acute T. cruzi infections usually seen in infants are asymptomatic, unperceived by the patient, and unrecognized by the physician; (iii) why high morbidity and mortality rates are found for chronically infected patients usually 3 decades or later after the onset of the infections; (iv) why the parasite is not found in some tissues, particularly those in close physical proximity to the destructive lesions in the heart and digestive tube of chronically infected cases; and (v) what is the mechanism triggering the multifaceted clinical and pathological manifestations of Chagas' disease.
In the previous section it was shown that heart pathology is a hallmark present in 94.5% of the cases who succumbed to Chagas' disease, and therefore, this common denominator will be the “gold standard” for the evaluation of experimental procedures aimed at an understanding of the pathogenesis of the disease. Therefore, myocarditis will be the hallmark to determine the pathogenesis of human disease (Table 3).
Table 3.
Theories on the pathogenesis of Chagas' diseasea
| Theory | Antiparasite immunity | Pathology | Concept of disease | Proposed mechanism of tissue injury | Experimental animal |
|---|---|---|---|---|---|
| Parasite persistence | Tissue parasitism | Mechanical rupture of parasitized cells and inflammation | Rupture of parasitized cells | Mammal | |
| Neurogenic | Acquired/nonsterile | Loss of parasite-free neurons | Toxin action and lysis of neurons | Parasite neurotoxinb | Mammal |
| Autoimmune | Acquired/nonsterile | Loss of parasite-free target cells | Parasite antigen-dependent autoimmune target cell lysis | Cross-reaction due to molecular mimicry | Mammal |
| Innate/sterile | Parasite-induced, genetically driven autoimmune rejection of target cells | Clonal cytotoxicity | Avesc |
Mechanisms of tissue injury in the mouse model of parasite-antigen-dependent autoimmunity and in the chicken model of parasite-induced genetically driven autoimmune Chagas' disease.
A T. cruzi toxin has not been demonstrated.
The T. cruzi kDNA-mutated chicken shows a grossly enlarged heart, which is not seen in mammals after multiple immunizations with T. cruzi antigens.
Parasite Persistence
The theory of parasite persistence is a consequence of the early detection of nests of T. cruzi amastigotes in the heart of a child who died of acute Chagas' disease, which suggests that the disease stems directly from a microbial infection (436). Accordingly, the dividing amastigotes produce a mechanical rupture of the host cell and the release of parasite residue attractants into inflammatory cells. This theory suggests that the inflammatory cells infiltrating the heart are important to alleviate the consequence of the toxic actions of the parasite antigens (423). Regardless of the interpretations of causes and consequences, an early report of acute Chagas' disease myocarditis showed quiescent parasitism contrasting with active inflammatory lesions (424). The absence of parasitism in the tissue lesions endangers the theory of parasite persistence; a great majority of chronic chagasic patients (∼90%) who succumb to the disease do not show parasitic nests in histological sections of the heart (410). The failure to demonstrate the parasite in the tissues of chagasic patients is a major objection against the theory of parasite persistence (Table 3).
The epidemiological importance of Chagas' disease was denied in the early period after its discovery because the parasitic nest was difficult to demonstrate in heart sections and because a suitable test to detect anti-T. cruzi antibodies did not exist. This is considered a major drawback to the recognition of the disease and to the acceptance of the high mortality rates resulting from the devastating chronic Chagas' heart disease in hinterland populations. In this regard, the absence of parasites in in situ myocarditis was attributed previously to an “allergic state” of the host (423). This interpretation considers that myocarditis would develop following repeated T. cruzi inoculations, and the parasite's transitory existence is considered a consequence of the host's immune state. This early concept prevails in some review papers (67, 423–425) and in textbooks (44). Further important updates of the theory of parasite persistence are encountered in papers by contemporaneous authors (18, 35, 259).
T. cruzi infection of the mammal host usually proceeds through repeated intracellular cycles, and the dividing amastigotes reach exponential growth in short periods of time. Interestingly, repeated inoculations of T. cruzi do not produce a reactivation of the acute disease in an immunocompetent host. Furthermore, field and laboratory studies showed that reinfections do not influence the outcome of late-stage Chagas' disease in humans (250). T. cruzi infections of beagle puppies revealed that variations in the source of infecting trypanosomes and the route of inoculation did not interfere with the late outcome of the disease. The inoculation of 10 infective trypomastigotes delayed the onset of acute infection, but the late chronic phase is similar to that of animals receiving 1,000 trypanosomes (264).
Actually, the use of a highly sensitive enzyme-linked immunosorbent assay (ELISA), indirect immunofluorescence, and hemagglutination tests, showing specific anti-T. cruzi antibodies in the sera of chagasic patients, confirmed data from early clinical-epidemiological studies that documented the hyperendemicity of chronic Chagas' disease in Latin America. Currently, DNA tests extend the epidemiological importance of this major neglected tropical disease (173, 218, 430–432), and consistently, the parasite nuclear DNA test (42, 224) has attested to the persistence of cryptic T. cruzi infections in chronic intermediate patients, in Chagas' heart disease, and in cases of megacolon and megaesophagus. These findings are fundamental for any further discussions on the pathogenesis of Chagas' disease.
Unified Neurogenic Theory
The detection of significant losses of neuronal cells in the sympathetic and parasympathetic nervous systems of Chagas' disease cases, in the absence of T. cruzi in situ, is the basis for the hypothesis of neurotoxin release from the parasite nest hidden somewhere in the host body (211, 212). However, the hypothetical neurotoxin has never been demonstrated. In the absence of a neurotoxin or any kind of toxin release by the parasite, the documentation of the lysis of neurons by palisade cytotoxic lymphocytes and target cell depopulations is significant (74, 388). This finding suggests that neuronal cell loss correlates with the impairment of neurohormonal circuits (251, 298, 325, 388). Therefore, the neurogenic theory suggests that autoimmune-dependent abnormalities in the autonomous nervous system can perpetuate the cycle of catecholamine cardiotoxicity, myocytolysis, and heart failure (93–97). This theory reminds the physician about the extraordinary variability of clinical and pathological manifestations of Chagas' disease affecting many tissues and organs in the human body (304). However, the neurogenic theory does not explain the origin of autoimmunity in Chagas' disease, and therefore, further fundamental knowledge on the pathogenesis of the disease is required (Table 3).
Autoimmunity
The autoimmune theory of Chagas' disease is based on the demonstration of the accelerated cytotoxic interaction of T. cruzi-immune lymphocytes with nonparasitized allogeneic heart cells (344). Lymphocytes from rabbits with cryptic T. cruzi infections produce accelerated rejection with the destruction of embryonic heart tissue. These immune lymphocytes adhere to myofibers and lyse parasite-free target heart cells in 2 h, whereas control rabbit nonimmune lymphocytes require 72 h and overadhering to the target cells. Also, immune lymphocytes attach to and destroy the neurons of living segments of the large bowel of rabbits (408). These immunological attacks are organ specific, because the cytotoxicity does not affect equally the target cells of other vital structures of the experimental rabbit. Both the heart myofibers and the neurons of the parasympathetic nervous system are selected targets in rabbits with Chagas' disease (401, 416). These findings suggest that the immunocompetent lymphocytes of the chagasic rabbit have a preformed capacity to readily destroy the target cell (Table 4).
Table 4.
Main findings for parasite-induced genetically driven autoimmune Chagas' disease
| Feature of kDNA-mutated T cells | Clonala proliferation | Enhanced capacity | Pathologyb |
|---|---|---|---|
| Genetically modified T cell clonesc | Yes | Attack of target cells | Severe lymphocyte infiltrates in the lesions |
| Accelerated rejection of target cells | Yes | Rejection of heart cells and neurons | Lysis of target cells |
| Preformed capacity to lyse target cells | Yes | Production of clinical manifestations | Cardiomegaly and megasyndromes |
Each T cell performing target cell lysis is a cytotoxic T lymphocyte clone.
Parasite-free pathological lesions in the chicken model.
Rupture of ORFs encoding cell growth and differentiation, immune factors, and others genes.
ECG and chest X-ray alterations have been reported for an isogenic III/J rabbit model system, which are considered similar to those described for chagasic patients (397). Chest X rays show an enlarged cardiac silhouette in the chronic phase of the disease; grossly, the chagasic rabbit that succumbs to the disease shows cardiomegaly, and microscopically, the myocardium retains inflammatory infiltrations in the ventricles, in the atrioventricular node, and in the conduction system, where immune effector lymphocytes adhere to and destroy the specialized myofibers (397, 415). Direct evidence of cytotoxicity in the isogenic III/J rabbit model system stems from immune effector lymphocytes against isogenic cardiac myofibers (397, 416). In these experiments 73.5% of beating cardiac fibers in the colony completely ceased to pulse in vitro after 1 h of incubation. In control experiments, cardiac cells did not cease the beatings after incubation with nonimmune lymphocytes (416). However, these findings leave the following fundamental questions unanswered: (i) what the preformed self-destructive mechanism is that triggers the cytotoxic lymphocytes and (ii) what role it plays in the progressively severe Chagas' disease chronic-phase lesions.
A current belief suggests that in the acute phase of the infection autoimmunity can be triggered by an initial host-parasite contact and that the ensuing tissue damage can be caused directly by T. cruzi or by a cross-anti-self reaction; overloads of parasitic antigens dampening the immune system cause a loss of self-tolerance (126, 227). However, in the absence of disease manifestations for over 95% of acute infections, it is conceivable that autoimmunity unfolds at a later stage, or it is possible that autoimmune responses are subclinical and become highly pathogenic mainly in the chronic phase. In this regard, antibody-dependent cytotoxicity and/or the direct activation of autoreactive T cells is a candidate mechanism for the triggering of autoimmunity (40, 228).
Attempts to produce the gross and the microscopic pathologies of Chagas' disease by the immunization of rabbits, which received injections of particulate antigens from T. cruzi trypomastigotes emulsified with complete Freund's adjuvant (CFA) (mineral oil plus heat-killed delipidated mycobacterium), have failed (40, 401, 416, 417). The microscopic examination of the tissues revealed small, focal inflammatory infiltrates in the heart of the immunized rabbit. The results suggest that cross-reactive antigens in the heart may induce inconspicuous lymphocytic infiltrates, because the immunization of rabbits with parasitic antigens runs in the absence of clinical symptoms and of gross lesions, contrasting with the central hemodynamic progressive deficit and cardiomegaly in T. cruzi-infected rabbits that succumb to Chagas' heart disease (110, 224). Furthermore, the T. cruzi antigen-driven immune reactions that could generate either protection from or pathology in Chagas' disease are difficult to dissociate (152).
The production of lesions in the absence of active T. cruzi infection in mice immunized with cross-reacting shed acute-phase antigen (SAPA), a parasite acute-phase immunodominant antigen emulsified with CFA, was attempted, but tissue damage remained unclear because target heart cell lysis and the gross cardiomyopathy of Chagas' disease were not demonstrated (152). However, it was suggested that the discrete inflammatory infiltrates seen in the heart can be adoptively transferred to naive recipients. Also, the production of chagasic myocarditis by means of repeated inoculations of the parasite antigens emulsified in CFA is considered inadequate. First, the small lymphocytic infiltrates in the heart do not translate into the typical target cell rejection; similar inflammatory infiltrates were present in control rabbits receiving CFA alone albeit to a lesser degree (416, 417). Second, the gross lesions typical of Chagas' disease were not produced (40, 199).
Several reports described candidate mechanisms to explain the clinical and pathological manifestations of Chagas' disease (4, 36, 38, 40, 82, 91, 126, 150–153, 156, 182, 183, 197–200, 227–231, 232, 262, 265, 302, 347, 391, 393–395, 461). Generally speaking, all proposed mechanisms have been used to try to explain how the infectious agent can initiate and perpetuate the autoimmune disease (73, 209). One such mechanism is molecular mimicry, a situation in which an antigen-specific T or B cell's anti-self response can be initiated in the presence of a pathogen (83, 132, 152, 153). Accordingly, a cross-reaction of parasite antigen-stimulated immunocompetent cells against a self-protein with putative similar amino acid motifs or three-dimensional epitopes is required to trigger the rejection of a self-tissue. Thus, immune lymphocyte recognition of the heart tissue may produce a delayed-type hypersensitivity response to a tissue-specific component bearing structural similarities to a given T. cruzi antigen (85, 227). In this regard, the molecular mimicry between the cardiac myosin heavy chain (residues 1442 to 1447, AAALDK) and T. cruzi protein B13 (residues AAAGDK) suggests that the lesions involved in chagasic cardiomyopathy may be triggered by the parasitic antigen-specific T cell effector response to cardiac myosin (1, 36). However, myosin autoimmunity is not essential for cardiac inflammation in Chagas' heart disease (229). The antimyosin reaction in response to immunization with T. cruzi proteins does not induce cardiac damage (231). Also, the T. cruzi antigens Cha, cruzipain (119), and 45-kDa calreticulin, highly conserved in humans, rabbits, and mice, have been reported to cross-react with host antigens. The challenge of mice with immunogenic recombinant calreticulin induces an inflammatory reaction against a host 45-kDa protein. It was suggested that calreticulin-induced inflammatory infiltrates foster autoimmunity in Chagas' heart disease (102, 324).
A putative role is given to self-antigen immunizations. Considering that the inflammatory environment induces a differential process of self-epitope release, a high level of myocardial antigen favors the presentation of self-peptides and the expansion of autoreactive cells (227). The proinflammatory environment in the host tissue during parasite infection may activate autoreactive T cells. In such an environment, some proteins that are usually sequestered and shielded from immune recognition are exposed to immune system cells. Supposedly, self-protein cryptic peptides usually are not antigenic, unless they become accessible to autoreactive T lymphocytes escaping from central and peripheral tolerance mechanisms (358). In the course of Chagas' disease, tissue inflammation exposes cryptic epitopes to antigen-presenting cells. In the inflammation setting the cellular content of proteases appears to foster the production and modification of antigenic peptides that stimulate immune system inflammatory cells and the release of IFN-γ. Conceivably, the supply of antigenic peptides that increases with peptidase activity is possibly augmenting the amount of antigens available for presentation (457).
In seropositive dogs, high IgG1 and high IgG2 levels have been associated with myocarditis, suggesting that a humoral immune response may lead to an increasing severity of the disease (80). The development of chronic chagasic cardiomyopathy in dogs correlates with high levels of IFN-γ and TNF-α and low levels of IL-10 production (161). In the T. cruzi-infected rodent Calomys callosus histopathological lesions in the heart and skeletal muscles have been associated with high levels of the cytokines TNF-α, IFN-γ, and TGF-β (254). In this context, the presence of amastigote nests and alterations in the extracellular matrix have been reported (57).
A considerable amount of attention has been given to the importance of iTreg and Th17 cells in the development and progression of inflammatory autoimmune diseases (447, 463). The treatment of naive peripheral CD4 T cells with TGF-β plus IL-2 and a TCR stimulant increases Treg cells in the thymus. iTreg cells are involved in immune modulation with anti-inflammatory properties and a deceleration of autoimmunity by recomposing self-tolerance (8, 464).
The proposed mechanisms of autoimmunity in Chagas' disease suggested that T. cruzi antigens induce self-reactive immunocompetent cells, which produce anti-self humoral factors. On the one hand, active immunization with a parasitic subcellular antigen produces discrete inflammatory lesions (416), but a documentation of clinical manifestations and cardiomegaly has not been not obtained (Table 4). On the other hand, the passive transfer of immunocompetent donor cells renders small, focal lymphocytic infiltrates in the heart of the syngeneic recipient but no gross lesion (152, 153). Furthermore, reports showing the passive transfer of autoimmune heart lesions are consistently challenged because parasite persistence among donor immune system cells from chagasic patients has not been ruled out. Thus, the typical gross and microscopic inflammatory chagasic cardiomyopathies are not obtained by conventional antigenic stimulation, and therefore, the primary cause of autoimmune-driven lesions of Chagas' disease is missing.
Autoimmunity can be triggered by antigen-specific immunocompetent cells or by bystander activation. This mode of activation is obtained by the exposure of damaged tissue to antigens, which cause the sensitization of immunocompetent cells in an inflammatory environment. In this regard, bystander activation may result from myocardial cytolysis due to T. cruzi infection, which leads to the release of self-proteins. The processing and presentation of self-antigens can be increased by bystander activation, which induces epitope spreading and the expansion of the immune responses toward different self-antigens (358). This polyclonal activation mechanism may be involved in the pathogenesis of Chagas' disease (305, 339, 443, 448). However, it does not withstand the clinical-epidemiological data showing that acute-phase chagasic infections in humans, with high parasitic burdens, are frequently asymptomatic regardless of the high-level release of self-proteins from heart cells damaged by the mechanical actions of intracellular parasitic forms.
The autoimmune theory of Chagas' disease continues to be challenged in different ways, because the anti-self direct mechanism triggering the inflammatory effector lymphocytes is unknown (197–200, 232, 393–396). On the one hand, tissue lesions stemming from a putative autoimmune mechanism of variable intensities are regulated by the genetics of the host and the parasite (426, 427). On the other hand, multiple genes that regulate the host's susceptibility and resistance to T. cruzi may not be involved directly in autoimmunity (311, 332). Thus, whether inflammatory infiltrates are driven by a T. cruzi antigen or by antigen-independent autoimmunity remains controversial (152, 227). Moreover, the treatment of infections with a trypanocidal drug reduces cardiac myosin-specific delayed-type reactions and antibody production, but the curtailment of the T. cruzi infection does not diminish the prospects for autoimmunity in Chagas' disease (182, 183). Actually, autoimmune humoral factors are, possibly, a consequence rather than a cause of the heart disease (73, 91, 229, 332). A conclusive answer to the question of the origin of autoimmune chagasic myocardiopathy is not within reach by any canonical explanation (126, 223).
Various environmental factors can contribute to the development of an anti-self immune response, among which the infectious agent is considered a major factor (209, 293 [see comments regarding this retracted article at first citation]). Chagas' disease with multifaceted (407, 453) clinical presentations involving primarily the muscles and the neuroendocrine and peripheral nervous systems with ominous repercussions for the cardiovascular system cannot be explained by the actions of the parasite encrypted in the tissues of the human host. The immune system must be persistently activated, adapted, and improved for several decades to face the eradication of the pathogen in a highly efficient way. A host tolerance mechanism distinguishes between self-tissue targets and nonself parasitic antigens throughout the life span of the majority of chronically infected chagasic patients. This so-called immune surveillance is fundamental to keep the self-constituents free of the destructive reactions from the body's self-defense apparatus. In this regard, autoimmunity means the deregulation of the surveillance mechanism and the breakdown of self-tolerance, leading to an attack of the immune system against normal self-tissues. Within this context, T. cruzi may interact in the conventional way with the host's immune system to evoke resistance, or it may evoke genotype modifications in the host's immune system effector cells and autoimmunity.
PROGRESSIVE CHAGAS' HEART DISEASE IN DRUG-TREATED CASES
The treatment of human Chagas' disease with the anti-T. cruzi drugs nifurtimox [4-(5-nitro-furylidenoamine)-tetrahydro-4-4-1,4-thiazine-1-1-dioxide] or benznidazole (N-benzyl-2-nitro-imidazoleacetamide) has had limited success (184, 224, 253, 306, 365, 373). First, these nitroderivatives do not eradicate acute human T. cruzi infections, and an indication of parasite persistence is given by positive after-treatment immunological and nucleic acid tests (407). Second, acutely infected patients treated with a nitroderivative drug show signs of myocarditis in biopsy specimens (184). Third, the after-treatment progression of the disease is not halted in chronically infected cases (224). Moreover, ECG alterations in nitroderivative-treated cases do not differ significantly from those of similar lesions recorded for a placebo-treated cohort of Chagas' disease cases (365). The ECGs showed severe alterations in Chagas' heart disease 24 years after treatment with a nitroderivative compound (373). In conjunction, clinical and field studies suggested that the treatment of human Chagas' disease with trypanocidal nitroderivatives does not prevent the onset of severe heart lesions (224, 407). In conclusion, currently, treatments available for Chagas' disease are unsatisfactory.
Studies of experimental Chagas' disease in laboratory rabbits revealed that the inoculation of virulent T. cruzi trypomastigotes into 1-month-old rabbits results in parasitemias during a period of 3 months, but the animals survived the acute phase of the infection. Interestingly, when the rabbits died, usually after the infection reached the chronic stage, they showed the typical cardiomegaly associated with chronic Chagas' disease. A histopathological study with rabbits that succumbed to Chagas' disease revealed the rejection of heart cells by immune system lymphocytic infiltrates (408).
The standardization of the course of T. cruzi infections in outbred New Zealand White rabbits, which die with reproducible clinical and pathological lesions, raises another important question: can treatment with a trypanocidal drug halt the progression of Chagas' heart disease in rabbits? In order to answer this question, experiments were carried out to show that treatment with a trypanocidal drug curtails parasitemias, but the lesions in the heart of the treated animals remained as severe as those seen for T. cruzi-infected but untreated rabbits (415–417, 419). What can be sustaining the severe destructive lesions in the hearts of the treated rabbits? Is it possible to answer satisfactorily the above-mentioned question and shed light on the origin of autoimmunity in Chagas' disease? These overlapping questions merit further investigations with a basis on the hypothesis that T. cruzi DNA can be retained in the host's body.
TRANSFER OF TRYPANOSOMA CRUZI kDNA MINICIRCLES TO THE GENOME OF CHAGASIC PATIENTS
Lateral kDNA Transfer
Lateral DNA transfer is defined as the process and the successful outcome of the transfer of genetic material from one organism to another far apart in the kingdom (173). Retrospectively, molecular evolutionary biology investigations suggested that ancient lateral DNA transfer events have contributed to the evolution of eukaryotic organisms (118). Lateral DNA transfer is considered pivotal to current paradigm changes in evolutionary biology, playing an important role in the vertical inheritance of genes and thus creating increasing genetic diversity, evolution, and pathology (173, 293 [see comments regarding this retracted article at first citation], 402). Our hypothesis is that T. cruzi DNA can be retained in the host's body and that DNA transfer-induced genotype alterations can lead to the origin of autoimmunity with the multifaceted features of human Chagas' disease. At the very beginning, in the absence of a previously defined pathway for the acquisition of T. cruzi DNA and of adequate tools to identifying a putative exogenous element supposedly transferred between eukaryote organisms far apart in the kingdom, the investigation was spurred by chance. A cytogenetic analysis of peritoneal macrophages from mice acutely infected with virulent T. cruzi trypomastigotes yielded the first reliable information: the Giemsa-stained metaphasic plates revealed an accessory exogenous DNA associated with the macrophage chromosome, which was not present in the control mouse macrophage chromosome. The fluorescent biotin-labeled fluorescence in situ hybridization (FISH) technique using parasite mitochondrial DNA (kDNA) and nuclear DNA (nDNA) probes was employed to identify the origin of the accessory DNA (412, 413). The investigation demonstrated that the exogenous element associated with the mouse chromosome is the kDNA minicircle sequence (Fig. 8A).
Fig. 8.

Insertion of the Trypanosoma cruzi kDNA minicircle into the human macrophage genome. (A) Identification of the kDNA in a metaphase plate chromosome by the FISH method. Fluorescence (arrow) is seen in a chromosome probed with a biotin-labeled minicircle. (Reprinted from reference 63 with permission of the publisher.) (B) Southern hybridization of NsiI digests of 7-day-postinfection (pi) macrophages with a kDNA probe on blots of 0.8% agarose gels. The T. cruzi kDNA minicircle forms a single 0.36-kb band, whereas the early-infection macrophage DNAs show upper 1.2-, 1.8-, and 2.2-kb bands. (Reprinted from reference 408 with permission of the publisher.) (C) Hybridization of NsiI digests of 30-day (postinfection) macrophages with a kDNA probe. The absence of the 0.36-kb band in the Southern blot and the presence of the 1.8- and 2.2-kb PCR products in the 30-day-postinfection macrophage DNA indicate that the T. cruzi infection is eradicated and that the minicircle is inserted into the host cell genome. (Reprinted from reference 408 with permission of the publisher.)
The possibility of existing lateral kDNA transfer (LkDT) is advanced with the use of an ex vivo simplified model system in the T. cruzi-infected macrophage U937 cell line, which is able to eradicate the infection but retains the parasite kDNA, forming specific bands shown by Southern blot analysis: the NsiI digest made as a single cut in the DNA from acutely infected macrophages (7 days postinfection) showed the 0.36-kb parasite kDNA band and upper bands averaging 1.2, 1.8, and 2.2 kb revealed by a specific radiolabeled probe (Fig. 8B). Moreover, the NsiI digests of genomic DNA from chronically infected U937 macrophages that eradicate live T. cruzi infections (30 days postinfection) retained the upper bands, but the 0.36-kb kDNA band was absent (Fig. 8C). These findings suggest that the kDNA minicircle is integrated into the macrophage genome (412). The topology of the minicircle at the integration junction site is as yet unknown, but again, the chance of two integration events occurring at a short distance favors the PCR amplification of the host DNA flanked by the kDNAs at both ends (366). In this context, the amplification of the chimeric kDNA-host DNA is obtained with primer sets annealing to the flanking kDNAs at both ends. Interestingly, the minicircle sequence was localized in long interspersed nuclear element 1 (LINE-1), located at chromosomes Y, 4, and 13. The perpetuation of kDNA integration into the macrophage genome was demonstrated for the clones of macrophages that continued to replicate in culture after several years. Observations throughout this long period of time showed that minicircle integration within LINE-1 creates the potential for foreign DNA mobility within the host genome via the machinery associated with the retrotransposon; in a tracked LkDT event, the minicircle integrated into LINE-1 was subsequently relocated to another chromosome. The travel of the minicircle within the host genome is associated with active LINE-1 characteristics. The original minicircle integration relocated to the p15 locus on chromosome 5 by hitchhiking along with a LINE-1 element originating on chromosome 4. The documentation of this event serves as an indicator of what happens on the broader scale of decades of persistent cryptic infections in chronic cases of Chagas' disease. The demonstration of a molecular pathology stemming from the mobilization of a kDNA/LINE-1 mutation is consistent with the hypothesis that minicircle integration can be a component of the parasite-induced, autoimmune-driven lesions seen in the heart and other target tissues in Chagas' disease (366).
Investigations into the role played by LkDT events in the development of progressive Chagas' heart disease continued with infected rabbits treated with antitrypanosomal nitroderivatives, which showed progressively lethal cardiomyopathy (293 [see comments regarding this retracted article at first citation], 408). The digest of chagasic rabbit DNA from blood, heart, skeletal muscle, liver, intestine, and kidney hybridized with a constant-region minicircle probe revealed an altered configuration of the band pattern of kDNA integrated into the tissues, which was distinct from the minicircle unit size (1.4 kb) seen for parasite DNA alone. This finding shows that the kDNA is integrated into the rabbit's genome (Fig. 9A). Sequencing of the 2-kb band revealed that the rabbit DNA flanks the minicircle insertion at sites of direct CACCAACC repeats within rabbit DNA. The flanking DNA, showing homology with the LINE-1 clone LBNL-1125D4 (Fig. 9B), contains short interspersed nuclear element (SINE) repeats. In a specific case, the insertion mutation showed truncated kDNA fragments of diverse sizes and structures, comprising a grand total of 10.8 kb (293 [see comments regarding this retracted article at first citation]). These findings are a gateway to the exploration of multiple LkDT events in the genomes of Chagas' disease patients. The LkDT seen at multiple sites and the accumulation of kDNA integrations (parasite-induced mutations) can be forces naturally driving the variable clinical manifestations and pathogenesis in the rabbit model of human Chagas' disease.
Fig. 9.

Integration of the kDNA minicircle sequence into the genome of a rabbit with Chagas' disease. (A) Hybridization of rabbit DNA with a specific kDNA probe. A total of 20 μg of EcoRI-digested DNA separated on a 0.7% agarose gel was used for Southern hybridization with 1 μg of a T. cruzi cloned kDNA constant region (kCR) probe. (B) Schematic representation of kDNA integration into rabbit DNA. Arrows show primers used for 5′ RACE to detect the kDNA insertion into the rabbit genomic clone LBNL1. The integration of CCA/ACC-rich kDNA (bp 1255 to 1907) occurred within rabbit DNA, showing attachment sites of direct short CACCAACC repeats. An ORF spans the chimeric sequence at bp 1217 to 1582. (Reprinted from reference 293 with permission of the publisher [see comments regarding this retracted article at first citation].)
Persistence of kDNA in tissues of newborn rabbits.
The congenital transmission of T. cruzi infection occurs when the parasite trypomastigote forms transverse the placental barrier and, through the umbilical cord, reach the fetus (349, 399). The transmission of T. cruzi via the placenta has been demonstrated with litters from rabbits chronically infected with Chagas' disease (293 [see comments regarding this retracted article at first citation]). The crossbreeding of the sexually mature does and bucks during the course of a chronic infection (293 [see comments regarding this retracted article at first citation]) generated chagasic litters; PCR on the genomic DNA from tissues of stillborn offspring or from blood cells of offspring survivors were analyzed for both T. cruzi nDNA and kDNA. The PCR assays showed that those offspring retained nDNA and kDNA but that some retained kDNA alone (Fig. 10A). This finding is confirmed because DNA from heart, skeletal muscle, liver, spleen, and large and small intestines yielded amplification products, which formed specific bands with the kDNA probe. No bands were detected with nDNA or maxicircle probes. The offspring of chagasic does showed kDNA bands that were larger than the unit minicircle in the Southern blots of EcoRI-digested genomic DNA and hybridization with a specific probe, and this finding suggests an integration event. Genomic DNAs from kDNA-positive offspring were subjected to 5′ rapid amplification of cDNA ends (RACE), cloning, and sequencing and revealed the topology of the integration sites of the kDNA minicircles (293 [see comments regarding this retracted article at first citation]).
Fig. 10.

Genetic markers of T. cruzi infection in offspring of rabbits with Chagas' disease with evident pathology. (A) Specific hybridization of PCR amplification products from template DNA obtained from offspring of doe C with Chagas' disease using specific sets of T. cruzi nDNA and kDNA primers. DNA products were resolved on 1% agarose gels. (1) Analysis of kDNA amplification showing bands of 330 bp and its catemers from parasite DNA and from genomic DNA of six progeny with hybridization with the kCR probe. (2) Analysis of nDNA amplification showing bands of 188 bp and its catemers formed with parasite DNA and from genomic DNA of offspring 2 after hybridization with the specific internal probe. (B) Destructive myocarditis and ganglionitis present in 2-week-old offspring. (1) Histopathological section showing mononuclear cell infiltration and lysis of target heart cells. Note the round lymphocytes adhering to the surface of the target cells. (2) Normal histological features of myocardium. (3) Intracardiac ganglion cells from a control offspring of a noninfected rabbit. The circle depicts a neuron. (4) Intracardiac parasympathetic ganglion showing mononuclear cell infiltration and neuron dropout (circle). (Reprinted from reference 293 with permission of the publisher [see comments regarding this retracted article at first citation].)
In the rabbit model of congenital Chagas' disease, active T. cruzi infection is needed for the kDNA to integrate into the host genome. The heat-killed trypomastigotes and the naked kDNA minicircle sequences, which were inoculated intravenously into rabbits and monitored weekly for 3 months, did not integrate: kDNA products were amplified from blood DNA from these rabbits up to but not beyond the third week postinoculation. The control, uninfected rabbit DNA showed the absence of bands that hybridized with nDNA or kDNA probes.
Tissue-specific histopathological lesions are usually present in the hearts and in the peripheral nervous systems of stillborn T. cruzi-infected rabbits. The typical myocarditis of Chagas' disease was shown (293 [see comments regarding this retracted article at first citation], 403) for offspring of a chagasic doe (Fig. 10B). This set of experiments demonstrates the high frequency of horizontal kDNA transfer in vivo into the vertebrate host genome. The offspring harbored persistent living infections (399), but the presence of kDNA fragments in various tissues suggests that some integration occurs shortly after parasite invasion, resulting in the possibility of integrated kDNA throughout the germ line of the host (293 [see comments regarding this retracted article at first citation]).
LINE-1 is the main hot spot of kDNA integrations.
LINE-1 is the most common active retroelement in Homo sapiens, which emerged about 120 mya and continues to expand in mammalian genomes (226). These retrotransposons are segments of DNA that can mobilize to a new location in a chromosome at a distance. The sequencing of the human genome showed that LINE-1 accounts for 17% of the total genome (256). LINEs are freeloaders of repeat-rich short interspersed nuclear elements (SINEs) (Alu-like and mammalian interspersed repetitive [MIR] elements), endogenous retroviral (ERV) elements, mammalian apparent LTR (MaLR) retrotransposons, and intracisternal A particle (IAP) retrotransposons. Full-copy LINEs are about 6 kb in length and have a 5′ untranslated region (UTR), two open reading frames (ORFs), and a polyadenylated 3′ UTR. ORF1 encodes an RNA binding protein with nuclear acid chaperone activity, which is essential for retrotransposition (190, 266). ORF1p is also capable of interacting with other proteins, like cytoskeleton proteins. The protein encoded by ORF2 is ∼150 kDa and harbors endonuclease (EN) and reverse transcriptase (RT) domains (133, 270). The ORF1 and ORF2 proteins form a ribonucleoprotein (RNP) complex, which is responsible for the transport of L1 RNA to the nucleus (117). LINE-1 integrates through reactions of DNA cleavage named target primed reverse transcription (193). Alternative pathways of retrotransposition, like internal-prime (376) and endonuclease-independent (287, 357) retrotransposition events, have also been described. It is believed that various cellular factors, such as DNA repair enzymes, participate in the L1 retrotransposition process (284, 384); however, the entire mechanism of retrotransposition has not been deciphered (117).
The mobilization of retroelements requires DNA double-strand-break (DSB) repair (261, 377, 384, 467). A DSB is a genotoxic lesion that can be caused by a microorganism's invasion. The cells have repair mechanisms, such as homologous recombination, which uses homologous sequences of the genome to repair the DNA damage (362). A microhomology-mediated end-joining (MMEJ) error-prone mechanism has been described (294), which inter-mediates exogenous DNA integration. Various enzymes participate in repair, including the heterodimer KU70/80, DNA protein kinases (DNA-PKs), DNA ligase, poly(ADP-ribose) polymerase 1 (PARP-1), topoisomerase, and others (78, 362). This end-joining pathway uses sequences with 5- to 25-bp microhomologies to repair DSBs (275, 458). Nonhomologous end joining (NHEJ) is a different pathway of DNA repair, whereby the DSB is sealed without reliance on a template. Interestingly, extrachromosomal DNA, mainly mitochondrial DNA, is used in NHEJ and MMEJ to restore genome integrity (100, 172).
LkDT and Human Chagas' Disease
The documentation of contemporaneous LDT in humans used to be prevented by a lack of suitable technologies. In a study on the pathogenesis of Chagas' disease, this problem was circumvented by the addition of specific host DNA primers used in combination with kDNA primer sets (173, 241). The documentation of LkDT in chagasic patients has now been obtained by modified targeted-primer thermal asymmetric interlaced PCR (tpTAIL-PCR), which consists of the use of host LINE-1 DNA primer sets L1-1 to L1-6 to anneal at the 5′ UTR, ORF2, and the 3′ UTR in combination with nested kDNA primers by three consecutive nested PCRs (42, 173, 402). The tpTAIL-PCR used to detect kDNA minicircle integrations into the human genome yielded amplification products, and BLASTn sequence analysis revealed chimeras displaying kDNA linked to host DNA. In one case of acute Chagas' disease with T. cruzi detected in the blood smear, kDNA integration was shown in LINE-1 at chromosome 9. Of great interest, chronic chagasic patients show multiple independent LkDT events targeting retrotransposable elements and coding regions at various chromosomes with highly significant expectation E-value scores.
The tpTAIL-PCR technique, which was used to map T. cruzi kDNA minicircle integration into the human genome in a family study (173), confirming and expanding previous observations (293 [see comments regarding this retracted article at first citation]), has consistently shown the link between the research results and the conclusions. In cases of chronic Chagas' disease, kDNA minicircle footprints are found on several chromosomes, and the main targets for kDNA integration into the human genome are LINEs in 65% of overall LkDT events. Multiple integrations have been observed for chagasic patients, and a minimum of 10 independent events were found in six different chromosomes from a case. In a control group, five individuals of European origin were both nDNA and kDNA free. Because all the chagasic patients from a large group showed LkDT events, the phenomenon holds a promising role in the pathogenesis of Chagas' disease (173, 293 [see comments regarding this retracted article at first citation], 255).
LkDTs are distributed among various loci at different chromosomes. The observed preference of kDNA integrations in unique CArsb repeats shared by kDNA minicircles and retrotransposable elements suggests integration hot spots. CArsbs in the kDNA constant regions are associated with minicircle replication, transcription, and recombination (173, 178, 382). An analysis of the structure of chimeric sequences at the junction between the kDNA minicircle and the host DNA revealed intermediate CArsb stretches (CCCAAAACCA/CCCAAAACC/ACACCAACCCCAA/ACCAACCCC/CCAACCCCAA) sharing alignment similarities. In about 270 chimeric sequences obtained from 54 chagasic patients and descendants, there were over 900 CArsbs, which showed kDNA minicircles and LINE sequences (173). The insertion of kDNA at CArsbs shared by kDNA minicircles and retrotransposable elements suggests that the T. cruzi kDNA minicircle integrates into the host genome by an end-joining homologous recombination mechanism. Altogether, these findings suggest that LkDT is a constant biological process associated with cells during replication and growth. The T. cruzi invasion-induced stress continues during parasite and host cell divisions, creating opportunities for the exchange of genetic material, and homologous recombination is likely the main mechanism mediating the integration of kDNA (173).
The integration of kDNA sequences in LINE-1 in various chromosomes was confirmed by in situ hybridization on host DNA in a metaphase plate of blood mononuclear cells from Chagas' patients and progeny. By using specific host DNA and parasite DNA probes, the kDNA sequences colocalized within LINE-1 on several chromosomes (173). The specificity of the finding is supported by the absence of hybridization in Leishmania braziliensis, a distantly related kinetoplastid, and the LINE-1 probe did not label parasite DNA in control hybridizations with T. cruzi cells (173, 366, 413).
In addition, LkDT events have been detected in nonautonomous retroelement SINEs (Alu and MIR) (61%), MER, ERV, and MalR (39%), also constantly found in coding regions showing sequence stretches complementary to LINE primers (115). The kDNA integration events promote the rupture of ORFs of different genes, and therefore, the transfer of mitochondrion DNA from T. cruzi to Chagas' disease patients can have broad physiopathological significance.
The kDNA integrates into specific loci encoding proteins with important functions: the PARP-1, CLEC5, CITb-109, and HLA haplotype genes, encoding a major histocompatibility complex class 1 antigen, are silenced by the kDNA integrations (173). The rupture of the ORFs of the ADAM gene, involved in myogenesis and neurogenesis, has been observed (402). Additionally, the kDNA integrates into a LINE present at the ORF of the CLEC5A gene 7 q.33 locus in the chromosome illustrated in Fig. 11, which encodes a member of the C-type lectin/C-type lectin-like domain (CTL/CTLD) family, which promotes cell adhesion, cell-cell signaling, glycoprotein turnover, and proimmune response-mediated proinflammatory cytokines (71). The knockouts of the PARP-1, CNTNAP2, ADAM, and CLEC5 genes, which are detected in cases with evident disease manifestations, sustain a correlation with Chagas' heart disease (72).
Fig. 11.

Topology of the Trypanosoma cruzi kDNA minicircle integrated into the genome of a Chagas' case and knockout of the CLEC5A gene. The scheme shows kDNA integration into the chromosome 7 CLEC5A gene open reading frame in continuity with LINE-1 at RP11-707F14. The tpTAIL-PCR amplicon that was cloned and sequenced shows that the s67 reverse kDNA primer anneals at the 5′ end and that the L1-5 primer anneals at the 3′ end. The kDNA (light blue, variable region) extends from nucleotides 1 to 286, and the host DNA (green) extends from nucleotides 270 to 1230. E values for kDNA and for host DNA are highly statistically significant. (Scheme reproduced from reference 174a with permission of the publisher.)
The LkDT events in association with clinical and pathological presentations of Chagas' disease suggest autoimmunity by the following intermediates: (i) the gradual accumulation of kDNA sequences integrated into somatic cells, (ii) the rupture of open reading frames of important genes regulating cell growth and differentiation and the immune responses, (iii) heart damage produced by lymphocytic infiltrates and lysis of target cells, (iv) clonal proliferation of lymphocytes associated with the rejection of heart cells, and (v) age-specific high rates of the autoimmune disease. Consistently, these changes can explain the origin of the pathogenesis of Chagas' disease, which cannot be explained by any other conventional means. To associate main mutations with the multifaceted clinical and pathological features of Chagas' disease, a genetic epidemiologic study comprised of large groups of chagasic patients from different ecosystems is under way. It is postulated that each different locus in the host's genome affected by the kDNA integrations bears a distinct pathophysiological importance. A prospective genetic-epidemiologic field study can show whether the accumulation of kDNA mutations, associating genotype-phenotype modifications, will drive the autoimmune rejection of target tissues and the variable clinical manifestations of Chagas' disease. Here, we formulate a new paradigm for the etiology of chronic Chagas' disease symptoms, namely, genotype modification resulting from the integration of parasite DNA into the host genome as a driver of an autoimmune pathology.
Vertical kDNA Transfer
The permissiveness of embryonic stem cells to T. cruzi infection is an indication that differentiating germ line cells in the genital crest early during gestation retain kDNA minicircles (293 [see comments regarding this retracted article at first citation], 408). Investigations aiming at the demonstration of the integration of the kDNA minicircle into the germ line also showed that it can be transferred vertically to progeny (173, 293 [see comments regarding this retracted article at first citation], 402). Thus, vertical kDNA transfer (VkDT) has been demonstrated in germ line cells from Chagas' disease patient F1 and F2 progeny sperm donors of template material for the amplification of nDNA and kDNA. Both of these markers were positive in nine samples, kDNA alone was detected in six samples, and five samples were nDNA and kDNA free. Therefore, LkDT and VkDT events are largely independent, as parasitic kDNA integrations occur via germ line or transplacental live-infection transmission. This finding is consistent with host sexual reproduction being a streamline of kDNA inheritance: kDNA integrations are found at several loci, and the haploid cell-kDNA amplifications from the progeny yield novel mutation events. The main targets for kDNA integration in the male gametes are LINEs. Multiple integrations are seen in many cases, and various independent events can be captured for a chagasic patient. VkDTs have been detected in the germ line cells of males with active T. cruzi infections and also in infection-free F1 and F2 progeny within the population.
The integration of the kDNA minicircles into the genome of a healthy patient can occur via live infections and via the germ line. In this regard, it is expected that kDNA mutations spread in the human population from T. cruzi infections have been endemic to Latin America for several centuries. As a consequence, kDNA minicircle integrations have been obtained from healthy patients (our unpublished data). In order to determine the prevalence of kDNA mutations in the population, a clinical genetic-epidemiological study is under way. Further specific information on the kDNA mutations will possibly be disclosed as human genomes are sequenced.
Heritability and Fixation of kDNA Mutations
A thorough documentation of vertical kDNA transfer in a chagasic family can be achieved by the sequencing of a specific hot spot chromosome that concentrates most of the kDNA integrations or by the sequencing of genomes of chagasic patients. In a family study, chagasic progenitors showing the highest integration insertion frequencies were found to be natural candidates for full sequencing (173, 465). Because of a lack of these studies, which are deemed necessary to demonstrate further the heritability and fixation of the kDNA mutations in the progeny, the documentation available stems from random tpTAIL-PCR. This technique reveals that 26% of the VkDTs integrate into LINE-1 at nucleotide (nt) 77363 of locus AL732374.14 (173). Bioinformatics analyses showed that a 3′ host LINE consensus sequence clone anneals exclusively at locus AL732374.14, representing a type-specific hot spot in chromosome X. The heritability of the kDNA mutations and their fixation in the progeny are shown explicitly by the alignment of chimeric sequences derived from somatic cells and gametes of a father and from the somatic cells of his daughters at locus AL732374.14. Validation of these findings was provided by Southern analysis of EcoRI digestions from the father and his daughters (173). Interestingly, in the absence of specific anti-T. cruzi antibodies, the germ line-transmitted kDNA mutations are consistently seen in immune-tolerized patients.
We anticipate that the high rate of disease manifestations seen for insect-transmitted T. cruzi infections will tend to discontinue. On the one hand, since the populations affected have obtained an increased level of education, the people will no longer be exposed to insect-transmitted T. cruzi infections, and on the other hand, the cohorts harboring congenitally transmitted (approximately 10% of chagasic mothers) infections passing through placental filters tend to diminish gradually. Only kDNA mutations via the germ line will pass without interruption to descendants; solely, kDNA mutations will testify to evolution over time.
MOSAIC RECOMBINATION AND HITCHHIKING: CHANGING PARADIGM
LkDT- and VkDT-induced mutations can undergo mosaic-type recombination involving two or more DNA stretches from different loci. Approximately 15% of the somatic cell mutations involve truncated stretches of a minicircle interspersed with variable-length fragments hitchhiking from a second-party locus. Approximately 40% of germ line mosaics show variegated patterns linking three different loci with minicircle sequences. These germ line mosaics have minicircle sequences and two host DNA stretches from different loci. The observed variable distances among interspaced microhomologies might explain the mosaic recombination and the hitchhiking events (173, 366, 372).
LkDT and VkDT are considered unexpected findings, and therefore, some concerns that were brought into the discussion (293 [see comments regarding this retracted article at first citation]) demand further data and information to clarify specific points addressing to the broad significance of the phenomena (173, 402). LkDT and VkDT (293 [see comments regarding this retracted article at first citation]) represent a major paradigm change essential to an explanation of the pathogenesis of Chagas' disease, here considered a fortuitous share of negative selection. In order to secure this challenging proposal, the elimination of a residual cryptic infection, which would mask the LkDT and VkDT events in chagasic patients, is required to confirm the major conceptual change. First, the instrumental limitation to unraveling LkDT and VkDT events is unfolded. In this regard, the substitution of degenerate primers by host DNA-specific primers in modified targeted-primer thermal asymmetric interlaced PCR (tpTAIL-PCR) is a step forward (173). Second, having shown that tpTAIL-PCR consistently shows host DNA-kDNA chimeras in chagasic patients, the method can be used in large-scale clinical genetic-epidemiological studies to associate specific mutations with the manifestations of Chagas' disease.
In the context of Chagas' disease, autoimmunity is proposed on the basis of a preformed capacity of immune lymphocytes to carry out an accelerated destruction of nonparasitized target heart cells, which is triggered upon contact between effector and target cells (344, 401). However, the clinical and pathological manifestations of the disease have not been obtained by conventional immunizations with wild or with recombinant T. cruzi antigens (386), and therefore, the origin of the autoimmunity in Chagas' disease is unresolved. This explains the necessity to reproduce experimentally the clinical and pathological gross lesions and the microscopic rejection of the target cells in in vivo myocarditis in a parasite-free model system, which cannot be obtained by the injection of antigens into laboratory animals (40, 119, 152, 311, 324, 344, 400, 401, 417). Within this context, the challenge is to demonstrate that LkDT and VkDT are triggers of the genetically driven autoimmune inflammatory cardiomyopathy in human Chagas' disease.
The identification of kDNA mutations in several loci in the host's genome proposes potential conceptual changes regarding the pathogenesis and also the epidemiology and social importance of Chagas' disease (173, 282, 293 [see comments regarding this retracted article at first citation]). To substantiate the suggestion that lateral kDNA transfer can explain the pathogenesis of Chagas' disease, further original studies in a “clean” model system were undertaken with a cross-kingdom animal (402).
AUTOIMMUNE CHAGAS' DISEASE-LIKE CARDIOMYOPATHY IN kDNA-MUTATED GALLUS GALLUS
In humans, rabbits, mice, dogs, and baboons chronically infected with T. cruzi, the chagasic heart weakens due to destructive myocarditis (33, 407). Chagas' disease is considered incurable because the treatment of T. cruzi infections by the administration of an antitrypanosome nitroderivative does not prevent heart lesions and death of chagasic patients (33, 397, 411, 415, 419). This observation suggests that an effective treatment for Chagas' disease requires further knowledge about parasite-host relationships and the pathogenesis of the disease (407). On the one hand, the pathogenesis of Chagas' disease has been attributed to parasite persistence with the rupture of parasitized cells and the release of parasitic antigens that attract inflammatory cells infiltrates (165, 461), and on the other hand, it has been attributed to the autoimmune rejection of the heart by immune system inflammatory effector cytotoxic lymphocytes (152, 229, 344, 408). However, a clear demonstration of the mechanism of and the part that autoimmunity plays in the development of Chagas' heart disease is essential for the effective delivery of treatment. To demonstrate the important role played independently by autoimmunity in the pathogenesis of Chagas' heart disease, coexisting tissue inflammation with active infection needs to be eliminated (40, 203, 408, 434). An approach to answering this question requires the abrogation of the parasite infection. In order to fulfill this requirement, investigations proceeded with the chicken, which is refractory to T. cruzi infections.
These investigations were initiated by the demonstration that T. cruzi penetrates the stem cells of the chick epiblast and that the parasite can multiply in the early-growing embryo (293 [see comments regarding this retracted article at first citation], 402). A few T. cruzi trypomastigotes were inoculated into the air chamber of embryonated stage X chicken eggs prior to incubation at 37.5°C at a humidity of 65% (Fig. 12). The infection was established in the embryo tissues, and T. cruzi-infected 5-day-old embryos that were ground (5 mg/ml in phosphate-buffered saline [PBS] [pH 7.4]) and seeded in axenic cultured medium yielded the epimastigote forms of the parasite. Also, the inoculation of the macerated tissue suspension in weaning mice allowed the recovery of blood trypomastigotes after 2 weeks. Additionally, the T. cruzi-infected embryo tissue DNA collected on the second, fourth, and eighth days postinfection produced nDNA and kDNA PCR amplifications with specific primer sets; however, tissue collected on the 10th, 12th, 18th, and 20th days (Fig. 13A) yielded PCR amplification products only for kDNA (293 [see comments regarding this retracted article at first citation], 402). Although the refractory nature of the response of birds to T. cruzi is well known (281, 402, 408), the possibility of any residual infection is excluded by the highly sensible nDNA PCR test, which detects 10 fg of the parasite DNA (157, 288), which is 24-fold below the total amount of DNA from a single parasite (Fig. 13B). These findings show that T. cruzi infection is eliminated by the innate immune response upon the development of the immune system after the first week of embryo growth and confirm that kDNA alone is retained in the chicken genome (122, 233, 402). In a T. cruzi-free control experiment, embryonated chicken eggs subjected to PCR showed neither nDNA nor kDNA.
Fig. 12.

Trypanosoma cruzi infection is established in a Gallus gallus embryo. (A) Positive-control T. cruzi trypomastigote fluorescein labeled with antibody from a Chagas' disease patient. The inset shows a fluorescein-labeled amastigote stained with Chagas' disease patient antibody diluted 1:128 in PBS (pH 7.4). (B) Section (HE stained) (magnification, ×100) from an uninfected chicken embryo which remained unstained with the anti-T. cruzi antibody diluted 1:128. (C) Background staining of the same control section treated with fluorescein-labeled anti-T. cruzi antibody. (D) Infected chicken embryo endoderm and mesoderm parasitized cells stained green by the fluorescein-labeled anti-T. cruzi antibody diluted 1:128. (E) X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) blue-stained β-galactosidase-expressing T. cruzi Tulahuen-infected chicken embryo endoderm and mesoderm cells. Control serum from an uninfected donor lacking anti-T. cruzi antibody did not stain parasitized embryo cells. (Reprinted from PLoS Neglected Tropical Diseases [402].)
Fig. 13.

Evidence of kDNA integration into germ line cells and tissues from birds hatched from T. cruzi-infected eggs, with accompanying pathology. (A) Establishment of T. cruzi infection early in embryonic development followed by elimination early in Gallus gallus embryonic development. (Top) Bands (330 bp) formed by PCR-amplified minicircle kDNA templates harvested at several stages of chicken embryonic development, after hybridization with a specific probe. (Bottom) Bands formed by PCR amplified from the same embryos after separation on a 1% agarose gel and hybridization with a specific nDNA probe. The 188-bp nDNA band is diagnostic of the parasite persistence in the host tissue. (B) Sensitivity of PCR with nDNA primers Tcz1 and Tcz2. Lanes 1 and 2, control DNA from kDNA-negative and from kDNA-mutated chickens; lanes 3 to 7, mix of 200 ng of control chicken DNA with increasing amounts of T. cruzi DNA, 1 fg, 10 fg, 1 pg, 100 pg, and 1 ng, respectively. Hybridization with the radiolabeled 188-bp probe improved the sensitivity of the technique (10 fg), which is 24-fold below that for single T. cruzi total DNA. (C) Destructive myocarditis and ganglionitis in a 2-week-old F1 chick. (1) Histopathological section showing mononuclear cell infiltration and lysis of target heart cells. (2) Normal histological features of the myocardium. (3) Intracardiac ganglion cells from a control offspring of a noninfected chick. (4) Section of an intracardiac parasympathetic ganglion showing lymphocytic infiltration and dropout (circle) of neuronal cells. (Modified from reference 293 with permission of the publisher [see comments regarding this retracted article at first citation] and from PLoS Neglected Tropical Diseases [402].)
LkDT in Somatic Cells
The establishment of T. cruzi infections in the chicken embryo is possible during the first week of growth, prior to the development of the immune system; an immunological window is open for parasite growth in the epiblast stem cells in the mesodermal, endodermal, neural, and genital crests of the growing embryo. The T. cruzi infection is eradicated by the innate immune response after the first week of growth, and the parasite mitochondrial kDNA alone is retained in the chicken embryo (293 [see comments regarding this retracted article at first citation], 402). Moreover, the retention of the mitochondrial kDNA minicircle sequences in the genome of chicks hatched from T. cruzi-infected eggs defines a parasite-free cross-kingdom “clean” model system. The chicken model system is adequate to study the relationship between genotype alterations resulting from kDNA integrations and autoimmune heart lesions, and parasite persistence is avoided.
Interestingly, chicks hatched from T. cruzi-infected eggs, retaining the kDNA integrated into the genome, showed inflammatory lesions in the heart and in the parasympathetic ganglion, whereby target cells were lysed by immune system lymphocytes (Fig. 13C). Furthermore, the chicks that died early after hatching had pronounced cardiomegaly and failure, with features similar to those described for Chagas' disease in humans (293 [see comments regarding this retracted article at first citation], 408).
The documentation of parasite-free gross and microscopic inflammatory cardiomyopathy in chicks hatched from eggs that received the T. cruzi inoculations suggests that the chicken model system is suitable for studies of the multifaceted clinical and pathological manifestations of Chagas' disease. Additionally, the DNA templates obtained from those adult chickens showed T. cruzi-kDNA amplicons in the absence of parasite nDNA. In the T. cruzi-free control experiment, embryonated chicken eggs and mock-infected eggs subjected to PCR showed neither nDNA nor kDNA. Therefore, it was shown that kDNA alone is transferred to the chicken genome during transient T. cruzi embryonic infections. The horizontal transfer of kDNA minicircle sequences to parental (F0) chicken genomes has physiopathological consequences. The crossbreeding of F0 birds generated F1, F2, and F3 progeny, and they all tested positive for kDNA without nDNA. T. cruzi infection early in embryonic growth generates mature chickens with kDNA integrated into gonadal tissue, and therefore, its vertical transfer to progeny can be tested (402).
VkDT in Germ Line Cells
Sperm and ova from birds hatched from T. cruzi-infected eggs were obtained to confirm the vertical transfer of minicircle sequences to progeny via the germ line (293 [see comments regarding this retracted article at first citation]). DNA templates of germ line cells from roosters and hens yielded PCR amplicons with kDNA primers but lacked nDNA amplification with the specific primer sets. Crossings of kDNA-mutated F0 birds generated F1, F2, and F3 progeny, and each sibling showed amplicons of the minicircle alone. In control experiments template DNAs were subjected to PCR, and neither nDNA nor kDNA was detected. Thus, chickens with kDNA integrated into their germ line, and somatic cells in the absence of infection, are obtained. The detection of kDNA with signals of distinct sizes combined with the absence of T. cruzi nDNA attests to the success of the integration event and of the subsequent eradication of the infection (402).
Use of tpTAIL-PCR To Map kDNA Mutations in the Chicken Genome
A previous investigation conducted with the human genome relied on the chance of two kDNA integration events occurring in proximity to LINE-1, which can be copied by direct PCR with specific primers annealing to the kDNA ends (366). In the parasite-free chicken model system, however, a similar event showing the putative sites of kDNA minicircle integrations has not been obtained. In this regard, by the time the investigation on the cross-kingdom model was initiated (293 [see comments regarding this retracted article at first citation]), the chicken genome project was under way. The publication of this project revealed a haploid content of 1.2 × 109 bp (20,000 to 23,000 genes) divided among 39 chromosomes. The autosomes are classified into macrochromosomes 1 through 5, intermediate chromosomes 6 through 10, and microchromosomes 11 through 32. The sex chromosomes are denominated Z and W, with homogametic males (Z/Z) and heterogametic females (Z/W). Repetitive elements made up 10% of the chicken genome, compared with 40 to 50% in the genomes of most mammals. A relatively compact genome structure is the result of the limited accumulation of repetitive elements. Unlike other vertebrate genomes, active short interspersed nuclear elements (SINEs) are not found in the chicken genome. Most retroelements found in G+C-rich regions and many of the chicken repeat 1 (CR-1) elements flank multiple genes, but CR-1 elements may also accumulate within A+T-rich satellite regions (408).
An obstacle precluding the investigation of LkDT events in the cross-kingdom model is the scarcity of information about candidate sites for exogenous kDNA integration. If, on the one hand, information on the chicken genome potentiates an understanding of topological features of the chicken genome, on the other hand, the demonstration of LkDT in the chicken model system continues to be limited amazingly by the lack of a suitable technique. The trial-and-error approach included testing by the 5′ RACE technique then used for the PCR amplification of integration events in the chagasic rabbit, combining the kDNA primer sets with degenerate primers, which may anneal to cognate repeats in proximity to the integration sites (335; data not shown). By using this technique (http://www.plosntds.org/article/info%3Adoi%2F10.1371%2Fjournal.pntd.0001000), chance favored the amplification of a chimera sequence with kDNA integration close to a CR-1 retrotransposon (GenBank accession number AY237306) within the locus NW_001471687.1 at chromosome 4 of Gallus gallus (293 [see comments regarding this retracted article at first citation]). Likewise, the problem can be resolved by tpTAIL-PCR modification with the use of specific chicken DNA primer sets, and the mapping is shown in Fig. 14. A bioinformatic search of the kDNA-mutated chick sequences revealed CArsb repeats between kDNA and host DNA junctions. These repeats are concentrated in coding regions of chicken macrochromosomes 1 to 4. CArsbs are also present in long-terminal-repeat (LTR) Hitchcock transposons and in CR-1 non-LTR retrotransposons (402). As in the family study described above, the consensus microhomologies present in coding regions and in minicircle end joining mediate the integration of the exogenous sequence into the chicken chromosomes (275).
Fig. 14.
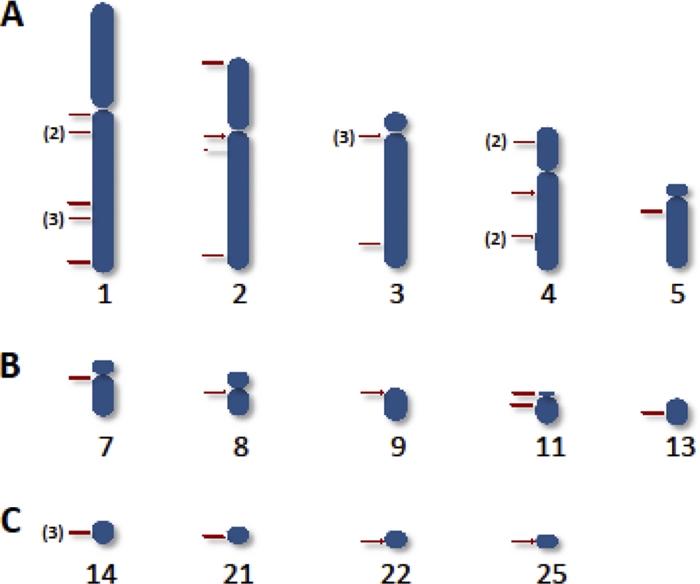
Heredity of integrations of Trypanosoma cruzi kDNA minicircles into several loci of the chicken genome. Rows A, B, and C show integrations into the macrochromosomes, into the intermediate chromosomes, and into the microchromosomes, respectively. The numbers in parentheses indicate the total numbers of times that an insertion (red bar) was present at a chromosomal locus from animal source. (Reprinted from PLoS Neglected Tropical Diseases [402].)
Crossbreeding generates F1, F2, and F3 progeny, and they all test positive for kDNA with a lack of nDNA. This finding indicates that T. cruzi infection, occurring early in embryonic development, produces mature chickens with kDNA integrated into gonadal tissue. Twenty-six chimeras with an average of 555 ± 153 nt have been documented in 14 chromosomes, and the scores attained for each of the chimeras are statistically significant (P < 0.001). Overall, 61.6% of kDNA mutations enter into the macrochromosomes. The alignment of chimeric sequences from F0 and F1 chickens shows the vertical transfer of the kDNA mutation in the noncoding locus of chromosome 4. Additionally, kDNA mutations are present in the dystrophin gene, which encodes a high-molecular-weight protein connecting the cytoskeleton to muscle and the nervous cell membrane (296). Interestingly, clinical and pathological manifestations are clearly associated with the kDNA mutations in the locus of the dystrophin gene in F1 and F2 chickens, with muscle weakness, cardiomegaly, and heart failure (293 [see comments regarding this retracted article at first citation]). The control chickens did not test positive with the specific kDNA assay.
The chimeric sequences that were obtained from F0, F1, F2, and F3 kDNA-mutated chickens enclosed ORFs with the potential for the translation of hybrid proteins. Among these ORFs, 43.3% comprise kDNA alone, and 56.7% are chimeras formed by kDNA-host DNA. A majority of these ORFs encode proteins without significant similarity, but 20% of them translate hypothetical proteins with significant similarities with others from our human study (173). However, a functional role for the ORF-encoded neoantigen in the pathogenesis of Chagas' disease can be excluded in the absence of humoral autoimmune factors in the actively tolerized kDNA-mutated chicken (402).
kDNA integrations are seen frequently in genes encoding protein kinases playing important roles in cell division, differentiation, and growth factors, transcription factors, and immune factors (402). Other important genes encoding GTPase, adenylate cyclase, and adhesion molecules, which are involved in macrophage recruitment and blood vessel maturation, are disrupted. A gene expressed in blood mononuclear cells from patients with systemic lupus erythematosus is ruptured by kDNA integration. A minimum of 12 mutations can be observed for chickens with severe inflammatory cardiomyopathy. These mutations can skew coding regions of chromosomes, with subsequent functional alterations such as cell cycle regulation, clonal proliferation of immune system cells, and tissue injury (72, 185, 196, 205, 455). The complex intragenomic interactions affecting immune effectors and target cells may explain the variegated pathology in Chagas' disease.
Clinical and Pathological Manifestations of Chagas' Disease in kDNA-Mutated Chickens
In the cross-kingdom model of Chagas' disease, the experimental T. cruzi inoculation of the chicken embryo highlights the innate immune system exclusion of infection. Parasite-free adult kDNA-mutated chickens can develop shortness of breath and impairment of blood oxygenation, which evolves to severe cyanosis (Fig. 15A). The electrocardiogram recordings from kDNA-positive birds showed a consistent change of the axis to the frontal left positions as the heart increased in size over time. Control chickens never exposed to T. cruzi kept the electric axis unchanged. The heart/body weight indexes for kDNA-positive F2, F1, and F0 birds increased over time, from 6 ± 2 to 6.7 ± 2 and to 12 ± 5, respectively, whereas the control group index was 4.2 ± 2. The heart indexes from F0 and F1 kDNA-mutated chickens were significantly different from those of the controls. Lengths of survival time for the F0 and F1 kDNA-positive birds were shorter for F0 (12 ± 4 months) and F1 (13 ± 2 months) birds than for those in the control group (19 ± 5 months), and these differences were statistically significant. Cardiomegaly and heart failure in F0 and F1 kDNA-positive birds were attenuated in F2 and F3 progeny. Thus, kDNA integrations into macrochromosome coding regions, generating skewing, instability, and clonality (72, 185, 375), can undergo long-range intragenomic signaling interactions (236). Investigations on the intragenomic mechanisms associating kDNA mutations and autoimmunity, which tend to achieve physiological balance in the descendants, are under way.
Fig. 15.

Clinical and pathological findings for Gallus gallus with Trypanosoma cruzi kDNA mutations. (A) Nine-month-old F1 hen with heart insufficiency, showing cyanosis of the comb, and a control hen of the same age, showing a bright red comb. (B) Cardiomegaly (30 g) in a 9-month-old hen that died of heart failure. (C) Control heart (8 g) from a 9-month-old hen. (D) Myocarditis showing immune system mononuclear cell infiltrates and lysis of target heart cells. The red circle depicts a minimal rejection unit, whereby effector lymphocytes destroy a target heart cell. The inset shows control heart histology. (E) CD45+ lymphocytes (arrows) identified in heart lesions by a phycoerythrin-labeled specific monoclonal antibody. (F) CD8+ γδ immune lymphocytes (arrows) involved in severe destruction of the heart. (G) Abundant CD8α+ T cells present in severe lesions with heart cell lysis. (Modified from PLoS Neglected Tropical Diseases [402].)
kDNA-mutated adult chickens develop clinical manifestations and gross cardiomegaly similar to those described for human disease (110, 224, 408). The lethal cardiomyopathy in the parasite-free chicken model system, for which the destruction of heart cells by lymphocytes has been documented, covalidates the autoimmune pathogenesis of human Chagas' disease. Heart lesions are never seen in mock-infected control chickens. The inflammatory Chagas' disease-like cardiomyopathy hallmark of human disease translates to the multifaceted clinical presentations in kDNA-mutated chickens, involving primarily the heart and skeletal muscles along with the peripheral nervous system, leading to severe repercussions for the cardiovascular system.
Cardiomegaly is present in kDNA-mutated adult birds and absent in control chickens of the same age. The heart weight of a kDNA-mutated chicken can reach over three times that of a control bird of the same gender and age (Fig. 15B and C). Pleural and peritoneal effusions can be found in kDNA-positive birds with heart failure. Microscopic examination of sections from the myocardium shows infiltrates of immune system effector lymphocytes and target cell lysis (Fig. 15D). The destruction of parasite-free target fibers by effector cells characterizes a minimal rejection unit in the heart of a kDNA-mutated chicken. The coalescence of several rejection units creates diffuse myocarditis with a massive destruction of the myocardium. The intracardiac parasympathetic ganglion shows mononuclear cell infiltrates and the destruction of neurons. None of these findings were seen for the control chickens.
The kDNA-mutated chickens died early compared to controls. When these chickens died, they had a destruction of target tissues by immune system cytotoxic T lymphocytes (402). The phenotype of immune system cells in the inflammatory infiltrates revealed a thymus-dependent immune response. The CD45, CD8+ γδ, and CD8α+ immune lymphocytes carry out the lysis of target heart cells in kDNA-positive birds (Fig. 15E, F, and G), and each kDNA-integrated immunocyte producing “self”-tissue destruction is essentially a mutated clone (50). By the phenotyping definition, a clone not withstanding thymic selection is considered an autoreactive T lymphocyte repertoire producing the heart lesion, which is an important risk factor for disease outcomes (313, 358, 402). Sections from myocardia of healthy, control birds showed an absence of inflammation and an absence of tissue destruction.
The inflammatory autoimmune myocarditis observed for F0 and F1 chickens also appears to a lesser degree in F2 progeny (Fig. 16). In this regard, functional categories of kDNA mutations have been described (402) as follows: (i) high-lethality mutations that generate abortions, congenital inflammatory cardiomyopathy, and early death, in which the genotype modifications generate pathology incompatible with life; (ii) age-group-specific mutations, which attenuate in a majority of chickens that succumb to the Chagas' disease-like inflammatory cardiomyopathy late in adult life; (iii) neutral kDNA mutations probably present in chickens not compromised by heart disease (these neutral mutations contribute to genome growth and positive selection); and (iv) beneficial mutations which may exist theoretically but which cannot be identified for a few generations of kDNA-mutated chickens (206).
Fig. 16.

Chagas' disease-like dilated inflammatory cardiomyopathy in an F2 chicken with a kDNA mutation in the dystrophin gene. (A) Dilated heart occupying most of the thoracic cavity. (B) Dark round mononuclear cells infiltrate and destroy the myocardium of the kDNA-mutated hen. (C) Normal heart size (weight, 7 g) of a 10-month-old control chicken. (D) Normal histology of a control chicken heart. (Reprinted from PLoS Neglected Tropical Diseases [402].)
Human Chagas' disease is not considered a risk factor for the development of cancer (116, 407). Contrarily, an anticancer profile of T. cruzi infections has been reported (194). In this regard, the key environmental factor contributing to the development of autoimmunity and self-heart destruction in the chicken model and in human Chagas' disease is the T. cruzi kDNA minicircle that integrates into specific sites of the host's genome. The generation of autoimmune disease in mammals and in birds is an intrinsic feature stemming from the kDNA sequences leading to insertional mutations in specific CArsb loci spread in the genomes and, therefore, concerted intragenomic interactions.
Actually, investigations have shown that the interactions of the chicken immune system with the environmental agent result in two independent types of responses: (i) innate immunity that eradicates the T. cruzi infection after the first week of embryonic growth and (ii) thymus-dependent autoimmune reactions carried out by genotypically modified T cells, producing clonal cytotoxicity and heart cell destruction. In this regard, the innate and the autoimmune responses involved in the control of the parasitic infection and in the production of heart disease, respectively, are clearly identified.
The kDNA minicircles integrated into the chicken genome show variable regions with the potential for gRNA transcription (402). Analyses of those regions revealed significant alignments with T. brucei edited maxicircle gene sequences (257). Cognate gRNAs, adequately positioned in the variable region of the minicircles, predict highly significant amino acid similarities for NADH dehydrogenase subunit 7 (NAD7)-, ATPase 6-, and ND8-edited T. brucei- and T. cruzi-matched genes. However, gRNA editing is a mechanism specific to trypanosomes; in the absence of T. cruzi-specific enzymatic activities to introduce transcript modification, the chance of achieving functional editing in a host nuclear environment can be considered essentially nil.
The scattered nature of the minicircle integrations into CA-rich sites in the chicken genome indicates that a large number of host loci are susceptible to kDNA mutagenesis (275). The determination of the full extent of this phenomenon requires the sequencing of a kDNA-mutated chicken genome. It is postulated that some classes of minicircle integrations are associated with specific mutation events; each new class of minicircle give rise to a unique integration for a given individual. In this regard, the diversity of classes of minicircles calls out a broad spectrum of clinical consequences stemming from autoimmune disease.
The genetic control of immune tolerance in healthy chickens is impaired in kDNA-mutated birds with inflammatory cardiomyopathy. The disruption of multiple genes results in self-tolerance and permissiveness to the autoimmune rejection of target tissue, inflammatory cardiomyopathy, and heart failure, and several integration events are associated with modified T cell phenotypes. Thus, groups of integration mutations and combinations thereof explain the pathogenesis and the variegated clinical manifestations of the Chagas' disease-like inflammatory cardiomyopathy in the chicken model. Accordingly, 20 genes and five X-linked disorders have been correlated with manifestations of failure in the genetic etiology of the heterogeneous group of cardiomyopathy in humans (110, 227, 231).
Interspecies DNA transfer used to be refuted or considered an unusual phenomenon (299), but the onslaught of lateral DNA transfer among eukaryotes (149, 173, 293 [see comments regarding this retracted article at first citation], 402) demands a major shift in paradigms, and there now stands a new paradigm accurately called transfer all the time (TAT). Here, DNA transfer stands as a major force of evolution because the TAT saga can be confirmed each time a mammalian host is infected by T. cruzi. The rapidly expanding pool of knowledge in this field indicates that we are just recognizing Chagas' disease as part of an endless evolutionary process (173, 293 [see comments regarding this retracted article at first citation], 402).
CONCLUDING REMARKS AND PERSPECTIVE ON TREATMENT
The treatment of T. cruzi infections with currently available nitroarenes does not prevent the onset of severe destructive inflammatory cardiomyopathy in late-phase chronic Chagas' disease. This observation suggests that in addition to a nontoxic trypanocidal drug to eliminate the infection, an effective treatment for the disease requires a further understanding of host-parasite relationships and pathogenesis. A mechanical or toxic role played by the parasite in the outcome of the disease is persistently challenged, because it does not interfere directly with the destruction of the target heart, skeletal muscles, digestive tube, and neuroendocrine systems, with repercussions on the variable clinical and pathological manifestations.
The phenomenon of the lateral transfer of the T. cruzi kDNA minicircle sequences to several loci in the chromosomes of chagasic patients, which can accumulate over time as the parasite encrypts in the human body, is described. A clear demonstration of the important role played by LkDT in the pathogenesis of the chagasic lesions is shown in a cross-kingdom model system, which eliminates the infection.
The inoculation of T. cruzi into embryonated eggs generates parasite-free chicks that retain the kDNA minicircles in the body. Chicks hatched from T. cruzi-infected eggs retain the minicircle sequences in the absence of parasite nuclear DNA. The kDNA integrates into the genome of somatic and germ line cells, from where it can be vertically transmitted to progeny; crossbreeding generates kDNA-positive progeny. The kDNA mutations are detected mainly in coding regions on several chromosomes. Interestingly, kDNA-mutated chickens develop gross cardiomegaly with an inflammatory myocarditis similar to that of Chagas' disease in humans, in which parasite-free myofibers are destroyed by immune system effector cells, as well as heart failure. The progression from a physiological state to a physiopathological state is associated with kDNA-mutated multigenic-dependent clonal T cell proliferation in the chicken model system, showing the rupture of multiple genes, accelerated anti-self cytotoxicity, and autoimmune Chagas' heart disease.
Severe myocarditis is present in F0, F1, and, to a lesser extent, F2 kDNA-mutated birds. The inflammatory cardiomyopathy in the kDNA-mutated birds shows the typical age-group-dependent genetic familiar autoimmune disease. The attenuation of the disease outcome in F2 and F3 progeny, which is detected in the chicken model, can be explained by the enormous plasticity of genomes that counterbalance positively the effects of mutations in the benefit of the fitness of extant species. It is postulated that the intragenomic signaling-associated balance can modulate the kDNA insertion mutations, which become neutral and favor species evolution. The genotype alterations recorded for the chicken model system explain satisfactorily the multifaceted clinical and pathological manifestations of the genetically driven autoimmune Chagas' disease. The attenuation of pathological effects in chicken progeny with genotype modifications has been demonstrated. The cross-kingdom model reveals a spectrum of age-prone pathologies stemming from kDNA minicircle integrations into the chicken genome. The genotype modifications in the kDNA-mutated chicken translate to the autoimmune inflammatory cardiomyopathy of Chagas' disease. So far, this is the first time that gross and microscopic features, which correlate with the clinical manifestations of Chagas' disease, have been produced experimentally in a parasite-free cross-kingdom model.
The study suggests that the treatment of human Chagas' disease requires multiple drugs in order to eliminate the cryptic infection and to prevent the autoimmune rejection of the target tissues. In this respect, an effective trypanocidal drug treatment without side effects is deemed necessary. Meanwhile, the ablation of the progenitors of clones of immune lymphocytes that carry out tissue destruction by treatment with antimetabolite and cytostatic drugs prior to histocompatible healthy bone marrow transplantation can interrupt the progressive destruction of the heart in cases of severe heart disease. The testing of this therapeutic regime is under way with congenic birds of Prague, in order to demonstrate that Chagas' heart disease can halt after the transplantation of normal bone marrow cells from healthy donors. Thus, an effective therapy can be attempted by the passive transfer of naive bone marrow cells from a healthy donor to a severely ill chagasic patient. The treatment cannot be delayed in view of the demonstration of genotype modifications associated with the pathogenesis of Chagas' disease and because the prospects for gene therapy may be a long-range resource.
ACKNOWLEDGMENTS
We are indebted to colleagues that have contributed with comments and criticisms to this publication: Ciro Cordeiro, Perla F. Araujo, Roseneide M. Alves, Fernando C. Pimentel, and A. Cassia Rosa.
This work was supported by the National Research Council (CNPq), by the Ministry of Education (CAPES), and by the Foundation for Research Development (FAPDF), Federal District, Brazil.
Biographies
 Antonio R. L. Teixeira received his M.D. degree from the Faculty of Medicine of the Federal University of Bahia, Bahia, Brazil, in 1967. From 1971 to 1974 he was a Research Fellow in Pathology at the New York Hospital, Cornell University Medical College. He trained at the L'Institut d'Imunogénétique et Cancerologie in Villejuif, France, and received his Ph.D. in Pathology from the Federal University of Minas Gerais, Minas Gerais, Brazil. Dr. Teixeira is Full Professor of Parasitology at the Faculty of Medicine of the University of Brasilia. His main research interest is epidemiology, clinics, and pathogenesis of human Chagas' disease.
Antonio R. L. Teixeira received his M.D. degree from the Faculty of Medicine of the Federal University of Bahia, Bahia, Brazil, in 1967. From 1971 to 1974 he was a Research Fellow in Pathology at the New York Hospital, Cornell University Medical College. He trained at the L'Institut d'Imunogénétique et Cancerologie in Villejuif, France, and received his Ph.D. in Pathology from the Federal University of Minas Gerais, Minas Gerais, Brazil. Dr. Teixeira is Full Professor of Parasitology at the Faculty of Medicine of the University of Brasilia. His main research interest is epidemiology, clinics, and pathogenesis of human Chagas' disease.
 Mariana M Hecht graduated in Biology at the University of Brasilia, Brazil, in 2002. During study for her M.Sc. degree she conducted studies about the allergens in the saliva of Triatoma infestans, the main transmitter of Trypanosoma cruzi, and she described a notable T. infestans preference for feeding upon fully immunized hosts. In 2008 she concluded her Ph.D. studies showing contemporaneous lateral transfer of mitochondrial kDNA from T. cruzi to human hosts. She is a Visiting Professor of Parasitology at the University of Brasilia.
Mariana M Hecht graduated in Biology at the University of Brasilia, Brazil, in 2002. During study for her M.Sc. degree she conducted studies about the allergens in the saliva of Triatoma infestans, the main transmitter of Trypanosoma cruzi, and she described a notable T. infestans preference for feeding upon fully immunized hosts. In 2008 she concluded her Ph.D. studies showing contemporaneous lateral transfer of mitochondrial kDNA from T. cruzi to human hosts. She is a Visiting Professor of Parasitology at the University of Brasilia.
 Maria C. Guimaro graduated in Pharmaceutical Sciences at the University of Brasília, Brazil, in 2007. She is a Ph.D. student with Professor Antonio Teixeira in the Chagas Disease Multidisciplinary Research Laboratory at the Faculty of Medicine. Her study is focused on the cross-kingdom Gallus gallus model system to determine the role that autoimmunity plays in the pathogenesis of Chagas' disease. Genetically driven autoimmune disease is her main research subject.
Maria C. Guimaro graduated in Pharmaceutical Sciences at the University of Brasília, Brazil, in 2007. She is a Ph.D. student with Professor Antonio Teixeira in the Chagas Disease Multidisciplinary Research Laboratory at the Faculty of Medicine. Her study is focused on the cross-kingdom Gallus gallus model system to determine the role that autoimmunity plays in the pathogenesis of Chagas' disease. Genetically driven autoimmune disease is her main research subject.
 Alessandro O. Sousa obtained his pharmaceutical sciences degree from the University of Brasilia in 2008. He is a Ph.D. student at the Chagas Disease Multidisciplinary Research Laboratory at the Faculty of Medicine, carrying on studies about the insertions of kDNA minicircles from Trypanosoma cruzi within LINE-1 in the murine genome. He is investigating the possible association between the kDNA mutation drivers of the thymus-dependent autoimmune responses in the experimental mouse model of Chagas' disease.
Alessandro O. Sousa obtained his pharmaceutical sciences degree from the University of Brasilia in 2008. He is a Ph.D. student at the Chagas Disease Multidisciplinary Research Laboratory at the Faculty of Medicine, carrying on studies about the insertions of kDNA minicircles from Trypanosoma cruzi within LINE-1 in the murine genome. He is investigating the possible association between the kDNA mutation drivers of the thymus-dependent autoimmune responses in the experimental mouse model of Chagas' disease.
 Nadjar Nitz graduated in Biomedical Sciences from the Federal University of Pernambuco, Pernambuco, Brazil, in 1996. She pursued Ph.D. studies in 2001 and Postdoctoral studies in 2009 at University of Brasilia, Brazil, on the transfer of mitochondrial kDNA minicircles from T. cruzi to vertebrate hosts. She is mostly interested in Molecular Biology, Molecular Evolution, and Immunology. Currently, she is an Associate Professor of Parasitology at the Chagas Disease Multidisciplinary Research Laboratory, Faculty of Medicine, University of Brasilia.
Nadjar Nitz graduated in Biomedical Sciences from the Federal University of Pernambuco, Pernambuco, Brazil, in 1996. She pursued Ph.D. studies in 2001 and Postdoctoral studies in 2009 at University of Brasilia, Brazil, on the transfer of mitochondrial kDNA minicircles from T. cruzi to vertebrate hosts. She is mostly interested in Molecular Biology, Molecular Evolution, and Immunology. Currently, she is an Associate Professor of Parasitology at the Chagas Disease Multidisciplinary Research Laboratory, Faculty of Medicine, University of Brasilia.
REFERENCES
- 1. Abel L. C., et al. 2005. T cell epitope characterization in tandemly repetitive Trypanosoma cruzi B13 protein. Microbes Infect. 7:1184–1195 [DOI] [PubMed] [Google Scholar]
- 2. Abrahamsohn I. A., Silva W. D. 1977. Antibody dependent cell-mediated cytotoxicity against Trypanosoma cruzi. Parasitology 75:317–323 [DOI] [PubMed] [Google Scholar]
- 3. Ackermann A. A., Carmona S. J., Agüero F. 2009. TcSNP: a database of genetic variation in Trypanosoma cruzi. Nucleic Acids Res. 37:D544–D549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Acosta A. M., Santos-Buch C. A. 1985. Autoimmune myocarditis induced by Trypanosoma cruzi. Circulation 71:1255–1261 [DOI] [PubMed] [Google Scholar]
- 5. Acosta-Serrano A., Almeida I. C., Freitas L. H., Jr., Yoshida N., Schenkman S. 2001. The mucin-like glycoprotein super-family of Trypanosoma cruzi: structure and biological roles. Mol. Biochem. Parasitol. 114:143–150 [DOI] [PubMed] [Google Scholar]
- 6. Adad S. J., et al. 2001. Neuron count reevaluation in the myenteric plexus of chagasic megacolon after morphometric neuron analysis. Virchows Arch. 438:254–258 [DOI] [PubMed] [Google Scholar]
- 7. Adad S. J., Andrade D. C., Lopes E. R., Chapadeiro E. 1991. Pathological anatomy of chagasic megaesophagus.. Rev. Inst. Med. Trop. Sao Paulo 33:443–450 [PubMed] [Google Scholar]
- 8. Afzali B., Lombardi G., Lechler R. I., Lord G. M. 2007. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin. Exp. Immunol. 148:32–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Agüero F., Zheng W., Weatherly D. B., Mendes P., Kissinger J. C. 2006. TcruziDB: an integrated, post-genomics community resource for Trypanosoma cruzi. Nucleic Acids Res. 34:D428–D431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alarcón de Noya B., et al. 2010. Large urban outbreak of orally acquired acute Chagas disease at a school in Caracas, Venezuela. J. Infect. Dis. 201:1308–1315 [DOI] [PubMed] [Google Scholar]
- 11. Albareda M. C., et al. 2009. Chronic human infection with Trypanosoma cruzi drives CD4+ T cells to immune senescence. J. Immunol. 183:4103–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Almeida I. C., Gazzinelli R., Ferguson M. A., Travassos L. R. 1999. Trypanosoma cruzi mucins: potential functions of a complex structure. Mem. Inst. Oswaldo Cruz 94(Suppl. 1):173–176 [DOI] [PubMed] [Google Scholar]
- 13. Alves M. J., Colli W. 2008. Role of the gp85/trans-sialidase superfamily of glycoproteins in the interaction of Trypanosoma cruzi with host structures. Subcell. Biochem. 47:58–66 [DOI] [PubMed] [Google Scholar]
- 14. Andersson B., et al. 1998. Complete sequence of a 93.4-kb contig from chromosome 3 of Trypanosoma cruzi containing a strand-switch region. Genome Res. 8:809–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andrade L. O., Andrews N. W. 2004. Lysosomal fusion is essential for the retention of Trypanosoma cruzi inside host cells. J. Exp. Med. 200:1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andrade L. O., Andrews N. W. 2005. The Trypanosoma cruzi-host-cell interplay: location, invasion, retention. Nat. Rev. Microbiol. 3:819–823 [DOI] [PubMed] [Google Scholar]
- 17. Andrade S. G., Andrade Z. A. 1966. Chagas disease and neuronal alterations at the Auerbach's plexus.. Rev. Inst. Med. Trop. Sao Paulo 8:219–224 [PubMed] [Google Scholar]
- 18. Andrade Z. A., Andrade S. G. 1955. Pathogenesis of Chagas' chronic myocarditis; importance of ischemic lesions. Arq. Bras. Med. 45:279–288 [PubMed] [Google Scholar]
- 19. Andrews N. W. 2002. Lysosomes and the plasma membrane: trypanosomes reveal a secret relationship. J. Cell Biol. 158:389–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ansari N. A., Saluja S., Salotra P. 2006. Elevated levels of interferon-gamma, interleukin-10, and interleukin-6 during active disease in Indian kala azar. Clin. Immunol. 119:339–345 [DOI] [PubMed] [Google Scholar]
- 21. Ansari N. A., Ramesh V., Salotra P. 2006. Interferon (IFN)-gamma, tumor necrosis factor-alpha, interleukin-6, and IFN-gamma receptor 1 are the major immunological determinants associated with post-kala azar dermal leishmaniasis. J. Infect. Dis. 194:958–965 [DOI] [PubMed] [Google Scholar]
- 22. Antúnez M. I., Cardoni R. L. 2001. Early IFN-gamma production is related to the presence of interleukin (IL)-18 and the absence of IL-13 in experimental Trypanosoma cruzi infections. Immunol. Lett. 79:189–196 [DOI] [PubMed] [Google Scholar]
- 23. Aras R., da Matta J. A., Mota G., Gomes I., Melo A. 2003. Cerebral infarction in autopsies of chagasic patients with heart failure. Arq. Bras. Cardiol. 81:414–416 [DOI] [PubMed] [Google Scholar]
- 24. Araújo A., Jansen A. M., Reinhard K., Ferreira L. F. 2009. Paleoparasitology of Chagas disease—a review. Mem. Inst. Oswaldo Cruz 104:9–16 [DOI] [PubMed] [Google Scholar]
- 25. Araújo-Jorge T. C., et al. 2008. Pivotal role for TGF-beta in infectious heart disease: the case of Trypanosoma cruzi infection and consequent Chagasic myocardiopathy. Cytokine Growth Factor Rev. 19:405–413 [DOI] [PubMed] [Google Scholar]
- 26. Araújo-Jorge T. C., et al. 2002. Implication of transforming growth factor-beta1 in Chagas disease myocardiopathy. J. Infect. Dis. 186:1823–1828 [DOI] [PubMed] [Google Scholar]
- 27. Arner E., et al. 2007. Database of Trypanosoma cruzi repeated genes: 20,000 additional gene variants. BMC Genomics 8:391–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arsenijevic D., Girardier L., Seydoux J., Chang H. R., Dulloo A. G. 1997. Altered energy balance and cytokine gene expression in a murine model of chronic infection with Toxoplasma gondii. Am. J. Physiol. 272:E908–E917 [DOI] [PubMed] [Google Scholar]
- 29. Atwood J. A., III, et al. 2005. The Trypanosoma cruzi proteome. Science 309:473–476 [DOI] [PubMed] [Google Scholar]
- 30. Aufderheide A. C., et al. 2004. A 9,000-year record of Chagas' disease. Proc. Natl. Acad. Sci. U. S. A. 101:2034–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aviles H., et al. 2008. Kinetics of systemic cytokine and brain chemokine gene expression in murine toxoplasma infection. J. Parasitol. 94:1282–1288 [DOI] [PubMed] [Google Scholar]
- 32. Baldauf S. L., Roger A. J., Wenk-Siefert I., Doolittle W. F. 2000. A kingdom-level phylogeny of eukaryotes based on combined protein data. Science 290:972–977 [DOI] [PubMed] [Google Scholar]
- 33. Barr S. C. 2009. Canine Chagas' disease (American trypanosomiasis) in North America. Vet. Clin. North Am. Small Anim. Pract. 39:1055–1064 [DOI] [PubMed] [Google Scholar]
- 34. Barrias E. S., Dutra J. M., De Souza W., Carvalho T. M. 2007. Participation of macrophage membrane rafts in Trypanosoma cruzi invasion process. Biochem. Biophys. Res. Commun. 363:828–834 [DOI] [PubMed] [Google Scholar]
- 35. Benvenuti L. A., et al. 2008. Chronic American trypanosomiasis: parasite persistence in endomyocardial biopsies is associated with high-grade myocarditis. Ann. Trop. Med. Parasitol. 102:481–487 [DOI] [PubMed] [Google Scholar]
- 36. Bilate A. M., Cunha-Neto E. 2008. Chagas disease cardiomyopathy: current concepts of an old disease.. Rev. Inst. Med. Trop. Sao Paulo 50:67–74 [DOI] [PubMed] [Google Scholar]
- 37. Bittencourt A. L., Vieira G. O., Tavares H. C., Mota E., Maguire J. 1984. Esophageal involvement in congenital Chagas' disease. Report of a case with megaesophagus. Am. J. Trop. Med. Hyg. 33:30–33 [DOI] [PubMed] [Google Scholar]
- 38. Bixby L. M., Tarleton R. L. 2008. Stable CD8+ T cell memory during persistent Trypanosoma cruzi infection. J. Immunol. 181:2644–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bluestone J. A., Mackay C. R., O'Shea J. J., Stockinger B. 2009. The functional plasticity of T cell subsets. Nat. Rev. Immunol. 9:811–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bonney K. M., Taylor J. M., Daniels M. D., Epting C. L., Engman D. M. 2011. Heat-killed Trypanosoma cruzi induces acute cardiac damage and polyantigenic autoimmunity. PLoS One 6:e14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Borda E. S., et al. 1991. Trypanosoma cruzi attachment to lymphocyte muscarinic cholinergic and beta adrenergic receptors modulates intracellular signal transduction. Mol. Biochem. Parasitol. 47:91–100 [DOI] [PubMed] [Google Scholar]
- 42. Braga M. S., Lauria-Pires L., Arganaraz E. R., Nascimento R. J., Teixeira A. R. 2000. Persistent infections in chronic Chagas' disease patients treated with anti-Trypanosoma cruzi nitroderivatives.. Rev. Inst. Med. Trop. Sao Paulo 42:157–161 [DOI] [PubMed] [Google Scholar]
- 43. Brener Z., Chiari E. 1965. Aspects of early growth of different T. cruzi strains in culture medium. J. Parasitol. 51:922–926 [PubMed] [Google Scholar]
- 44. Brener Z., Andrade Z. A., Barral-Neto M. 2000. Trypanosoma cruzi and Chagas disease, 2nd ed. Guanabara Koogan, Rio de Janeiro, Brazil [Google Scholar]
- 45. Bringaud F., et al. 1998. Conserved organization of genes in trypanosomatids. Mol. Biochem. Parasitol. 94:249–264 [DOI] [PubMed] [Google Scholar]
- 46. Bringaud F., Ghedin E., El-Sayed N. M., Papadopoulou B. 2008. Role of transposable elements in trypanosomatids. Microbes Infect. 10:575–581 [DOI] [PubMed] [Google Scholar]
- 47. Brown D., Waneck G. L. 1992. Glycosyl-phosphatidylinositol-anchored membrane proteins. J. Am. Soc. Nephrol. 3:895–906 [DOI] [PubMed] [Google Scholar]
- 48. Brown E. L., et al. 18 December 2009, posting date Seroprevalence of Trypanosoma cruzi among eleven potential reservoir species from six states across the Southern United States. Vector Borne Zoonotic Dis. doi:10.1089/vbz.2009.0009. [DOI] [PMC free article] [PubMed]
- 49. Burleigh B. A., Woolsey A. M. 2002. Cell signaling and Trypanosoma cruzi invasion. Cell. Microbiol. 4:701–711 [DOI] [PubMed] [Google Scholar]
- 50. Burnet M. 1972. Auto-immunity and auto-immune disease. F. A. Davis Company, Philadelphia, PA [Google Scholar]
- 51. Caler E. V., Vaena de Avalos S., Haynes P. A., Andrews N. W., Burleigh B. A. 1998. Oligopeptidase B-dependent signaling mediates host cell invasion by Trypanosoma cruzi. EMBO J. 17:4975–4986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Campbell D. C., Sturm N. R. 2009. Trypanosoma cruzi nuclear DNA and its correlation with the parasite lifecycle, p. 70–82.In Teixeira A. R. L., Vinaud M. C., Castro A. M. (ed.), Emerging Chagas disease, 1st ed. Bentham Science Publishers, New York, NY [Google Scholar]
- 53. Campbell D., Thomas S., Sturm N. R. 2003. Transcription in the kinetoplastid protozoa: why be normal? Microbes Infect. 5:1231–1240 [DOI] [PubMed] [Google Scholar]
- 54. Candolfi E., Hunter C. A., Remington J. S. 1995. Roles of gamma interferon and other cytokines in suppression of the spleen cell proliferative response to concanavalin A and toxoplasma antigen during acute toxoplasmosis. Infect. Immun. 63:751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cardinal M. V., et al. 2008. Molecular epidemiology of domestic and sylvatic Trypanosoma cruzi infection in rural northwestern Argentina. Int. J. Parasitol. 38:1533–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cardoni R. L., Docampo R., Casellas A. M. 1982. Metabolic requirements for the damage of Trypanosoma cruzi epimastigotes by human polymorphonuclear leukocytes. J. Parasitol. 68:547–552 [PubMed] [Google Scholar]
- 57. Carvalho L. O., et al. 2009. Trypanosoma cruzi and myoid cells from seminiferous tubules: interaction and relation with fibrous components of extracellular matrix in experimental Chagas' disease. Int. J. Exp. Pathol. 90:52–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cavalier-Smith T. 2002. The phagotrophic origin of eukaryotes and phylogenetic classification of protozoa. Int. J. Syst. Evol. Microbiol. 52:297–354 [DOI] [PubMed] [Google Scholar]
- 59. Cavalier-Smith T. 2009. Predation and eukaryote cell origins: a coevolutionary perspective. Int. J. Biochem. Cell Biol. 41:307–322 [DOI] [PubMed] [Google Scholar]
- 60. Cavalier-Smith T. 2010. Deep phylogeny, ancestral groups and the four ages of life. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365:111–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cavalier-Smith T. 2010. Origin of the cell nucleus, mitosis and sex: roles of intracellular coevolution. Biol. Direct 5:77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cenini P., Berhe N., Hailu A., McGinnes K., Frommel D. 1993. Mononuclear cell subpopulations and cytokine levels in human visceral leishmaniasis before and after chemotherapy. J. Infect. Dis. 168:986–993 [DOI] [PubMed] [Google Scholar]
- 63. Chagas C. 1909. New human trypanosomiasis. Morphology and lifecycle of Schizotrypanum cruzi, the cause of a new human disease. Mem. Inst. Oswaldo Cruz 1:159–218 [Google Scholar]
- 64. Chagas C. 1910. Nova entidade mórbida do homem. Arch. Soc. Méd. Cirurg. S. Paulo 1:255–292 [Google Scholar]
- 65. Chagas C. 1911. A new human disease. Summary of ethiological and clinical studies. Mem. Inst. Oswaldo Cruz 3:219–275 [Google Scholar]
- 66. Chagas C. 1912. Sobre um trypanosoma do tatú. Tatusia novemcineta, transmitido pelo Triatoma geniculata Latr. (1811). Possibilidade de ser o tatú um depositário do Trypanosoma cruzi no mundo exterior. Bras. Med. 26:305–306 [Google Scholar]
- 67. Chagas C. 1934. Estado actual da tripanossomíase Americana. Rev. Biol. Hyg. 5:58–64 [Google Scholar]
- 68. Chagas C. 1916. Tripanosomiaze Americana. Forma aguda da moléstia. Mem. Inst. Oswaldo Cruz 8:37–65 [Google Scholar]
- 69. Chagas C., Villela E. 1922. Forma cardíaca da Tripanosomiase Americana. Mem. Inst. Oswaldo Cruz 14:5–61 [Google Scholar]
- 70. Chamizo C., Moreno J., Alvar J. 2005. Semi-quantitative analysis of cytokine expression in asymptomatic canine leishmaniasis. Vet. Immunol. Immunopathol. 103:67–75 [DOI] [PubMed] [Google Scholar]
- 71. Chen G. L., Prchal J. T. 2007. X-linked clonality testing: interpretation and limitations. Blood 110:1411–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen S. T., et al. 2008. CLEC5A is critical for dengue-virus-induced lethal disease. Nature 453:672–676 [DOI] [PubMed] [Google Scholar]
- 73. Cihakova D., Rose N. R. 2008. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv. Immunol. 99:95–114 [DOI] [PubMed] [Google Scholar]
- 74. Cillari E., et al. 1995. In vivo and in vitro cytokine profiles and mononuclear cell subsets in Sicilian patients with active visceral leishmaniasis. Cytokine 7:740–745 [DOI] [PubMed] [Google Scholar]
- 75. Coffman R. L. 2010. Immunology: the origin of TH2 responses. Science 328:1116–1117 [DOI] [PubMed] [Google Scholar]
- 76. Coura J. R. 2006. Transmission of chagasic infection by oral route in the natural history of Chagas disease. Rev. Soc. Bras. Med. Trop. 39(Suppl. 3):113–117 [PubMed] [Google Scholar]
- 77. Coura J. R., Junqueira A. C. V., Fernades O., Valente S. A. S., Miles M. A. 2002. Emerging Chagas disease in Amazonian Brazil. Trends Parasitol. 18:171–176 [DOI] [PubMed] [Google Scholar]
- 78. Covo S., de Villartay J. P., Jeggo P. A., Livneh Z. 2009. Translesion DNA synthesis-assisted non-homologous end-joining of complex double-strand breaks prevents loss of DNA sequences in mammalian cells. Nucleic Acids Res. 37:6737–6745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Crane M. S., Dvorak J. A. 1979. Trypanosoma cruzi: interaction with vertebrate cells. DNA synthesis and growth of intracellular amastigotes and their relationship to host cell DNA synthesis and growth. J. Protozool. 26:599–604 [DOI] [PubMed] [Google Scholar]
- 80. Cruz-Chan J. V., et al. 2009. Immunopathology of natural infection with Trypanosoma cruzi in dogs. Vet. Parasitol. 162:151–155 [DOI] [PubMed] [Google Scholar]
- 81. Cubillos-Garzon L. A., Casas J. P., Morillo C. A., Bautista L. E. 2004. Congestive heart failure in Latin America: the next epidemic. Am. Heart J. 147:412–417 [DOI] [PubMed] [Google Scholar]
- 82. Cunha-Neto E., et al. 2006. Induction of cardiac autoimmunity in Chagas heart disease: a case for molecular mimicry. Autoimmunity 39:41–54 [DOI] [PubMed] [Google Scholar]
- 83. Cunha-Neto E., Kalil J. 2001. Heart-infiltrating and peripheral T cells in the pathogenesis of human Chagas' disease cardiomyopathy. Autoimmunity 34:187–192 [DOI] [PubMed] [Google Scholar]
- 84. Cunha-Neto E., et al. 2009. Immunological and non-immunological effects of cytokines and chemokines in the pathogenesis of chronic Chagas disease cardiomyopathy. Mem. Inst. Oswaldo Cruz 104(Suppl. 1):252–258 [DOI] [PubMed] [Google Scholar]
- 85. Cunha-Neto E., et al. 1995. Autoimmunity in Chagas disease cardiopathy: biological relevance of a cardiac myosin-specific epitope crossreactive to an immunodominant Trypanosoma cruzi antigen. Proc. Natl. Acad. Sci. U. S. A. 92:3541–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cunningham D. S., Brewer T. E., Kuhn R. E., Craig W. H. 1981. Partial characterization of a Trypanosoma cruzi-released decomplementing factor. J. Parasitol. 67:475–480 [PubMed] [Google Scholar]
- 87. Cunningham D. S., Craig W. H., Kuhn R. E. 1978. Reduction of complement levels in mice infected with Trypanosoma cruzi. J. Parasitol. 64:1044–1049 [PubMed] [Google Scholar]
- 88. Da-Cruz A. M., et al. 2004. Long-term follow-up of co-infected HIV and Trypanosoma cruzi Brazilian patients. Trans. R. Soc. Trop. Med. Hyg. 98:728–733 [DOI] [PubMed] [Google Scholar]
- 89. da Mata J. R., Camargos E. R. S., Chiari E., Machado C. R. S. 2000. Trypanosoma cruzi infection and the rat central nervous system: proliferation of parasites in astrocytes and the brain reaction to parasitism. Brain Res. Bull. 53:153–162 [DOI] [PubMed] [Google Scholar]
- 90. da Matta Guedes P. M., et al. 2010. IL-17 produced during Trypanosoma cruzi infection plays a central role in regulating parasite-induced myocarditis. PLoS Negl. Trop. Dis. 4:e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Daniels M. D., Hyland K. V., Wang K., Engman D. M. 2008. Recombinant cardiac myosin fragment induces experimental autoimmune myocarditis via activation of Th1 and Th17 immunity. Autoimmunity 41:490–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Das A., Banday M., Bellofatto V. 2008. RNA polymerase transcription machinery in trypanosomes. Eukaryot. Cell 7:429–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Davila D. F. 2004. Sympathetic nervous system activation in chagasic patients with congestive heart failure. J. Am. Coll. Cardiol. 43:1723–1724 [DOI] [PubMed] [Google Scholar]
- 94. Dávila D. F., Donis J. H., Torres A., Ferrer J. A. 2004. A modified and unifying neurogenic hypothesis can explain the natural history of chronic Chagas heart disease. Int. J. Cardiol. 96:191–195 [DOI] [PubMed] [Google Scholar]
- 95. Dávila D. F., et al. 2008. Beta-adrenergic blockers in chronic systolic heart failure secondary to Chagas' disease. Int. J. Cardiol. 128:1–4 [DOI] [PubMed] [Google Scholar]
- 96. Dávila D. F., et al. 2008. Anti-muscarinic autoantibodies and vagal modulation in Chagas disease: positive allosteric modulators vs desensitization and downregulation of M2 cardiac acetylcholine receptors. Int. J. Cardiol. 123:328–329 [DOI] [PubMed] [Google Scholar]
- 97. Dávila D. F., Núñez T. J., Odreman R., de Dávila C. A. 2005. Mechanisms of neurohormonal activation in chronic congestive heart failure: pathophysiology and therapeutic implications. Int. J. Cardiol. 101:343–346 [DOI] [PubMed] [Google Scholar]
- 98. de Alencar B. C., et al. 2009. Perforin and gamma interferon expression are required for CD4+ and CD8+ T-cell-dependent protective immunity against a human parasite, Trypanosoma cruzi, elicited by heterologous plasmid DNA prime-recombinant adenovirus 5 boost vaccination. Infect. Immun. 77:4383–4395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Deane M. P., Lenzi H. L., Jansen A. M. 1986. Double development cycle of Trypanosoma cruzi in the opossum. Parasitol. Today 2:146–147 [DOI] [PubMed] [Google Scholar]
- 100. Decottignies A. 2007. Microhomology-mediated end joining in fission yeast is repressed by pku70 and relies on genes involved in homologous recombination. Genetics 176:1403–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Deem S. L., et al. 2009. Health assessment of free-ranging three-banded (Tolypeutes matacus) and nine-banded (Dasypus novemcinctus) armadillos in the Gran Chaco, Bolivia. J. Zoo Wildl. Med. 40:245–256 [DOI] [PubMed] [Google Scholar]
- 102. de Meis J., Morrot A., Farias-de-Oliveira D. A., Villa-Verde D. M., Savino W. 2009. Differential regional immune response in Chagas disease. PLoS Negl. Trop. Dis. 3:e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. de Souza W. 2009. Structural organization of Trypanosoma cruzi. Mem. Inst. Oswaldo Cruz 104:89–100 [DOI] [PubMed] [Google Scholar]
- 104. de Souza W., Cavalcanti D. P. 2008. DNA-containing organelles in pathogenic protozoa: a review. Trends Cell. Mol. Biol. 2:89–104 [Google Scholar]
- 105. de Souza W., Meyer H. 1974. On the fine structure of the nucleus in Trypanosoma cruzi in tissue culture forms. Spindle fibers in the dividing nucleus. J. Protozool. 21:48–52 [DOI] [PubMed] [Google Scholar]
- 106. de Thoisy B., Michel J. C., Vogel I., Vié J. C. 2000. A survey of hemoparasite infections in free-ranging mammals and reptiles in French Guiana. J. Parasitol. 86:1035–1040 [DOI] [PubMed] [Google Scholar]
- 107. Dias E. 1944. Non-receptivity of the domestic pigeon to the Schizotrypanum infection. Mem. Inst. Oswaldo Cruz 40:181 [Google Scholar]
- 108. Dias E. 1952. Chagas' disease in the Americas IV. Colombia, Venezuela and the Guianas. Rev. Bras. Malariol. Doencas Trop. 4:255–280 [PubMed] [Google Scholar]
- 109. Dias E. 1953. Chagas disease in the Americas VII. Chile. Rev. Bras. Malariol. Doencas Trop. 5:131–136 [PubMed] [Google Scholar]
- 110. Dias E., Laranja F. S., Miranda A., Nobrega G. 1956. Chagas disease; a clinical, epidemiologic, and pathologic study. Circulation 14:1035–1060 [DOI] [PubMed] [Google Scholar]
- 111. Dias J. C. P., Schofiel J. C. 2009. History of Chagas disease as a public health problem in Latin America, p. 1–9.In Teixeira A. R. L., Vinaud M. C., Castro A. M. (ed.), Emerging Chagas disease, 1st ed. Bentham Science Publishers, New York, NY [Google Scholar]
- 112. Diotaiuti L. 2009. Triatomine-vector of Trypanosoma cruzi infection, p. 24–39.In Teixeira A. R. L., Vinaud M. C., Castro A. M. (ed.), Emerging Chagas disease, 1st ed. Bentham Science Publishers, New York, NY [Google Scholar]
- 113. Dittmar K. 2002. Arthropod and helminth parasites of the wild guinea pig, Cavia aperea, from the Andes and the cordillera in Peru, South America. J. Parasitol. 88:409–411 [DOI] [PubMed] [Google Scholar]
- 114. Dittmar K., Jansen A. M., Araújo A., Reinhard K. 2003. Molecular diagnosis of prehistoric T. cruzi in the Texas-Coahuila border region. Thirteenth Annual Meeting of the Paleopathology Association, Tempe, Arizona: Paleopathol. News 1:4 [Google Scholar]
- 115. Dolezel D., Jirku M., Maslov D. A., Lukes J. 2000. Phylogeny of the bodonid flagellates (Kinetoplastida) based on small-subunit rRNA gene sequences. Int. J. Syst. Evol. Microbiol. 50:1943–1951 [DOI] [PubMed] [Google Scholar]
- 116. Dominical V. M., et al. 2010. Chagas disease and gynecologic neoplasias. Ann. Diagn. Pathol. 14:337–340 [DOI] [PubMed] [Google Scholar]
- 117. Dong C., Poulter T., Han J. S. 2009. LINE-like retrotransposition in Saccharomyces cerevisiae. Genetics 181:301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Doolitle W. F. 1998. A paradigm gets shifty. Nature 392:15–16 [DOI] [PubMed] [Google Scholar]
- 119. dos Reis F. C., et al. 2008. The role of conserved residues of chagasin in the inhibition of cysteine peptidases. FEBS Lett. 582:485–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Doyle P. S., Hsieh I., Engel J. C. 2000. Trypanosoma cruzi: of man, kissing-bugs, and frogs. Exp. Parasitol. 95:71–74 [DOI] [PubMed] [Google Scholar]
- 121. Dutra W. O., et al. 1997. Cytokine mRNA profile of peripheral blood mononuclear cells isolated from individuals with Trypanosoma cruzi chronic infection. Scand. J. Immunol. 45:74–80 [DOI] [PubMed] [Google Scholar]
- 122. Dvorak J. A. 1984. The natural heterogeneity of Trypanosoma cruzi: biological and medical implications. J. Cell. Biochem. 24:357–371 [DOI] [PubMed] [Google Scholar]
- 123. Dvorak J. A., et al. 1982. Trypanosoma cruzi: flow cytometric analysis. I. Analysis of total DNA/organism by means of mithramycin-induced fluorescence. J. Protozool. 29:430–437 [DOI] [PubMed] [Google Scholar]
- 124. Elias M. C., Marques-Porto R., Freymuller E., Schenckman S. 2001. Transcription rate modulation through the Trypanosoma cruzi life cycle occurs in parallel with changes in nuclear organization. Mol. Biochem. Parasitol. 112:79–90 [DOI] [PubMed] [Google Scholar]
- 125. El-Sayed N. M., et al. 2005. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309:409–415 [DOI] [PubMed] [Google Scholar]
- 126. Engman D. M., Leon J. S. 2002. Pathogenesis of Chagas heart disease: role of autoimmunity. Acta Trop. 81:123–132 [DOI] [PubMed] [Google Scholar]
- 127. Epting C. L., Coates B. M., Engman D. M. 18 June 2010, posting date. Molecular mechanisms of host cell invasion by Trypanosoma cruzi. Exp. Parasitol. doi:10.1016/j.exppara.2010.06.023. [DOI] [PMC free article] [PubMed]
- 128. Epting C. L., Bonney M., Olson C. L., Engman D. M. 2009. Cellular host-pathogen relationships, p. 83–93.In Teixeira A. R. L., Vinaud M. C., Castro A. M. (ed.), Emerging Chagas disease, 1st ed. Bentham Science Publishers, New York, NY [Google Scholar]
- 129. Fairweather D., Frisancho-Kiss S., Rose N. R. 2005. Viruses as adjuvants for autoimmunity: evidence from coxsackievirus-induced myocarditis. Rev. Med. Virol. 15:17–27 [DOI] [PubMed] [Google Scholar]
- 130. Fairweather D., et al. 2004. Mast cells and innate cytokines are associated with susceptibility to autoimmune heart disease following coxsackievirus B3 infection. Autoimmunity 37:131–145 [DOI] [PubMed] [Google Scholar]
- 131. Fairweather D., et al. 2003. IL-12 receptor beta 1 and Toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J. Immunol. 170:4731–4737 [DOI] [PubMed] [Google Scholar]
- 132. Felix J. C., von Kreuter B. F., Santos-Buch C. A. 1993. Mimicry of heart cell surface epitopes in primary anti-Trypanosoma cruzi Lyt 2+ T lymphocytes. Clin. Immunol. Immunopathol. 68:141–146 [DOI] [PubMed] [Google Scholar]
- 133. Feng Q., Moran J. V., Kazazian H. H., Jr., Boeke J. D. 1996. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 87:905–916 [DOI] [PubMed] [Google Scholar]
- 134. Ferguson M. A., Cross G. M. A. 1984. Myristylation of the membrane form of a Trypanosoma brucei variant surface glycoprotein. J. Biol. Chem. 259:3011–3015 [PubMed] [Google Scholar]
- 135. Ferguson M. A., et al. 1994. Glycosyl-phosphatidylinositol molecules of the parasite and the host. Parasitol. 108(Suppl.):S45–S54 [DOI] [PubMed] [Google Scholar]
- 136. Fernandes M. C., et al. 2007. Novel strategy in Trypanosoma cruzi cell invasion: implication of cholesterol and host cell microdomains. Int. J. Parasitol. 37:1431–1441 [DOI] [PubMed] [Google Scholar]
- 137. Field M. C., Medina-Acosta E., Cross G. A. 1991. Characterization of a glycosylphosphatidylinositol membrane protein anchor precursor in Leishmania mexicana. Mol. Biochem Parasitol. 48:227–229 [DOI] [PubMed] [Google Scholar]
- 138. Flynn J. J., Wyss A. R. 1998. Recent advances in South American mammalian paleontology. Tree 13:449–454 [DOI] [PubMed] [Google Scholar]
- 139. Forero D., Weirauch C., Baena M. 2004. Synonymy of the reduviid (Hemiptera: Heteroptera) genus Torrealbaia (Triatominae) with Amphibolus (Harpactorinae), with notes on Amphibolus venator (Klug, 1830). Zootaxa 670:1–12 [Google Scholar]
- 140. Fortes-Rego J., Macedo V. O., Prata A. R. 1980. Alterações neurológicas na doença de Chagas crônica. Arq. Neuropsi. 38:45–52 [DOI] [PubMed] [Google Scholar]
- 141. Francis J., Sankar V., Nair V. K., Priori S. G. 2005. Catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2:550–554 [DOI] [PubMed] [Google Scholar]
- 142. Freire-de-Lima L., et al. 2010. Trypanosoma cruzi subverts host cell sialylation and may compromise antigen-specific CD8+ T cell responses. J. Biol. Chem. 285:13388–13396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Galuppo S., et al. 2009. Predominance of Trypanosoma cruzi genotypes in two reservoirs infected by sylvatic Triatoma infestans of an endemic area of Chile. Acta Trop. 111:90–93 [DOI] [PubMed] [Google Scholar]
- 144. Gantt K. R., et al. 2003. Activation of TGF-beta by Leishmania chagasi: importance for parasite survival in macrophages. J. Immunol. 170:2613–2620 [DOI] [PubMed] [Google Scholar]
- 145. Gardner T. A., et al. 2008. The cost-effectiveness of biodiversity surveys in tropical forests. Ecol. Lett. 11:139–150 [DOI] [PubMed] [Google Scholar]
- 146. Gaunt M. W., Miles M. A. 2002. An insect molecular clock dates the origin of the insects and accords with palaeontological and biogeographic landmarks. Mol. Biol. Evol. 19:748–761 [DOI] [PubMed] [Google Scholar]
- 147. Geer D. A. 1955. Found: two cases of Chagas disease. Texas Health Bull. 9:11 [Google Scholar]
- 148. Ghedin E., et al. 2004. Gene synteny and evolution of genome architecture in trypanosomatids. Mol. Biochem. Parasitol. 134:183–191 [DOI] [PubMed] [Google Scholar]
- 149. Gilbert C., Schaack S., Pace J. K., Brindley P. J., Feschotte C. 2010. A role for host-parasite interactions in the horizontal transfer of transposons across phyla. Nature 464:1347–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Gironès N., et al. 2001. Antibodies to an epitope from the Cha human autoantigen are markers of Chagas' disease. Clin. Diagn. Lab. Immunol. 8:1039–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Gironès N., et al. 2001. Dominant T- and B-cell epitopes in an autoantigen linked to Chagas' disease. J. Clin. Invest. 107:985–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Gironès N., et al. 2007. Role of Trypanosoma cruzi autoreactive T cells in the generation of cardiac pathology. Ann. N. Y. Acad. Sci. 1107:434–444 [DOI] [PubMed] [Google Scholar]
- 153. Gironès N., Cuervo H., Fresno M. 2005. Trypanosoma cruzi-induced molecular mimicry and Chagas' disease. Curr. Top. Microbiol. Immunol. 296:89–123 [DOI] [PubMed] [Google Scholar]
- 154. Glück B., Schmidtke M., Merkle I., Stelzner A., Gemsa D. 2001. Persistent expression of cytokines in the chronic stage of CVB3-induced myocarditis in NMRI mice. J. Mol. Cell. Cardiol. 33:1615–1626 [DOI] [PubMed] [Google Scholar]
- 155. Goble F. C., Boyd J. L. 1962. Reticulo-endothelial blockade in experimental Chagas' disease. J. Parasitol. 48:223–228 [PubMed] [Google Scholar]
- 156. Godsel L. M., et al. 2001. Prevention of autoimmune myocarditis through the induction of antigen-specific peripheral immune tolerance. Circulation 103:1709–1714 [DOI] [PubMed] [Google Scholar]
- 157. Gomes Y. M., et al. 1995. A monoclonal antibody against blood forms of Trypanosoma cruzi lyses the parasite in vitro and inhibits host cell invasion. Appl. Biochem. Biotechnol. 50:57–69 [DOI] [PubMed] [Google Scholar]
- 158. Gopal S., Cross G. A., Gaasterland T. 2003. An organism-specific method to rank predicted coding regions in Trypanosoma brucei. Nucleic Acids Res. 31:5877–5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Goronzy J. J., Weyand C. M. 2009. Developments in the scientific understanding of rheumatoid arthritis. Arthritis Res. Ther. 11:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Goto H., Prianti M. G. 2009. Immunoactivation and immunopathogeny during active visceral leishmaniasis.. Rev. Inst. Med. Trop. Sao Paulo 51:241–246 [DOI] [PubMed] [Google Scholar]
- 161. Guedes P. M., et al. 2009. Development of chronic cardiomyopathy in canine Chagas disease correlates with high IFN-gamma, TNF-alpha, and low IL-10 production during the acute infection phase. Vet. Immunol. Immunopathol. 130:43–52 [DOI] [PubMed] [Google Scholar]
- 162. Guerra F. 1970. American trypanosomiasis. A historical and a human lesson. J. Trop. Med. Hyg. 73:105–118 [PubMed] [Google Scholar]
- 163. Günzl A. 2010. The pre-mRNA splicing machinery of trypanosomes: complex or simplified? Eukaryot. Cell 9:1159–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Gürtler R. E., et al. 2009. Strong host-feeding preferences of the vector Triatoma infestans modified by vector density: implications for the epidemiology of Chagas disease. PLoS Negl. Trop. Dis. 3:e447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Gutierrez F. R. S., Guedes P. M. M., Gazzinelli R. T., Silva J. S. 2009. The role of parasite persistence in pathogenesis of Chagas heart disease. Parasite Immunol. 31:673–685 [DOI] [PubMed] [Google Scholar]
- 166. Hagger R., et al. 2000. A deficiency of interstitial cells of Cajal in Chagasic megacolon. J. Auton. Nerv. Syst. 80:108–111 [DOI] [PubMed] [Google Scholar]
- 167. Hamilton P. B., Gibson W. C., Stevens J. R. 2007. Patterns of co-evolution between trypanosomes and their hosts deduced from ribosomal RNA and protein-coding gene phylogenies. Mol. Phylogenet. Evol. 44:15–25 [DOI] [PubMed] [Google Scholar]
- 168. Harms G., Feldmeier H. 2005. The impact of HIV infection on tropical diseases. Infect. Dis. Clin. North Am. 19:121–135 [DOI] [PubMed] [Google Scholar]
- 169. Harris T. H., et al. 2010. NF-kappaB1 contributes to T cell-mediated control of Toxoplasma gondii in the CNS. J. Neuroimmunol. 222:19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Hatcher F. M., Kuhn R. E. 1982. Destruction of Trypanosoma cruzi by natural killer cells. Science 218:295–296 [DOI] [PubMed] [Google Scholar]
- 171. Hatcher F. M., Kuhn R. E., Cerrone M. C., Burton R. C. 1981. Increased natural killer cell activity in experimental American trypanosomiasis. J. Immunol. 127:1126–1130 [PubMed] [Google Scholar]
- 172. Hazkani-Covo E., Covo S. 2008. Numt-mediated double-strand break repair mitigates deletions during primate genome evolution. PLoS Genet. 4:e1000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Hecht M. M., et al. 2010. Inheritance of DNA transferred from American trypanosomes to human hosts. PLoS One 5:e9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Hecht M. M., Bussacos A. C., Lozzi S. P., Santana J. M., Teixeira A. R. 2006. Triatoma infestans chooses to feed upon immune prey. Am. J. Trop. Med. Hyg. 75:893–900 [PubMed] [Google Scholar]
- 174a. Hecht M. M., Nitz N., Araujo P. F., Teixeira A. R. L. 2009. Pathogenesis of Chagas disease: lateral DNA transfer, genotype alterations and autoimmunity. Rev. Soc. Bras. Med. Trop. 42(Suppl.):87–93 [Google Scholar]
- 175. Heras S. R., Lopez M. C., Olivares M., Thomas M. C. 2007. The L1Tc non-LTR retrotransposon of Trypanosoma cruzi contains an internal RNA-pol II-dependent promoter that strongly activates gene transcription and generates unspliced transcripts. Nucleic Acids Res. 35:2199–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Herrera H. M., et al. 2008. The coati (Nasua nasua, Carnivora, Procyonidae) as a reservoir host for the main lineages of Trypanosoma cruzi in the Pantanal region, Brazil. Trans. R. Soc. Trop. Med. Hyg. 102:1133–1139 [DOI] [PubMed] [Google Scholar]
- 177. Hertz-Fowler C., et al. 2004. GeneDB: a resource for prokaryotic and eukaryotic organisms. Nucleic Acids Res. 32:D339–D343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Hines J. C., Ray D. S. 2008. Structure of discontinuities in kinetoplast DNA-associated minicircles during S phase in Crithidia fasciculata. Nucleic Acids Res. 36:444–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Hoff R., et al. 1985. Serologic surveillance of Chagas disease. Ann. Soc. Belg. Med. Trop. 65:187–196 [PubMed] [Google Scholar]
- 180. Huber S. A., Pfaeffle B. 1994. Differential Th1 and Th2 cell responses in male and female BALB/c mice infected with coxsackievirus group B type 3. J. Virol. 68:5126–5132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Hyde T. P., Dvorak J. A. 1973. Trypanosoma cruzi: interaction with vertebrate cells in vitro. 2. Quantitative analysis of the penetration phase. Exp. Parasitol. 34:284–294 [DOI] [PubMed] [Google Scholar]
- 182. Hyland K. V., Engman D. M. 2006. Further thoughts on where we stand on the autoimmunity hypothesis of Chagas disease. Trends Parasitol. 22:101–102 [DOI] [PubMed] [Google Scholar]
- 183. Hyland K. V., et al. 2007. Modulation of autoimmunity by treatment of an infectious disease. Infect. Immun. 75:3641–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Inglessis I., et al. 1998. Clinical, parasitological and histopathologic follow-up studies of acute Chagas patients treated with benznidazole. Arch. Inst. Cardiol. Mex. 68:405–410 [PubMed] [Google Scholar]
- 185. Invernizzi P., Pasini S., Selmi C., Miozzo M., Podda M. 2008. Skewing of X chromosome inactivation in autoimmunity. Autoimmunity 41:272–277 [DOI] [PubMed] [Google Scholar]
- 186. Inverso J. A., Song Y., Santos-Buch C. A. 1995. Plasma membrane ATP receptors in Trypanosoma cruzi trypomastigotes. Receptor 5:197–206 [PubMed] [Google Scholar]
- 187. Ivens A. C., et al. 2005. The genome of the kinetoplastid parasite, Leishmania major. Science 309:436–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Iwai L. K., Juliano M. A., Juliano L., Kalil J., Cunha-Neto E. 2005. T-cell molecular mimicry in Chagas disease: identification and partial structural analysis of multiple cross-reactive epitopes between Trypanosoma cruzi B13 and cardiac myosin heavy chain. J. Autoimmun. 24:111–117 [DOI] [PubMed] [Google Scholar]
- 189. James S., Kipnis T., Sher A., Hoff R. 1982. Enhanced resistance to acute infection with Trypanosoma cruzi in mice with an interferon inducer. Infect. Immun. 35:588–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Januszyk K., et al. 2007. Identification and solution structure of a highly conserved C-terminal domain required for the retrotransposition of long interspersed nuclear element-1. J. Biol. Chem. 282:24893–24904 [DOI] [PubMed] [Google Scholar]
- 191. Johnson P. J., Kooter J. M., Borst P. 1987. Inactivation of transcription by UV irradiation of T. brucei provides evidence for a multicistronic transcription unit including a VSG gene. Cell 51:273–281 [DOI] [PubMed] [Google Scholar]
- 192. Junqueira L. F., Jr., Soares J. D. 2002. Impaired autonomic control of heart interval changes to Valsalva manoeuvre in Chagas' disease without overt manifestation. Auton. Neurosci. 97:59–67 [DOI] [PubMed] [Google Scholar]
- 193. Jurka J. 1997. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc. Natl. Acad. Sci. U. S. A. 94:1872–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Kallinikova V. D., et al. 2001. Anticancer properties of flagellate protozoan Trypanosoma cruzi Chagas. Izv. Akad. Nauk. SSSR Biol. 3:299–311 [PubMed] [Google Scholar]
- 195. Karakhanova S., Meisel S., Ring S., Mahnke K., Enk A. H. 2010. ERK/p38 MAP-kinases and PI3K are involved in the differential regulation of B7-H1 expression in DC subsets. Eur. J. Immunol. 40:254–266 [DOI] [PubMed] [Google Scholar]
- 196. Kenworthy A. K., et al. 2004. Dynamics of putative raft-associated proteins at the cell surface. J. Cell Biol. 165:735–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Kierszenbaum F. 1996. Chronic chagasic tissue lesions in the absence of Trypanosoma cruzi: a proposed mechanism. Parasitol. Today 12:414–415 [DOI] [PubMed] [Google Scholar]
- 198. Kierszenbaum F. 1999. Chagas' disease and the autoimmunity hypothesis. Clin. Microbiol. Rev. 12:210–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Kierszenbaum F. 2003. Views on the autoimmunity hypothesis for Chagas disease pathogenesis. FEMS Immunol. Med. Microbiol. 37:1–11 [DOI] [PubMed] [Google Scholar]
- 200. Kierszenbaum F. 2005. Where do we stand on the autoimmunity hypothesis of Chagas disease? Trends Parasitol. 21:513–516 [DOI] [PubMed] [Google Scholar]
- 201. Kierszenbaum F., Sonnenfeld G. 1982. Characterization of the antiviral activity produced during Trypanosoma cruzi infection and protective effects of exogenous interferon against experimental Chagas' disease. J. Parasitol. 68:194–198 [PubMed] [Google Scholar]
- 202. Kierszenbaum F., Pienkowski M. M. 1979. Thymus-dependent control of host defense mechanisms against Trypanosoma cruzi infection. Infect. Immun. 24:117–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Kierszenbaum F., Gottlieb C. A., Budzko D. B. 1981. Antibody-independent, natural resistance of birds to Trypanosoma cruzi infection. J. Parasitol. 67:656–660 [PubMed] [Google Scholar]
- 204. Kierszenbaum F., Ivanyi J., Budzko D. B. 1976. Mechanisms of natural resistance to trypanosomal infection. Role of complement in avian resistance to Trypanosoma cruzi infection. Immunology 30:1–6 [PMC free article] [PubMed] [Google Scholar]
- 205. Kim H. B., Evans I., Smallwood R., Holcombe M., Qwarnstrom E. E. 2010. NIK and IKKbeta interdependence in NF-kappaB signalling—flux analysis of regulation through metabolites. Biosystems 99:140–149 [DOI] [PubMed] [Google Scholar]
- 206. Kimura M. 1983. From Lamarck to population genetics, p. 1–14.In Kimura M. (ed.), The neutral theory of molecular evolution, 1st ed. Cambridge University Press, New York, NY [Google Scholar]
- 207. Kipnis T. L., David J. R., Alper C. A., Sher A., da Silva W. D. 1981. Enzymatic treatment transforms trypomastigotes of Trypanosoma cruzi into activators of alternative complement pathway and potentiates their uptake by macrophages. Proc. Natl. Acad. Sci. U. S. A. 78:602–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Kipnis T. L., James S. L., Sher A., David J. R. 1981. Cell-mediated cytotoxicity to Trypanosoma cruzi. II. Antibody-dependent killing of bloodstream forms by mouse eosinophils and neutrophils. Am. J. Trop. Med. Hyg. 30:47–53 [PubMed] [Google Scholar]
- 209. Kivity S., Agmon-Levin N., Blank M., Shoenfeld Y. 2009. Infections and autoimmunity—friends or foes? Trends Immunol. 30:409–414 [DOI] [PubMed] [Google Scholar]
- 210. Knipe D. M., Howley P. M. (ed.). 2007. Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 211. Koeberle F. 1963. Etiologia e patogenia do megaesôfago no Brasil. Rev. Goiana Med. 9:79–116 [Google Scholar]
- 212. Koeberle F. 1970. The causation and importance of nervous lesions in American trypanosomiasis. Bull. World Health Organ. 42:739–743 [PMC free article] [PubMed] [Google Scholar]
- 213. Korený L., Lukes J., Oborník M. 2010. Evolution of the haem synthetic pathway in kinetoplastid flagellates: an essential pathway that is not essential after all? Int. J. Parasitol. 40:149–156 [DOI] [PubMed] [Google Scholar]
- 214. Kress Y., Bloom B. R., Whitner M., Rowen A., Tanowitz H. 1975. Resistance of Trypanosoma cruzi to killing by macrophages. Nature 257:394–396 [DOI] [PubMed] [Google Scholar]
- 215. Krettli A. U. 2009. The utility of anti-trypomastigote lytic antibodies for determining cure of Trypanosoma cruzi infections in treated patients: an overview and perspectives. Mem. Inst. Oswaldo Cruz 104(Suppl. 1):142–151 [DOI] [PubMed] [Google Scholar]
- 216. Kuhn R. E., Durum S. K. 1975. The onset of immune protection in acute experimental Chagas' disease in C3H(HE) mice. Int. J. Parasitol. 5:241–244 [DOI] [PubMed] [Google Scholar]
- 217. Kumar R., Kline I. K., Abelmann W. H. 1970. Immunosuppression in experimental acute and subacute Chagasic myocarditis. Am. J. Trop. Med. Hyg. 19:932–939 [DOI] [PubMed] [Google Scholar]
- 218. Lages-Silva E., et al. 2001. Relationship between Trypanosoma cruzi and human chagasic megaesophagus: blood and tissue parasitism. Am. J. Trop. Med. Hyg. 65:435–441 [DOI] [PubMed] [Google Scholar]
- 219. Laguens R. P., et al. 1975. Immunopathologic and morphologic studies of skeletal muscle in Chagas' disease. Am. J. Pathol. 80:153–162 [PMC free article] [PubMed] [Google Scholar]
- 220. Lake J. A., de la Cruz V. F., Ferreira P. C., Morel C., Simpson L. 1988. Evolution of parasitism: kinetoplastid protozoan history reconstructed from mitochondrial rRNA gene sequences. Proc. Natl. Acad. Sci. U. S. A. 85:4779–4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Lang C., et al. 2008. Connective tissue growth factor: a crucial cytokine-mediating cardiac fibrosis in ongoing enterovirus myocarditis. J. Mol. Med. 86:49–60 [DOI] [PubMed] [Google Scholar]
- 222. Laranja F. S. 1949. Evolução dos conhecimentos sobre a cardiopatia chagásica. Revisão crítica da literatura. Mem. Inst. Oswaldo Cruz 47:605–669 [Google Scholar]
- 223. Laucella S. A., et al. 2009. Changes in Trypanosoma cruzi-specific immune responses after treatment: surrogate markers of treatment efficacy. Clin. Infect. Dis. 49:1675–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Lauria-Pires L., et al. 2000. Progressive chronic Chagas heart disease ten years after treatment with anti-Trypanosoma cruzi nitroderivatives. Am. J. Trop. Med. Hyg. 63:111–118 [DOI] [PubMed] [Google Scholar]
- 225. Lee J. H., Jung H. S., Gunzl A. 2009. Transcriptionally active TFIIH of the early-diverged eukaryote Trypanosoma brucei harbors two novel core subunits but not a cyclin-activating kinase complex. Nucleic Acids Res. 37:3811–3820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Lee J., et al. 2007. Different evolutionary fates of recently integrated human and chimpanzee LINE-1 retrotransposons. Gene 390:18–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Leon J. S., Engman D. M. 2001. Autoimmunity in Chagas heart disease. Int. J. Parasitol. 31:555–561 [DOI] [PubMed] [Google Scholar]
- 228. Leon J. S., Engman D. M. 2003. The significance of autoimmunity in the pathogenesis of Chagas heart disease. Front. Biosci. 8:e315–e322 [DOI] [PubMed] [Google Scholar]
- 229. Leon J. S., Wang K., Engman D. M. 2003. Myosin autoimmunity is not essential for cardiac inflammation in acute Chagas' disease. J. Immunol. 171:4271–4277 [DOI] [PubMed] [Google Scholar]
- 230. Leon J. S., Godsel L. M., Wang K., Engman D. M. 2001. Cardiac myosin autoimmunity in acute Chagas' heart disease. Infect. Immun. 69:5643–5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Leon J. S., Daniels M. D., Toriello K. M., Wang K., Engman D. M. 2004. A cardiac myosin-specific autoimmune response is induced by immunization with Trypanosoma cruzi proteins. Infect. Immun. 72:3410–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Levin M. J. 1996. In chronic Chagas heart disease, don't forget the parasite. Parasitol. Today 12:415–416 [DOI] [PubMed] [Google Scholar]
- 233. Lewis M. D., et al. 2009. Flow cytometric analysis and microsatellite genotyping reveal extensive DNA content variation in Trypanosoma cruzi populations and expose contrasts between natural and experimental hybrids. Int. J. Parasitol. 39:1305–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Li K., et al. 2009. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ. Res. 105:353–364 [DOI] [PubMed] [Google Scholar]
- 235. Lieberman L. A., et al. 2004. IL-23 provides a limited mechanism of resistance to acute toxoplasmosis in the absence of IL-12. J. Immunol. 173:1887–1893 [DOI] [PubMed] [Google Scholar]
- 236. Lieberman-Aiden E., et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326:289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Lima A. P., et al. 2001. Cysteine protease isoforms from Trypanosoma cruzi, cruzipain 2 and cruzain, present different substrate preference and susceptibility to inhibitors. Mol. Biochem. Parasitol. 114:41–52 [DOI] [PubMed] [Google Scholar]
- 238. Lin S., Huang Y. L., Wu W. F., Li Y., Tang S. D. 2009. Role of interleukin 17 in viral myocarditis and dilated cardiomyopathy. Nan Fang Yi Ke Da Xue Xue Bao 29:1994–1999(In Chinese.) [PubMed] [Google Scholar]
- 239. Lisboa C. V., Xavier S. C., Herrera H. M., Jansen A. M. 2009. The ecology of the Trypanosoma cruzi transmission cycle: dispersion of zymodeme 3 (Z3) in wild hosts from Brazilian biomes. Vet. Parasitol. 165:19–24 [DOI] [PubMed] [Google Scholar]
- 240. Lischke A., et al. 2000. Isolation and characterization of glycosylphosphatidylinositol-anchored, mucin-like surface glycoproteins from bloodstream forms of the freshwater-fish parasite Trypanosoma carassii. Biochem. J. 345:693–700 [PMC free article] [PubMed] [Google Scholar]
- 241. Liu Y. G., Whittier R. F. 1995. Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 25:674–681 [DOI] [PubMed] [Google Scholar]
- 242. Logan L. L., Hanson W. L. 1974. Trypanosoma cruzi: morphogenesis in diffusion chambers in the mouse peritoneal cavity and attempted stimulation of host immunity. Exp. Parasitol. 36:439–454 [DOI] [PubMed] [Google Scholar]
- 243. Lopes E. R., Chapadeiro E., Borges M. C., Cançado M. A., Rocha A. 1982. Sudden death and Chagas' disease—analysis of predisposing factors of sudden death in chronic Chagas' patients. Mem. Inst. Oswaldo Cruz 77:255–262 [DOI] [PubMed] [Google Scholar]
- 244. López A. F., Moreno M. M., Sanderson C. J. 1978. The lysis of Trypanosoma cruzi epimastigotes by eosinophils and neutrophils. Int. J. Parasitol. 8:485–489 [DOI] [PubMed] [Google Scholar]
- 245. Lopez Kostka S., et al. 2009. IL-17 promotes progression of cutaneous leishmaniasis in susceptible mice. J. Immunol. 182:3039–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Loudon R. P., Moraska A. F., Huber S. A., Schwimmbeck P., Schultheiss P. 1991. An attenuated variant of coxsackievirus B3 preferentially induces immunoregulatory T cells in vivo. J. Virol. 65:5813–5819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Lugo-Hernández A. 1973. Morphology of Trypanosoma cruzi in cells of chicken and lizards (Tropidurus hispidus, Sauria: Iguanidae) cultured at 37 degrees and 33 degrees C. Rev. Bras. Biol. 33:561–573 [PubMed] [Google Scholar]
- 248. Lukeš J., Hashimi H., Zíková A. 2005. Unexplained complexity of the mitochondrial genome and transcriptome in kinetoplastid flagellates. Curr. Genet. 48:277–299 [DOI] [PubMed] [Google Scholar]
- 249. Lyons R. E., et al. 2001. Immunological studies of chronic ocular toxoplasmosis: up-regulation of major histocompatibility complex class I and transforming growth factor beta and a protective role for interleukin-6. Infect. Immun. 69:2589–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Macedo V. 1999. Intermediate form of Chagas disease. Mem. Inst. Oswaldo Cruz 94(Suppl. 1):311–316 [DOI] [PubMed] [Google Scholar]
- 251. Machado C. R., Camargos E. R., Guerra L. B., Moreira M. C. 2000. Cardiac autonomic denervation in congestive heart failure: comparison of Chagas' heart disease with other dilated cardiomyopathy. Hum. Pathol. 31:3–10 [DOI] [PubMed] [Google Scholar]
- 252. Mady C., de Moraes A. V., Galiano N., Decourt L. V. 1982. Hemodynamic study of the intermediate form of Chagas' disease. Arq. Bras. Cardiol. 38:271–275 [PubMed] [Google Scholar]
- 253. Mady C., Ianni B. M., de Souza J. L., Jr 2008. Benznidazole and Chagas disease: can an old drug be the answer to an old problem? Expert Opin. Invest. Drugs 17:1427–1433 [DOI] [PubMed] [Google Scholar]
- 254. Magalhães-Santos I. F., Andrade S. G. 2005. Participation of cytokines in the necrotic-inflammatory lesions in the heart and skeletal muscles of Calomys callosus infected with Trypanosoma cruzi. Mem. Inst. Oswaldo Cruz 100:555–561 [DOI] [PubMed] [Google Scholar]
- 255. Maisch B., Richter A., Sandmöller A., Portig I., Pankuweit S. 2005. Inflammatory dilated cardiomyopathy (DCMI). Herz 30:535–544 [DOI] [PubMed] [Google Scholar]
- 256. Mandal P. K., Kazazian H. H., Jr 2008. SnapShot: vertebrate transposons. Cell 135:192–192.e1 [DOI] [PubMed] [Google Scholar]
- 257. Mandison-Antenucci S., Grams J., Hajduk S. L. 2002. Editing machines: the complexities of trypanosome RNA editing. Cell 108:430–438 [DOI] [PubMed] [Google Scholar]
- 258. Marande W., Lukes J., Burger G. 2005. Unique mitochondrial genome structure in diplonemids, the sister group of kinetoplastids. Eukaryot. Cell 4:1137–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Marcili A., et al. 2009. Comparative phylogeography of Trypanosoma cruzi TCIIc: new hosts, association with terrestrial ecotopes, and spatial clustering. Infect. Genet. Evol. 9:1265–1274 [DOI] [PubMed] [Google Scholar]
- 259a. Marcus E. 2005. Retraction controversy. Cell 123:173–175 [DOI] [PubMed] [Google Scholar]
- 260. Margulis L., Sagan D. 2002. Acquiring genomes. A theory of the origin of species. Basic Books, New York, NY [Google Scholar]
- 261. Mariner P. D., et al. 2008. Human Alu RNA is a modular transacting repressor of mRNA transcription during heat shock. Mol. Cell 29:499–509 [DOI] [PubMed] [Google Scholar]
- 262. Marin-Neto J. A., Cunha-Neto E., Maciel B. C., Simões M. V. 2007. Pathogenesis of chronic Chagas heart disease. Circulation 115:1109–1123 [DOI] [PubMed] [Google Scholar]
- 263. Marino A. P., et al. 2005. CC-chemokine receptors: a potential therapeutic target for Trypanosoma cruzi-elicited myocarditis. Mem. Inst. Oswaldo Cruz 100:93–96 [DOI] [PubMed] [Google Scholar]
- 264. Marsden P. D., Hagstrom J. W. C. 1968. Experimental Trypanosoma cruzi infections in beagle puppies. The effect of variations in the dose and source of infecting trypanosomes and the route of inoculation on the course of the infection. Trans. R. Soc. Trop. Med. Hyg. 62:816–824 [DOI] [PubMed] [Google Scholar]
- 265. Martin D. L., Murali-Krishna K., Tarleton R. L. 2010. Generation of Trypanosoma cruzi-specific CD8+ T-cell immunity is unaffected by the absence of type I interferon signaling. Infect. Immun. 78:3154–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Martin S. L. 2006. The ORF1 protein encoded by LINE-1: structure and function during L1 retrotransposition. J. Biomed. Biotechnol. 2006:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Martínez-Calvillo S., Nguyen D., Stuart K., Myler P. J. 2004. Transcription initiation and termination on Leishmania major chromosome 3. Eukaryot. Cell 3:506–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Martínez-Calvillo S., et al. 2003. Transcription of Leishmania major Friedlin chromosome 1 initiates in both directions within a single region. Mol. Cell 11:1291–1299 [DOI] [PubMed] [Google Scholar]
- 269. Mathias C. J. 1997. Autonomic disorders and their recognition. N. Engl. J. Med. 336:721–724 [DOI] [PubMed] [Google Scholar]
- 270. Mathias S. L., Scott A. F., Kazazian H. H., Jr., Boeke J. D., Gabriel A. 1991. Reverse transcriptase encoded by a human transposable element. Science 254:1808–1810 [DOI] [PubMed] [Google Scholar]
- 271. Matowicka-Karna J., Dymicka-Piekarska V., Kemona H. 2009. Does Toxoplasma gondii infection affect the levels of IgE and cytokines (IL-5, IL-6, IL-10, IL-12, and TNF-alpha)? Clin. Dev. Immunol. 2009:374696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Mazza S. 1995. Cas de trypanosomose humaine observe dans la ville de Jujuy. Compt. Rend. Soc. Biol. 95:815 [Google Scholar]
- 273. Mazza S. 1995. Infeccion spontannée du chien par le Schizotrypanum cruzi. Compt. Rend. Soc. Biol. 270:809–811 [Google Scholar]
- 274. Mazza S. 1951. Exposition of contributions of M.E.P. R. A. to the knowledge of biopathology of Chagas' disease. Sem. Med. 58:296–300 [PubMed] [Google Scholar]
- 275. McVey M., Lee S. E. 2008. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 24:529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Meira W. S., et al. 2006. Evaluation of blood tests, complement-mediated lysis and polymerase chain reaction in the verification of therapeutic efficacy in Chagas disease. Rev. Soc. Bras. Med. Trop. 39(Suppl. 3):107–109 [PubMed] [Google Scholar]
- 277. Mendes D. G., et al. 2007. Exposure to mixed asymptomatic infections with Trypanosoma cruzi, Leishmania braziliensis and Leishmania chagasi in the human population of the greater Amazon. Trop. Med. Int. Health 12:629–636 [DOI] [PubMed] [Google Scholar]
- 278. Meneghelli U. G., et al. 1982. Basal motility of dilated and non-dilated sigmoid colon and rectum in Chagas' disease. Arq. Gastroenterol. 19:127–132 [PubMed] [Google Scholar]
- 279. Meyer H., Oliveira Musacchio M., Andrade Mendonça I. 1958. Electron microscopy study of Trypanosoma cruzi in thin sections of infected tissue cultures and blood agar forms. Parasitology 48:1–8 [DOI] [PubMed] [Google Scholar]
- 280. Middleton M. K., et al. 2009. 12/15-lipoxygenase-dependent myeloid production of interleukin-12 is essential for resistance to chronic toxoplasmosis. Infect. Immun. 77:5690–5700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Minter-Goedbloed E., Croon J. J. 1981. The susceptibility of chickens to Trypanosoma (Schizotrypanum) cruzi. Trans. R. Soc. Trop. Med. Hyg. 75:350–353 [DOI] [PubMed] [Google Scholar]
- 282. Moncayo A., Silveira A. C. 2009. Current epidemiological trends for Chagas disease in Latin America and future challenges in epidemiology, surveillance and health policy. Mem. Inst. Oswaldo Cruz 104:17–30 [DOI] [PubMed] [Google Scholar]
- 283. Monteiro R. V., Dietz J. M., Jansen A. M. 2010. The impact of concomitant infections by Trypanosoma cruzi and intestinal helminths on the health of wild golden and golden-headed lion tamarins. Res. Vet. Sci. 89:27–35 [DOI] [PubMed] [Google Scholar]
- 284. Montoya-Durango D. E., et al. 2009. Epigenetic control of mammalian LINE-1 retrotransposon by retinoblastoma proteins. Mutat. Res. 665:20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Mordue D. G., Monroy F., La Regina M., Dinarello C. A., Sibley L. D. 2001. Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J. Immunol. 167:4574–4584 [DOI] [PubMed] [Google Scholar]
- 286. Moretti E., Basso B., Cervetta L., Brigada A., Barbieri G. 2002. Patterns of cytokines and soluble cellular receptors in the sera of children with acute Chagas' disease. Clin. Diagn. Lab. Immunol. 9:1324–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Morrish T. A., et al. 2007. Endonuclease-independent LINE-1 retrotransposition at mammalian telomeres. Nature 446:208–212 [DOI] [PubMed] [Google Scholar]
- 288. Moser D. R., Kirchhoff L. V., Donelson J. E. 1989. Detection of Trypanosoma cruzi by DNA amplification using the polymerase chain reaction. J. Clin. Microbiol. 27:1477–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Muñoz M., et al. 2009. Interleukin (IL)-23 mediates Toxoplasma gondii-induced immunopathology in the gut via matrixmetalloproteinase-2 and IL-22 but independent of IL-17. J. Exp. Med. 206:3047–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Nagineni C. N., Detrick B., Hooks J. J. 2002. Transforming growth factor-beta expression in human retinal pigment epithelial cells is enhanced by Toxoplasma gondii: a possible role in the immunopathogenesis of retinochoroiditis. Clin. Exp. Immunol. 128:372–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Nery-Guimarães F., Venâncio I., Grynberg N. 1974. Refractoriness of hens towards Trypanosoma (Schizotrypanum) cruzi. III. Dissociation of phenomena of the refractory state and lysis of the epimastigotes by fowl serum. Mem. Inst. Oswaldo Cruz 72:131–136 [DOI] [PubMed] [Google Scholar]
- 292. Nilsson D., Andersson B. 2005. Strand asymmetry patterns in trypanosomatid parasites. Exp. Parasitol. 109:143–149 [DOI] [PubMed] [Google Scholar]
- 293. Nitz N., et al. 2004. Heritable integration of kDNA minicircle sequences from Trypanosoma cruzi into the avian genome: insights into human Chagas disease. Cell 118:175–186 [DOI] [PubMed] [Google Scholar]
- 294. Nussenzweig A., Nussenzweig M. C. 2007. A backup DNA repair pathway moves to the forefront. Cell 131:223–225 [DOI] [PubMed] [Google Scholar]
- 295. Obado S. O., Bot C., Nilsson D., Andersson B., Kelly J. M. 2007. Repetitive DNA is associated with centromeric domains in Trypanosoma brucei but not Trypanosoma cruzi. Genome Biol. 8:R37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Obler D., et al. 2010. Familial dilated cardiomyopathy secondary to dystrophin splice site mutation. J. Card. Fail. 16:194–199 [DOI] [PubMed] [Google Scholar]
- 297. Ogbadoiyi E. O., Robinson D. R., Gull K. 2003. A high-order trans-membrane structural linkage is responsible for mitochondrial genome positioning and segregation by flagellar basal bodies in trypanosomes. Mol. Biol. Cell 14:1769–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Oliveira J. S. 1985. A natural human model of intrinsic heart nervous system denervation: Chagas' cardiopathy. Hum. Pathol. 110:1092–1098 [DOI] [PubMed] [Google Scholar]
- 299. O'Malley M. A., Boucher Y. 2005. Paradigm change in evolutionary microbiology. Stud. Hist. Philos. Biol. Biomed. Sci. 36:183–208 [DOI] [PubMed] [Google Scholar]
- 300. Paba J., et al. 2004. Proteomic analysis of Trypanosoma cruzi developmental stages using isotope-coded affinity tag reagents. J. Proteome Res. 3:517–524 [DOI] [PubMed] [Google Scholar]
- 301. Paba J., et al. 2004. Proteomic analysis of the human pathogen Trypanosoma cruzi. Proteomics 4:1052–1059 [DOI] [PubMed] [Google Scholar]
- 302. Padilla A. M., Bustamante J. M., Tarleton R. L. 2009. CD8+ T cells in Trypanosoma cruzi infection. Curr. Opin. Immunol. 21:385–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Paiva C. N., et al. 2007. Unraveling the lethal synergism between Trypanosoma cruzi infection and LPS: a role for increased macrophage reactivity. Eur. J. Immunol. 37:1355–1364 [DOI] [PubMed] [Google Scholar]
- 304. Pan-American Health Organization. 1994. Chagas disease and the nervous system. Scientific publication 547. Pan-American Health Organization, Washington, DC [Google Scholar]
- 305. Paque R. E., Miller R. 1991. Autoanti-idiotypes exhibit mimicry of myocyte antigens in virus-induced myocarditis. J. Virol. 65:16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Parada H., Carrasco H. A., Añez N., Fuenmayor C., Inglessis I. 1997. Cardiac involvement is a constant finding in acute Chagas' disease: a clinical, parasitological and histopathological study. Int. J. Cardiol. 60:49–54 [DOI] [PubMed] [Google Scholar]
- 307. Parodi-Talice A., et al. 2007. Proteomic analysis of metacyclic trypomastigotes undergoing Trypanosoma cruzi metacyclogenesis. J. Mass Spectrom. 42:1422–1432 [DOI] [PubMed] [Google Scholar]
- 308. Passos S. T., et al. 2010. IL-6 promotes NK cell production of IL-17 during toxoplasmosis. J. Immunol. 184:1776–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Paulin J. 1975. The chondriome of selected trypanosomatids. A three-dimensional study based on serial thick sections and high voltage electron microscopy. J. Cell Biol. 66:404–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Pauschinger M., et al. 2005. Carvedilol improves left ventricular function in murine coxsackievirus-induced acute myocarditis association with reduced myocardial interleukin-1beta and MMP-8 expression and a modulated immune response. Eur. J. Heart Fail. 7:444–452 [DOI] [PubMed] [Google Scholar]
- 311. Pellegrini A., et al. 2007. Spleen B cells from BALB/c are more prone to activation than spleen B cells from C57BL/6 mice during a secondary immune response to cruzipain. Int. Immunol. 19:1395–1402 [DOI] [PubMed] [Google Scholar]
- 312. Pena S. D., Machado C. R., Macedo A. M. 2009. Trypanosoma cruzi: ancestral genomes and population structure. Mem. Inst. Oswaldo Cruz 104:108–114 [DOI] [PubMed] [Google Scholar]
- 313. Pereira K. S., et al. 2009. Chagas' disease as a foodborne illness. J. Food. Prot. 72:441–446 [DOI] [PubMed] [Google Scholar]
- 314. Pinheiro S. W., et al. 2003. Morphometric study of the fibrosis and mast cell count in the circular colon musculature of chronic Chagas patients with and without megacolon. Rev. Soc. Bras. Med. Trop. 36:461–466 [DOI] [PubMed] [Google Scholar]
- 315. Pitta M. G., et al. 2009. IL-17 and IL-22 are associated with protection against human kala azar caused by Leishmania donovani. J. Clin. Invest. 119:2379–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Poinar G., Jr 2005. Triatoma dominicana sp. n. (Hemíptera: Reduviidae: Triatominae), and Trypannosoma antiquus sp. n. (Stercoraria: Trypanosomatidae), the first fossil evidence of a triatomine-trypanosomatid vector association. Vector Borne Zoonotic Dis. 5:72–81 [DOI] [PubMed] [Google Scholar]
- 317. Prata A. 1994. Chagas' disease. Infect. Dis. Clin. North Am. 8:61–76 [PubMed] [Google Scholar]
- 318. Prata A. 2001. Clinical and epidemiological aspects of Chagas disease. Lancet Infect. Dis. 1:92–100 [DOI] [PubMed] [Google Scholar]
- 319. Ramos B., Urdaneta-Morales S. 1977. Hematophagous insects as vectors for frog trypanosomes. Rev. Biol. Trop. 25:209–217 [PubMed] [Google Scholar]
- 320. Ramos S. G., Matturri L., Rossi L., Rossi M. A. 1995. Sudden cardiac death in the intermediate phase of Chagas' disease associated with acute infarction of the right carotid body. Int. J. Cardiol. 52:265–268 [DOI] [PubMed] [Google Scholar]
- 321. Rassi A., Jr., Rassi S. G., Rassi A. 2001. Sudden death in Chagas' disease. Arq. Bras. Cardiol. 76:75–96 [DOI] [PubMed] [Google Scholar]
- 322. Reed S. G. 1980. Adoptive transfer of resistance to acute Trypanosoma cruzi infection with T-lymphocyte-enriched spleen cells. Infect. Immun. 28:404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Reinhard K., Fink M., Skiles J. 2003. A case of megacolon in Rio Grande Valley as a possible case of Chagas disease. Mem. Inst. Oswaldo Cruz 98(Suppl. I):165–172 [DOI] [PubMed] [Google Scholar]
- 324. Ribeiro C. H., et al. 2009. Trypanosoma cruzi calreticulin: a possible role in Chagas' disease autoimmunity. Mol. Immunol. 46:1092–1099 [DOI] [PubMed] [Google Scholar]
- 325. Ribeiro L. C., Barbosa A. A., Jr., Andrade Z. A. 2002. Pathology of intracardiac nerves in experimental Chagas disease. Mem. Inst. Oswaldo Cruz 97:1019–1025 [DOI] [PubMed] [Google Scholar]
- 326. Riou G., Yot P. 1967. Heterogeneity of the kinetoplast DNA molecules of Trypanosoma cruzi. Biochemistry 16:2390–2396 [DOI] [PubMed] [Google Scholar]
- 327. Roberson E. L., Hanson W. L. 1974. Transfer of immunity to T. cruzi. Trans. R. Soc. Trop. Med. Hyg. 68:338. [DOI] [PubMed] [Google Scholar]
- 328. Roberson E. L., Chapman W. L., Jr., Hanson W. L. 1973. The effects of total-body X-irradiation on Trypanosoma cruzi infection (Chagas' disease) in mice and rats. Z. Parasitenkd. 41:83–94 [DOI] [PubMed] [Google Scholar]
- 329. Rocha A., et al. 1987. ELISA immunoenzymatic assay in the pericardial fluid: a new method for the post-mortem diagnosis of Chagas disease. Rev. Soc. Bras. Med. Trop. 20:213–216 [DOI] [PubMed] [Google Scholar]
- 330. Rodrigues M. M., Alencar B. C., Vasconcelos J. R. 2009. Acquired immunity against Trypanosoma cruzi infection and vaccine development, p. 94–103.In Teixeira A. R. L., Vinaud M. C., Castro A. M. (ed.), Emerging Chagas disease, 1st ed. Bentham Science Publishers, New York, NY [Google Scholar]
- 331. Rodriguez A. M., Santoro F., Afchain D., Bazin H., Capron A. 1981. Trypanosoma cruzi infection in B-cell-deficient rats. Infect. Immun. 31:524–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Rose N. R., Herskowitz A., Neumann D. A. 1993. Autoimmunity in myocarditis: models and mechanisms. Clin. Immunol. Immunopathol. 68:95–99 [DOI] [PubMed] [Google Scholar]
- 333. Rossi M. A. 1990. Microvascular changes as a cause of chronic cardiomyopathy in Chagas' disease. Am. Heart J. 120:233–236 [DOI] [PubMed] [Google Scholar]
- 334. Rossi M. A. 2001. Connective tissue skeleton in the normal left ventricle and in hypertensive left ventricular hypertrophy and chronic chagasic myocarditis. Med. Sci. Monit. 7:820–832 [PubMed] [Google Scholar]
- 335. Rudi K., Fossheim T., Jakobsen K. S. 1999. Restriction cutting independent method for cloning genomic DNA segments outside the boundaries of known sequences. Biotechniques 27:1170–1172,1176-1177 [DOI] [PubMed] [Google Scholar]
- 336. Rutschow S., et al. 2010. Left ventricular enlargement in coxsackievirus-B3 induced chronic myocarditis—ongoing inflammation and an imbalance of the matrix degrading system. Eur. J. Pharmacol. 630:145–151 [DOI] [PubMed] [Google Scholar]
- 337. Ryckman R. E. 1965. Epizootiology of Trypanosoma cruzi in Soutwestern North America. IV. Lizards, laboratory hosts for Trypanosoma cruzi and triatominae. J. Med. Entomol. 2:93. [DOI] [PubMed] [Google Scholar]
- 338. Sadigursky M., Santos-Buch C. A. 1997. A novel receptor mediated ATP transport system regulated by tyrosine and serine/threonine phosphokinases in Trypanosoma cruzi trypomastigotes. Recept. Signal Transduct. 7:29–43 [PubMed] [Google Scholar]
- 339. Sadigursky M., Von Kreuter B. F., Santos-Buch C. A. 1988. Development of chagasic autoimmune myocarditis associated with anti-idiotype reaction. Mem. Inst. Oswaldo Cruz 83:363–366 [DOI] [PubMed] [Google Scholar]
- 340. Sales P. A., Jr., et al. 2008. The regulatory CD4+CD25+ T cells have a limited role on pathogenesis of infection with Trypanosoma cruzi. Microbes Infect. 10:680–688 [DOI] [PubMed] [Google Scholar]
- 341. Samudio M., et al. 1999. Local and systemic cytokine expression during experimental chronic Trypanosoma cruzi infection in a Cebus monkey model. Parasite Immunol. 21:451–460 [DOI] [PubMed] [Google Scholar]
- 342. Sanderson C. J., Lopez A. F., Moreno M. M. 1977. Eosinophils and not lymphoid K cells kill Trypanosoma cruzi epimastigotes. Nature 268:340–341 [DOI] [PubMed] [Google Scholar]
- 343. Sanderson C. J., de Souza W. 1979. A morphological study of the interaction between Trypanosoma cruzi and rat eosinophils, neutrophils and macrophages in vitro. J. Cell Sci. 37:275–286 [DOI] [PubMed] [Google Scholar]
- 344. Santos-Buch C. A., Teixeira A. R. L. 1974. The immunology of experimental Chagas' disease. 3. Rejection of allogeneic heart cells in vitro. J. Exp. Med. 140:38–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Scharfstein J., Monteiro A. C., Schmitz V., Svensjo E. 2008. Angiotensin-converting enzyme limits inflammation elicited by Trypanosoma cruzi cysteine proteases: a peripheral mechanism regulating adaptive immunity via the innate kinin pathway. Biol. Chem. 389:1015–1024 [DOI] [PubMed] [Google Scholar]
- 346. Scharfstein J., Lima A. P. 2008. Roles of naturally occurring protease inhibitors in the modulation of host cell signaling and cellular invasion by Trypanosoma cruzi. Subcell. Biochem. 47:140–154 [DOI] [PubMed] [Google Scholar]
- 347. Scharfstein J., Gomes J. A., Correa-Oliveira R. 2009. Back to the future in Chagas disease: from animal models to patient cohort studies, progress in immunopathogenesis research. Mem. Inst. Oswaldo Cruz 104(Suppl. 1):187–198 [DOI] [PubMed] [Google Scholar]
- 348. Scharfstein J., et al. 2000. Host cell invasion by Trypanosoma cruzi is potentiated by activation of bradykinin B(2) receptors. J. Exp. Med. 192:1289–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Schenone H., Gaggero M., Sapúnar J., Contreras M. C., Rojas A. 2001. Congenital Chagas disease of second generation in Santiago, Chile. Report of two cases.. Rev. Inst. Med. Trop. Sao Paulo 43:231–232 [DOI] [PubMed] [Google Scholar]
- 350. Schiffler R. J., Mansur G. P., Navin T. R., Limpakarn-Janarat K. 1984. Indigenous Chagas disease (American trypanosomiasis) in California. JAMA 251:2983–2984 [PubMed] [Google Scholar]
- 351. Schlüter D., Kaefer N., Hof H., Wiestler O. D., Deckert-Schlüter M. 1997. Expression pattern and cellular origin of cytokines in the normal and Toxoplasma gondii-infected murine brain. Am. J. Pathol. 150:1021–1035 [PMC free article] [PubMed] [Google Scholar]
- 352. Schmidtke M., et al. 2000. Cytokine profiles in heart, spleen, and thymus during the acute stage of experimental coxsackievirus B3-induced chronic myocarditis. J. Med. Virol. 61:518–526 [PubMed] [Google Scholar]
- 353. Schmitz V., Svensjö E., Serra R. R., Teixeira M. M., Scharfstein J. 2009. Proteolytic generation of kinins in tissues infected by Trypanosoma cruzi depends on CXC chemokine secretion by macrophages activated via Toll-like 2 receptors. J. Leukoc. Biol. 85:1005–1014 [DOI] [PubMed] [Google Scholar]
- 354. Schmunis G. A., Yadon Z. E. 2010. Chagas disease: a Latin American health problem becoming a world health problem. Acta Trop. 115:14–21 [DOI] [PubMed] [Google Scholar]
- 355. Seder R. A., Paul W. E. 1994. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu. Rev. Immunol. 12:635–673 [DOI] [PubMed] [Google Scholar]
- 356. Seko Y., Takahashi N., Yagita H., Okumura K., Yazaki Y. 1997. Expression of cytokine mRNAs in murine hearts with acute myocarditis caused by coxsackievirus B3. J. Pathol. 183:105–108 [DOI] [PubMed] [Google Scholar]
- 357. Sen S. K., Huang C. T., Han K., Batzer M. A. 2007. Endonuclease-independent insertion provides an alternative pathway for L1 retrotransposition in the human genome. Nucleic Acids Res. 35:3741–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Sfriso P., et al. 2010. Infections and autoimmunity: the multifaceted relationship. J. Leukoc. Biol. 87:385–395 [DOI] [PubMed] [Google Scholar]
- 359. Shapira S., Harb O. S., Caamano J., Hunter C. A. 2004. The NF-kappaB signaling pathway: immune evasion and immunoregulation during toxoplasmosis. Int. J. Parasitol. 34:393–400 [DOI] [PubMed] [Google Scholar]
- 360. Shapiro T. A., Englund P. T. 1995. The structure and replication of kinetoplast DNA. Annu. Rev. Microbiol. 49:117–143 [DOI] [PubMed] [Google Scholar]
- 361. Sharma U., Singh S. 2009. Immunobiology of leishmaniasis. Indian J. Exp. Biol. 47:412–423 [PubMed] [Google Scholar]
- 362. Shrivastav M., De Haro L. P., Nickoloff J. A. 2008. Regulation of DNA double-strand break repair pathway choice. Cell Res. 18:134–147 [DOI] [PubMed] [Google Scholar]
- 363. Sica R. E., Gonzalez Cappa S. M., Sanz O. P., Mirkin G. 1995. Peripheral nervous system involvement in human and experimental chronic American trypanosomiasis. Bull. Soc. Pathol. Exot. 88:156–163 [PubMed] [Google Scholar]
- 364. Silva J. J., et al. 2010. Novel ruthenium complexes as potential drugs for Chagas's disease: enzyme inhibition and in vitro/in vivo trypanocidal activity. Br. J. Pharmacol. 60:260–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Silveira C. A., Castillo E., Castro C. 2000. Evaluation of an specific treatment for Trypanosoma cruzi in children, in the evolution of the intermediate phase. Rev. Soc. Bras. Med. Trop. 33:191–196 [DOI] [PubMed] [Google Scholar]
- 366. Simões-Barbosa A., et al. 2006. Hitchhiking Trypanosoma cruzi minicircle DNA affects gene expression in human host cells via LINE-1 retrotransposon. Mem. Inst. Oswaldo Cruz 101:833–843 [DOI] [PubMed] [Google Scholar]
- 367. Simpson A. G. B., Stevens J. R., Lukes J. 2006. The evolution and diversity of kinetoplastid flagellates. Trends Parasitol. 22:168–174 [DOI] [PubMed] [Google Scholar]
- 368. Simpson A. G., Perley T. A., Lara E. 2008. Lateral transfer of the gene for a widely used marker, alpha-tubulin, indicated by a multi-protein study of the phylogenetic position of Andalucia (Excavata). Mol. Phylogenet. Evol. 47:366–377 [DOI] [PubMed] [Google Scholar]
- 369. Soares C. M., et al. 1989. Alpha- and beta-tubulin mRNAs of Trypanosoma cruzi originate from a single multicistronic transcript. FEBS Lett. 250:497–502 [DOI] [PubMed] [Google Scholar]
- 370. Sodré C. L., et al. 2009. Proteomic map of Trypanosoma cruzi CL Brener: the reference strain of the genome project. Arch. Microbiol. 191:177–184 [DOI] [PubMed] [Google Scholar]
- 371. Solari A. J. 1995. Mitosis and genome partition in trypanosomes. Biocell 19:65–84 [PubMed] [Google Scholar]
- 372. Song M., Boissinot S. 2007. Selection against LINE-1 retrotransposons results principally from their ability to mediate ectopic recombination. Gene 390:206–213 [DOI] [PubMed] [Google Scholar]
- 373. Sosa Estani S., et al. 1998. Efficacy of chemotherapy with benznidazole in children in the intermediate phase of Chagas' disease. Am. J. Trop. Med. Hyg. 59:526–529 [DOI] [PubMed] [Google Scholar]
- 374. Souza P. E., et al. 2007. Trypanosoma cruzi infection induces differential modulation of costimulatory molecules and cytokines by monocytes and T cells from patients with intermediate and cardiac Chagas' disease. Infect. Immun. 75:1886–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Spivak J. L. 2004. The chronic myeloproliferative disorders: clonality and clinical heterogeneity. Semin. Hematol. 41(2 Suppl. 3):1–5 [DOI] [PubMed] [Google Scholar]
- 376. Srikanta D., et al. 2009. An alternative pathway for Alu retrotransposition suggests a role in DNA double-strand break repair. Genomics 93:205–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Srikanta D., Sen S. K., Conlin E. M., Batzer M. A. 2009. Internal priming: an opportunistic pathway for L1 and Alu retrotransposition in hominins. Gene 448:233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Steinert G., Firket H., Steinert M. 1958. Synthèse d'acide désoxyribonucléique dans le corps parabasal de Trypanosoma mega. Exp. Cell Res. 15:632–635 [DOI] [PubMed] [Google Scholar]
- 379. Stevens J. R., Gibson W. C. 1999. The evolution of pathogenic trypanosomes. Cad. Saude Publica 15:673–684 [DOI] [PubMed] [Google Scholar]
- 380. Stevens J. R., Noyes H. A., Schofield C. J., Gibson W. 2001. The molecular evolution of trypanosomatidae. Adv. Parasitol. 48:1–56 [DOI] [PubMed] [Google Scholar]
- 381. Stimpert K. K., Montgomery S. P. 2010. Physician awareness of Chagas disease, USA. Emerg. Infect. Dis. 16:871–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Sturm N. R. 2009. Trypanosoma cruzi mitochondrial DNA and the parasite lifecycle, p. 63–69.In Teixeira A. R. L., Vinaud M. C., Castro A. M. (ed.), Emerging Chagas disease, 1st ed. Bentham Science Publishers, New York, NY [Google Scholar]
- 383. Sturm N. R., Degrave W., Morel C., Simpson L. 1989. Sensitive detection and schizodeme classification of Trypanosoma cruzi cells by amplification of kinetoplast minicircle DNA sequences: use in diagnosis of Chagas' disease. Mol. Biochem. Parasitol. 33:205–214 [DOI] [PubMed] [Google Scholar]
- 384. Suzuki J., et al. 2009. Genetic evidence that the non-homologous end-joining repair pathway is involved in LINE retrotransposition. PLoS Genet. 5:e1000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Suzuki Y., et al. 2000. IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J. Immunol. 164:5375–5382 [DOI] [PubMed] [Google Scholar]
- 386. Tafuri L., Maria T. A., Lopes E. R. 1970. Pathogenesis of lesions of the autonomic nervous system of the mouse in experimental acute Chagas' disease. Light and electron microscope studies. Am. J. Trop. Med. Hyg. 19:405–417 [DOI] [PubMed] [Google Scholar]
- 387. Tafuri W. L. 1971. Light and electron microscope studies of the autonomic nervous system in experimental and human American trypanosomiasis. Virchows Arch. A Pathol. Anat. Histol. 354:136–349 [DOI] [PubMed] [Google Scholar]
- 388. Tafuri W. L., Maria T. A., Lopes E. R., Chapadeiro E. 1973. Electron microscopy of the myocardium in human trypanosomiasis cruzi. Rev. Inst. Med. Trop. Sao Paulo 15:347–370 [PubMed] [Google Scholar]
- 389. Takehara H. A., Perini A., da Silva M. H., Mota I. 1981. Trypanosoma cruzi: role of different antibody classes in protection against infection in the mouse. Exp. Parasitol. 52:137–146 [DOI] [PubMed] [Google Scholar]
- 390. Takle G. B., Young A., Snary D., Hudson L., Nicholls S. C. 1989. Cloning and expression of a trypomastigote-specific 85-kilodalton surface antigen gene from Trypanosoma cruzi. Mol. Biochem. Parasitol. 37:57–64 [DOI] [PubMed] [Google Scholar]
- 391. Tanowitz H. B., et al. 2009. Perspectives on Trypanosoma cruzi-induced heart disease (Chagas disease). Prog. Cardiovasc. Dis. 51:524–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Tanowitz H. B., et al. 1992. Chagas' disease. Clin. Microbiol. Rev. 5:400–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Tarleton R. L. 2001. Parasite persistence in the aetiology of Chagas disease. Int. J. Parasitol. 31:550–554 [DOI] [PubMed] [Google Scholar]
- 394. Tarleton R. L. 2003. Chagas disease: a role for autoimmunity? Trends Parasitol. 19:447–451 [DOI] [PubMed] [Google Scholar]
- 395. Tarleton R. L., Zhang L. 1999. Chagas disease etiology: autoimmunity or parasite persistence? Parasitol. Today 15:94–99 [DOI] [PubMed] [Google Scholar]
- 396. Tarleton R. L., Zhang L., Downs M. O. 1997. “Autoimmune rejection” of neonatal heart transplants in experimental Chagas disease is a parasite-specific response to infected host tissue. Proc. Natl. Acad. Sci. U. S. A. 94:3932–3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Teixeira A. R. 1986. Chagas' disease in inbred III/J rabbits. Am. J. Pathol. 124:363–365 [PMC free article] [PubMed] [Google Scholar]
- 398. Teixeira A. R. L., Ripoll C. M., Santos-Buch C. A. 1996. Autoimmunity in Chagas disease, p. 233–255.In Friedman H., Rose N. R., Bendinelli M. (ed.), Microorganisms and autoimmune diseases. Plenum Press, New York, NY [Google Scholar]
- 399. Teixeira A. R. L. 1987. Immunoprophylaxis against Chagas disease, p. 243–280.In Miller L. H., Pino J. A, McKelvey J. J., Jr. (ed.), Immunity to blood parasites of animals and man. Plenum Press, New York, NY [Google Scholar]
- 400. Teixeira A. R. L., Santos-Buch C. A. 1974. The immunology of experimental Chagas' disease. I. Preparation of Trypanosoma cruzi antigens and humoral antibody response to these antigens. J. Immunol. 113:859–869 [PubMed] [Google Scholar]
- 401. Teixeira A. R. L., Santos-Buch C. A. 1975. The immunology of experimental Chagas' disease. II. Delayed hypersensitivity to Trypanosoma cruzi antigens. Immunology 28:401–410 [PMC free article] [PubMed] [Google Scholar]
- 402. Teixeira A. R. L., et al. 2011. Trypanosoma cruzi in the chicken model: Chagas-like heart disease in the absence of parasitism. PLoS Negl. Trop. Dis. 5:e1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Teixeira A. R. L., Jabur E., Córdoba J. C., Mayor I. C. S., Solorzano E. 1983. Alteração da resposta imune mediada por células durante o tratamento com benzonidazol. Rev. Soc. Bras. Med. Trop. 16:11–22 [Google Scholar]
- 404. Teixeira A. R. L., Pimentel F., Cordeiro C. 2009. The pathology of Chagas disease, p. 110–121.In Teixeira A. R. L., Vinaud M. C., Castro A. M. (ed.), Emerging Chagas disease, 1st ed. Bentham Science Publishers, New York, NY [Google Scholar]
- 405. Teixeira A. R. L., Roters F., Mott K. E. 1970. Acute Chagas disease. Gaz. Med. Bahia 3:176–186 [Google Scholar]
- 406. Teixeira A. R. L., Vinaud M. C., Castro A. M. 2009. Chagas disease: a global health problem, p. 18–23.In Teixeira A. R. L., Vinaud M. C., Castro A. M. (ed.), Emerging Chagas disease, 1st ed. Bentham Science Publishers, New York, NY [Google Scholar]
- 407. Teixeira A. R. L., Nitz N., Guimaro M. C., Gomes C., Santos-Buch C. A. 2006. Chagas disease. Postgrad. Med. J. 82:788–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Teixeira A. R. L., Nascimento R., Sturm N. R. 2006. Evolution and pathology in Chagas disease. Mem. Inst. Oswaldo Cruz 101:463–491 [DOI] [PubMed] [Google Scholar]
- 409. Teixeira A. R., Teixeira M. G. 1995. Delayed hypersensitivity to Trypanosoma cruzi antigen. III. Sensitivity of the skin test with T12E antigen in the diagnosis of Chagas disease in hospitalized patients. Rev. Soc. Bras. Med. Trop. 28:267–271 [DOI] [PubMed] [Google Scholar]
- 410. Teixeira A. R., et al. 2009. Environment, interactions between Trypanosoma cruzi and its host, and health. Cad. Saude Publica 25(Suppl. 1):S32–S44 [DOI] [PubMed] [Google Scholar]
- 411. Teixeira A. R., Cunha Neto E., Rizzo L. V., Silva R. 1990. Trypanocidal nitroarene treatment of experimental Trypanosoma cruzi infection does not prevent progression of chronic-phase heart lesions in rabbits. J. Infect. Dis. 162:1420. [DOI] [PubMed] [Google Scholar]
- 412. Teixeira A. R., Lacava Z., Santana J. M., Luna H. 1991. Insertion of Trypanosoma cruzi DNA in the genome of mammal host cell through infection. Rev. Soc. Bras. Med. Trop. 24:55–58 [DOI] [PubMed] [Google Scholar]
- 413. Teixeira A. R., et al. 1994. Possible integration of Trypanosoma cruzi kDNA minicircles into the host cell genome by infection. Mutat. Res. 305:197–209 [DOI] [PubMed] [Google Scholar]
- 414. Teixeira A. R., Teixeira G., Macedo V., Prata A. 1978. Acquired cell-mediated immunodepression in acute Chagas' disease. J. Clin. Invest. 62:1132–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Teixeira A. R., Cordoba J. C., Souto Maior I., Solorzano E. 1990. Chagas' disease: lymphoma growth in rabbits treated with benznidazole. Am. J. Trop. Med. Hyg. 43:146–158 [DOI] [PubMed] [Google Scholar]
- 416. Teixeira A. R., Junqueira L. F., Jr., Solorzano E., Zappala M. 1983. Experimental Chagas' disease in isogenic III/J rabbits. I. Physiopathology of arrhythmia and sudden death in Chagas' disease patients. Rev. Assoc. Med. Bras. 29:77–83 [PubMed] [Google Scholar]
- 417. Teixeira A. R., Teixeira M. L., Santos-Buch C. A. 1975. The immunology of experimental Chagas' disease. IV. Production of lesions in rabbits similar to those of chronic Chagas' disease in man. Am. J. Pathol. 80:163–180 [PMC free article] [PubMed] [Google Scholar]
- 418. Teixeira A. R., et al. 2001. Emerging Chagas disease: trophic network and cycle of transmission of Trypanosoma cruzi from palm trees in the Amazon. Emerg. Infect. Dis. 7:100–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Teixeira A. R., Silva R., Cunha Neto E., Santana J. M., Rizzo L. V. 1990. Malignant, non-Hodgkin's lymphomas in Trypanosoma cruzi-infected rabbits treated with nitroarenes. J. Comp. Pathol. 103:37–48 [DOI] [PubMed] [Google Scholar]
- 420. Tiede A., Bastisch I., Schubert J., Orlean P., Schmidt R. E. 1999. Biosynthesis of glycosylphosphatidylinositols in mammals and unicellular microbes. Biol. Chem. 380:503–523 [DOI] [PubMed] [Google Scholar]
- 421. Todorov A. G., et al. 2003. Trypanosoma cruzi induces edematogenic responses in mice and invades cardiomyocytes and endothelial cells in vitro by activating distinct kinin receptor (B1/B2) subtypes. FASEB J. 17:73–75 [DOI] [PubMed] [Google Scholar]
- 422. Tomlinson S., Vandekerckhove F., Frevert U., Nussenzweig V. 1995. The induction of Trypanosoma cruzi trypomastigote to amastigote transformation by low pH. Parasitology 110:547–554 [DOI] [PubMed] [Google Scholar]
- 423. Torres C. M. 1930. Patogenia de la miocarditis cronica em la enfermedad de Chagas. Soc. Argent. Patol. Reg. Norte Quinta Reun. 2:902 [Google Scholar]
- 424. Torres C. M. 1960. Myocytolysis and fibrosis of the myocardium in Chagas' disease. Mem. Inst. Oswaldo Cruz 58:161–182 [DOI] [PubMed] [Google Scholar]
- 425. Torres S. H., et al. 2004. Capillary damage in skeletal muscle in advanced Chagas disease patients. Parasitol. Res. 93:364–368 [DOI] [PubMed] [Google Scholar]
- 426. Trischmann T., Tanowitz H., Wittner M., Bloom B. 1978. Trypanosoma cruzi: role of the immune response in the natural resistance of inbred strains of mice. Exp. Parasitol. 45:160–168 [DOI] [PubMed] [Google Scholar]
- 427. Trischmann T. M. 1986. Trypanosoma cruzi: early parasite proliferation and host resistance in inbred strains of mice. Exp. Parasitol. 62:194–201 [DOI] [PubMed] [Google Scholar]
- 428. Tzelepis F., et al. 2008. Infection with Trypanosoma cruzi restricts the repertoire of parasite-specific CD8+ T cells leading to immunodominance. J. Immunol. 180:1737–1748 [DOI] [PubMed] [Google Scholar]
- 429. Urdaneta-Morales S., McLure I. 1981. Experimental infections in Venezuelan lizards by Trypanosoma cruzi. Acta Trop. 38:99–105 [PubMed] [Google Scholar]
- 430. Vago A. R., et al. 2000. Genetic characterization of Trypanosoma cruzi directly from tissues of patients with chronic Chagas disease: differential distribution of genetic types into diverse organs. Am. J. Pathol. 156:1805–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Vago A. R., et al. 1996. Kinetoplast DNA signatures of Trypanosoma cruzi strains obtained directly from infected tissues. Am. J. Pathol. 149:2153–2159 [PMC free article] [PubMed] [Google Scholar]
- 432. Vago A. R., Silva D. M., Adad S. J., Correa-Oliveira R., d'Avila Reis D. 2003. Chronic Chagas disease: presence of parasite DNA in the oesophagus of patients without megaoesophagus. Trans. R. Soc. Trop. Med. Hyg. 97:308–309 [DOI] [PubMed] [Google Scholar]
- 433. Vaidian A. K., Weiss L. M., Tanowitz H. B. 2004. Chagas' disease and AIDS. Kinetoplastid Biol. Dis. 3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Van Baarlen P., Van Belkum A., Summerbell R. C., Crous P. W., Thomma B. P. 2007. Molecular mechanisms of pathogenicity: how do pathogenic microorganisms develop cross-kingdom host jumps? FEMS Microbiol. Rev. 31:239–277 [DOI] [PubMed] [Google Scholar]
- 435. van der Poll T., Zijlstra E. E., Mevissen M. 1995. Interleukin 6 during active visceral leishmaniasis and after treatment. Clin. Immunol. Immunopathol. 77:111–114 [DOI] [PubMed] [Google Scholar]
- 436. Vianna G. O. 1911. On pathologic anatomy in Chagas disease. Mem. Inst. Oswaldo Cruz 3:276–294 [Google Scholar]
- 437. Vickerman K. 1969. On the surface coat and flagellar adhesion in trypanosomes. J. Cell Sci. 5:163–194 [DOI] [PubMed] [Google Scholar]
- 438. Vickerman K. 1976. The diversity of the kinetoplastid flagellates, p. 1–34.In Lumsden W. H. R., Evans D. A. (ed.), Biology of the Kinetoplastida. Academic Press, London, United Kingdom [Google Scholar]
- 439. Villalta F., et al. 2009. Perspectives on the Trypanosoma cruzi-host cell receptor interactions. Parasitol. Res. 104:1251–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440. Villalta F., Zhang Y., Bibb K. E., Burns J. M., Jr., Lima M. F. 1998. Signal transduction in human macrophages by gp83 ligand of Trypanosoma cruzi: trypomastigote gp83 ligand up-regulates trypanosome entry through the MAP kinase pathway. Biochem. Biophys. Res. Commun. 249:247–252 [DOI] [PubMed] [Google Scholar]
- 441. von der Heyden S., Chao E. E., Vickerman K., Cavalier-Smith T. 2004. Ribosomal RNA phylogeny of bodonid and diplonemid flagellates and the evolution of euglenozoa. J. Eukaryot. Microbiol. 51:402–416 [DOI] [PubMed] [Google Scholar]
- 442. Von Kreuter B. F., Walton B. L., Santos-Buch C. A. 1995. Attenuation of parasite cAMP levels in T. cruzi-host cell membrane interactions in vitro. J. Eukaryot. Microbiol. 42:20–26 [DOI] [PubMed] [Google Scholar]
- 443. Vossenkämper A., et al. 2004. Both IL-12 and IL-18 contribute to small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii, but IL-12 is dominant over IL-18 in parasite control. Eur. J. Immunol. 34:3197–3207 [DOI] [PubMed] [Google Scholar]
- 444. Votýpka J., et al. 2010. Probing into the diversity of trypanosomatid flagellates parasitizing insect hosts in South-West China reveal both endemism and global dispersal. Mol. Phylogenet. Evol. 54:243–253 [DOI] [PubMed] [Google Scholar]
- 445. Watanabe M. A., Schmidt G. 2004. Heart rate turbulence: a 5-year review. Heart Rhythm 1:732–738 [DOI] [PubMed] [Google Scholar]
- 446. Weatherly D. B., Boehlke C., Tarleton R. L. 2009. Chromosome level assembly of the hybrid Trypanosoma cruzi genome. BMC Genomics 10:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447. Weaver C. T., Hatton R. D., Mangan P. R., Harrington L. E. 2007. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 25:821–852 [DOI] [PubMed] [Google Scholar]
- 448. Weremeichik H., Moraska A., Herzum M., Weller A., Huber S. A. 1991. Naturally occurring anti-idiotypic antibodies—mechanisms for autoimmunity and immunoregulation? Eur. Heart J. 12(Suppl. D):154–157 [DOI] [PubMed] [Google Scholar]
- 449.Reference deleted.
- 450. Williams J. T., Dick E. J., Jr., VandeBerg J. L., Hubbard G. B. 2009. Natural Chagas disease in four baboons. J. Med. Primatol. 38:107–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Wilson M. S., et al. 2010. Redundant and pathogenic roles for IL-22 in mycobacterial, protozoan, and helminth infections. J. Immunol. 184:4378–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452. Woody N. C., Woody H. B. 1961. American Trypanosomiasis. I. Clinical and epidemiologic background of Chagas disease in the United States. J. Pediatr. 58:568. [DOI] [PubMed] [Google Scholar]
- 453. World Health Organization. 2002. Control of Chagas' disease: second report of a WHO Expert Committee. World Health Organ. Tech. Rep. Ser. 905:1–109 [PubMed] [Google Scholar]
- 454. Xavier S. C., et al. 2007. Mapping of the distribution of Trypanosoma cruzi infection among small wild mammals in a conservation unit and its surroundings (Northeast-Brazil). Parasitol. Int. 56:119–128 [DOI] [PubMed] [Google Scholar]
- 455. Xing Z., Cardona C. J., Anunciacion J., Adams S., Dao N. 2010. Roles of the ERK MAPK in the regulation of proinflammatory and apoptotic responses in chicken macrophages infected with H9N2 avian influenza virus. J. Gen. Virol. 91:343–351 [DOI] [PubMed] [Google Scholar]
- 456. Yeo M., et al. 2005. Origins of Chagas disease: didelphis species are natural hosts of Trypanosoma cruzi I and armadillos hosts of Trypanosoma cruzi II, including hybrids. Int. J. Parasitol. 35:225–233 [DOI] [PubMed] [Google Scholar]
- 457. York I. A., Goldberg A. L., Mo X. Y., Rock K. L. 1999. Proteolysis and class I major histocompatibility complex antigen presentation. Immunol. Rev. 172:49–66 [DOI] [PubMed] [Google Scholar]
- 458. Yun M. H., Hiom K. 2009. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 459:460–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Zacks M. A., Garg N. 2006. Recent developments in the molecular, biochemical and functional characterization of GPI8 and the GPI-anchoring mechanism. Mol. Membr. Biol. 23:209–225 [DOI] [PubMed] [Google Scholar]
- 460. Zhang L., Tarleton R. L. 1996. Characterization of cytokine production in murine Trypanosoma cruzi infection by in situ immunocytochemistry: lack of association between susceptibility and type 2 cytokine production. Eur. J. Immunol. 26:102–109 [DOI] [PubMed] [Google Scholar]
- 461. Zhang L., Tarleton R. L. 1999. Parasite persistence correlates with disease severity and localization in chronic Chagas' disease. J. Infect. Dis. 180:480–486 [DOI] [PubMed] [Google Scholar]
- 462. Zhang M., et al. 2010. The Plasmodium eukaryotic initiation factor-2alpha kinase IK2 controls the latency of sporozoites in the mosquito salivary glands. J. Exp. Med. 207:1465–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463. Zhou L., Chong M. M., Littman D. R. 2009. Plasticity of CD4+ T cell lineage differentiation. Immunity 30:646–655 [DOI] [PubMed] [Google Scholar]
- 464. Zhu J., Paul W. E. 2010. Heterogeneity and plasticity of T helper cells. Cell Res. 20:4–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465. Ziegler A., Dohr G., Uchanska-Ziegler B. 2002. Possible roles for products of polymorphic MHC and linked olfactory receptor genes during selection processes in reproduction. Am. J. Reprod. Immunol. 48:34–42 [DOI] [PubMed] [Google Scholar]
- 466. Zíková A., et al. 2008. Trypanosoma brucei mitochondrial ribosomes: affinity purification and component identification by mass spectrometry. Mol. Cell. Proteomics 7:1286–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. Zingler N., et al. 2007. Analysis of 5′ junctions of human LINE-1 and Alu retrotransposons suggests an alternative model for 5′-end attachment requiring microhomology-mediated end-joining. Genome Res. 15:780–789 [DOI] [PMC free article] [PubMed] [Google Scholar]