

Summary
Peroxisome proliferator-activated receptor-gamma coactivator 1 beta (PGC-1β) is known to be a transcriptional coactivator for SREBP-1, the master regulator of hepatic lipogenesis. Here we evaluated the role of PGC-1β in the pathogenesis of fructose-induced insulin resistance by using an antisense oligonucletoide (ASO) to knockdown PGC-1 β in liver and adipose tissue. PGC-1 β ASO improved the metabolic phenotype induced by fructose feeding by reducing expression of SREBP-1 and downstream lipogenic genes in liver. PGC-1 β ASO also reversed hepatic insulin resistance induced by fructose in both basal and insulin stimulated states. Furthermore, PGC-1β ASO increased insulin-stimulated whole body glucose disposal due to a threefold increase in glucose uptake in white adipose tissue. These data support an important role for PGC-1 β in the pathogenesis of fructose-induced insulin resistance and suggest that PGC-1 β inhibition may be a novel therapeutic target for treatment of NAFLD, hypertriglyceridemia and insulin resistance associated with increased de novo lipogenesis.
Introduction
Insulin resistance is a common feature of the metabolic syndrome and type 2 diabetes mellitus (T2DM). Both have reached epidemic proportions worldwide (Zimmet et al., 2001) with the global adoption of the westernized diet along with increased consumption of fructose, stemming from the wide and increasing use of high fructose corn syrup sweeteners (Elliott et al., 2002; Basciano et al., 2005). It is well established that fructose is more lipogenic than glucose and high fructose diets have been linked to hypertriglyceridemia, nonalcoholic fatty liver disease (NAFLD) and insulin resistance (Daly et al., 1997; Sleder et al., 1980; Havel, 2005; Petersen et al., 2007). Despite a clear relationship between fructose and increased hepatic lipogenesis, the mechanisms responsible for this association remain poorly defined.
Hepatic lipogenesis is regulated by the action of sterol regulatory element binding proteins (SREBPs) transcription factors, especially SREBP-1c, the principal inducers of hepatic lipogenic gene expression (Shimano et al., 1997). Dietary fructose, per se, activates lipogenesis partly through inducing expression of SREBP-1 (Nagai et al., 2002). Among strains of inbred mice, a genetic polymorphism within the SREBP-1c promoter region underlies a differential sensitivity to fructose-induced lipid synthesis, further suggesting that this transcription factor mediates the lipogenic effects of fructose (Nagata et al., 2004). Although there is evidence linking induction of SREBP-1c to fructose induced lipogenesis in vivo, the mechanism responsible for fructose induced alterations in SREBP-1c transcription remains unknown.
Recent studies implicate the peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in regulating liver glucose and lipid metabolism. Though initially described as a co-activator of PPAR-γ (Lin et al., 2002), PGC-1 is now recognized to co-activate multiple other transcription factors. Through these interactions, they can coordinate both hepatic glucose metabolism, via forkhead transcription factor-1 (FOXO-1) (Puigserver et al., 2003), hepatocyte nuclear factor 4 alpha (HNF4α) (Rhee et al., 2003) and muscle-selective transcription factor-2 (MEF-2) (Michael et al., 2001), as well as hepatic lipid metabolism, via peroxisome proliferator-activated receptor-α (PPARα) (Vega et al., 2000), liver X receptor (LXR) (Lin et al., 2005), and SREBP (Lin et al., 2005). PGC-1 β, but not PGC-1α, activates the expression of genes involved in lipogenesis and triglyceride secretion via direct coactivation of SREBPs (Lin et al., 2005). Thus, PGC-1β, a regulator of both carbohydrate and lipid metabolism, is poised to play a pivotal role in fructose induced lipogenesis.
We hypothesized that PGC-1 β mediates fructose-induced lipogenesis via upregulation of SREBP-1c and knockdown of PGC-1β would prevent this from occurring. To test this hypothesis, we knocked down the expression of PGC-1 β using antisense oligonucleotides (ASO) in chronically (4 weeks) fructose-fed rats. Antisense oligonucleotides are ideal for these studies as they can target both liver and adipose tissues in adult animals. Our studies reveal an important role for PGC-1 β in the pathogenesis of fructose-induced insulin resistance and suggest that PGC-1 β inhibition may be a novel therapeutic target for treatment of NAFLD, hypertriglyceridemia and insulin resistance.
Results
PGC-1β ASO decreases PGC-1β expression in rat livers and white adipose tissue
Rats were divided into two diet groups and two ASO treatment groups. They received either a regular chow diet, which provided 60% calories from carbohydrate, predominantly in the form of starch or a high-fructose diet which provided 66.8% calories from fructose. Within each diet group, rats were treated with either control ASO or PGC-1β ASO at 25 mg/kg body weight twice weekly for 4 weeks. Caloric intake and weight gain were similar in all treated groups. Compared to control ASO, PGC-1β ASO treatment reduced PGC-1β mRNA expression by approximately 75% in the liver (Figure 1A) and 70% in the white adipose tissue (WAT) (Figure 1B). The reduction in mRNA with PGC-1β ASO translated into a similar reduction in PGC-1β protein (Figure 1C and 1D). In contrast, PGC-1β ASO did not significantly reduce expression of PGC-1β in either brown adipose tissue (BAT) or skeletal muscle (Figure 1E and 1F). Finally, rats remained free of hepatotoxicity, as reflected by the absence of alanine amino transaminase (ALT) elevation in all 4 groups (Table).
Figure 1. PGC-1β ASO is effective and well tolerated.
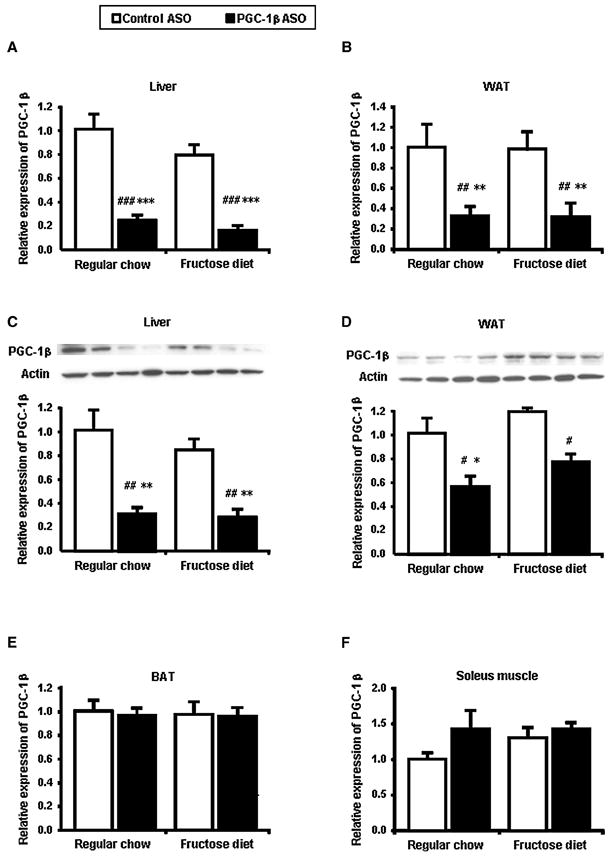
The amount of PGC-1β mRNA was determined by quantitative RT-PCR after 4 weeks of feeding with fructose and treatment with a control ASO, and an ASO against PGC-1β (PGC-1β ASO) in liver (A), epididymal white adipose tissue (WAT) (B), brown adipose tissue (BAT) (E) and soleus muscle (F). PGC-1β protein levels in liver (C) and WAT (D). Two separate samples are shown for each group fraction. Data are means ± SE. Data are expressed as relative to expression in regular chow fed control ASO treated rats (n=7-9 rats per treatment group). #P < 0.05, #P < 0.01, #P < 0.001 versus regular chow fed control ASO treated rats. *P < 0.05, **P < 0.01, ***P < 0.001 versus high-fructose fed control ASO treated rats.
Table.
Physiologic and plasma parameters in control and PGC-1β ASO treated rats
| Regular chow | Fructose chow | |||
|---|---|---|---|---|
| Control ASO | PGC-1 β ASO | Control ASO | PGC-1 β ASO | |
| Physiologic parameters
| ||||
| Body weight (g) | 356 ± 6 | 344 ± 7 | 363 ± 5 | 343 ± 6 |
| Epididymal fat mass (g) | 3.82 ± 0.21 | 4.57 ± 0.36* | 6.16 ± 0.40### | 5.33 ± 0.18# |
|
| ||||
| Plasma metabolites
| ||||
| Glucose (mg/dl) | 117.5 ± 1.8 | 112.9 ± 1.7*** | 129.2 ± 2.6### | 113.3 ± 1.6*** |
| Triglyceride (mg/dl) | 49.2 ± 4.1 | 52.4 ± 5.6** | 92.0 ± 10.4### | 55.2 ± 3.9** |
| Total cholesterol (mg/dl) | 64.1 ± 4.1 | 88.1 ± 5.8##,* | 68.9 ± 3.1 | 108.7 ± 5.4###,*** |
| HDL cholesterol (mg/dl) | 23.1 ± 0.9 | 32.1 ± 1.4##,* | 25.3 ± 0.9 | 41.3 ± 2.3###,*** |
| Nonesterified fatty acid (meq/l) | 1.04 ± 0.06 | 1.22 ± 0.10 | 1.50 ± 0.12## | 1.39 ± 0.10 |
| β-hydroxybutyrate (mmol/l) | 0.84 ± 0.05 | 1.18 ± 0.07# | 1.11 ± 0.08 | 1.23 ± 0.08## |
| ALT (units/l) | 47.1 ± 5.2 | 55.8 ± 6.0 | 38.9 ± 3.8 | 43.8 ± 3.4 |
|
| ||||
| Plasma hormones and adipocytokines
| ||||
| Insulin (mU/l) | 10.4 ± 1.1 | 8.0 ± 1.4*** | 19.8 ± 2.1### | 12.5 ± 1.0** |
| Glucagon (pg/ml) | 46.9 ± 8.8 | 51.9 ± 5.4 | 40.5 ± 5.0 | 54.6 ± 5.4 |
| Adiponectin (mg/ml) | 2.82 ± 0.07 | 3.11 ± 0.13 | 3.15 ± 0.04 | 2.78 ± 0.11 |
| Leptin (ng/ml) | 1.02 ± 0.19 | 0.74 ± 0.15 | 1.82 ± 0.28 | 0.91 ± 0.12* |
| RBP4(μg/ml) | 43.5 ± 4.33 | 35.1 ± 3.92 | 45.7 ± 2.99 | 53.2 ± 4.24 |
Data are means ± SE; n=~25 rats in each group.
P < 0.05,
P < 0.01,
P < 0.001 vs. regular chow fed control ASO treated rats.
P < 0.05,
P < 0.01,
P < 0.001 vs. high-fructose fed control ASO treated rats.
PGC-1β ASO improves metabolic parameters induced by high-fructose feeding
To evaluate the effect of knocking down PGC-1β on whole-body physiology, we performed detailed metabolic characterization, including measures of morphology, serum chemistry as well as glucose and lipid kinetics. As shown in the Table, epididymal fat mass and plasma concentrations of glucose, triglyceride, fatty acids, insulin, leptin were significantly higher in the high-fructose fed rats. Knockdown of PGC-1β protected rats from all these fructose-induced changes. Compared with the rats fed high-fructose diet, the plasma glucose and insulin levels were significantly lower in PGC-1β ASO treated rats by 12% and 37%, respectively. The plasma triglyceride and leptin levels and epididymal fat mass were also significantly lower by 40%, 50% and 13%, respectively. On the other hand, plasma concentrations of total cholesterol, HDL cholesterol and β-hydroxybutyrate were significantly higher in PGC-1β ASO treated rats by 58%, 63% and 11%, respectively. Finally, there were no differences in plasma concentrations of glucagon, adiponectin, or retinol binding protein 4 (RBP4).
The changes in plasma triglyceride and cholesterol concentrations seen with PGC-1β ASO treatment prompted further analysis of their respective pathways. Using primary hepatocytes, we quantified the impact of PGC-1β inhibition on lipid and sterol synthesis in vitro. Specifically, the synthesis was assessed by measuring the incorporation of 14C acetic acid into fatty acid and cholesterol. PGC-1β ASO inhibited the synthesis of fatty acids (Figure 2A) but not of cholesterol (Figure 2B). The decreased lipid synthesis in vitro was reflected in vivo where PGC-1β ASO prevented the accumulation of liver triglyceride observed with high fructose feeding (Figure 2C).
Figure 2. PGC-1β ASO ASO reduces fatty acid synthesis in vitro and triacylglycerol content in vivo.
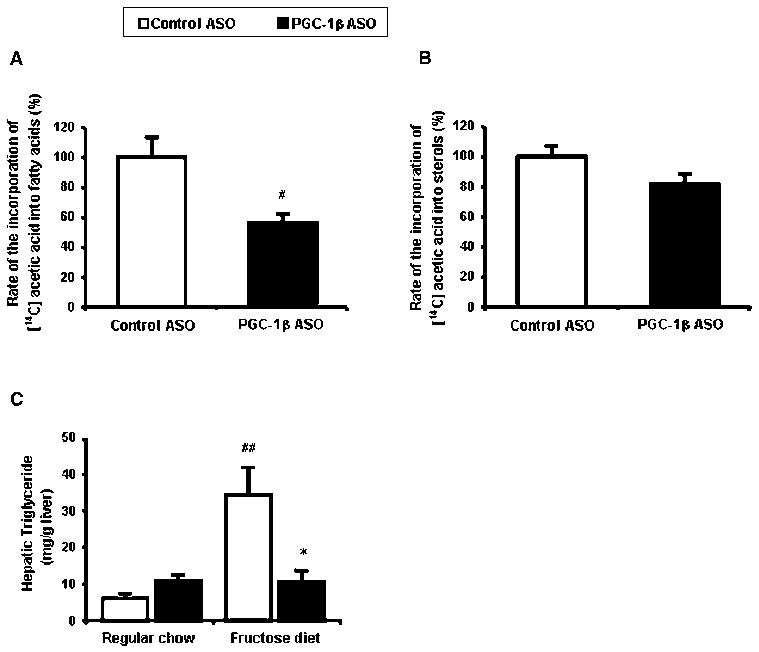
Synthesis of fatty acids (A) and sterols (B) in rat hepatocytes. #P < 0.05 versus control ASO. (C) Hepatic triglyceride content. Data are means ± SE. (n=11-14 rats per treatment group) #P < 0.01 versus regular chow fed control ASO treated rats, *P < 0.05 versus high-fructose fed control ASO treated rats.
PGC-1β ASO reduces lipigenic gene expression
Gene expression was profiled by RT-qPCR to determine what genes were affected by PGC-1β ASO in fructose fed rats. Under fasting condition, SREBP-1c and -a mRNA levels were similar among the four groups (Figure 3A and 3B). Though with refeeding, SREBP-1c expression increased in all groups and SREBP-1a expression increased in high-fructose fed groups; there were marked differences in the degree of induction. SREBP-1c and -1a expression increased with feeding over 5 times and 1.5 times over basal levels respectively. This exaggerated induction of SREBP-1c and -1a by fructose was attenuated by 40% with PGC-1β ASO treatment (Figure 3A). The changes in SREBP-1s expression were confirmed by quantifying SREBP-1 protein with western blot analysis (Figure 3C). The decreased induction of SREBP-1 with PGC-1β ASO was reflected in decreased activation of fatty acid synthase (FAS) gene by fructose (Figure 3D).
Figure 3. Reduced expression of lipogenic genes in liver of PGC-1β ASO treated rats.
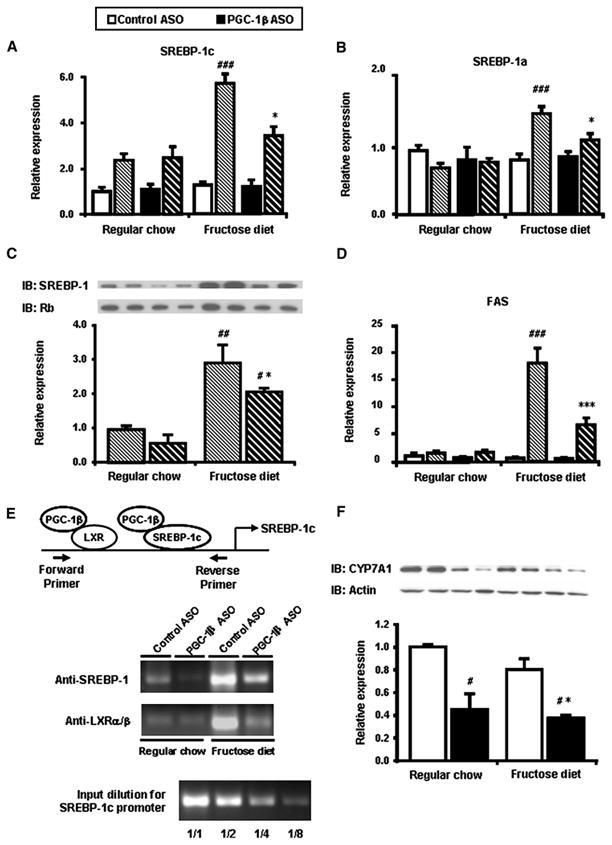
Total mRNA was isolated from fasted and fed liver and quantified using primers for SREBP-1c (A) and -1a (B). (C) Representative immnoblots of liver homogenates from liver in fed state. Two separate samples are shown for each group. (D) Total mRNA was isolated from fasted and fed liver and quantified using primers for key enzymes in fatty acid synthase (FAS). (E) Chromatin immunoprecipitation (ChIP) assay of SREBP-1c gene promoter. After cross-linking chromatin DNA to the interacting proteins, specific immunoprecipitation with anti-SREBP-1 or anti-LXRα/β antibodies was performed as described in EXPERIMENTAL PROCEDURES. PCR products of the SREBP-1c promoter were analyzed after PCR amplification (33 cycles). The lower part of the figure shows verification of the quantitative aspect of the PCR amplification using serial dilutions of the input. (F) Liver gene expression of CYP7A1. Data are means ± SE. (n=7-9 rats per treatment group) Solid bars represent fasted values and striped bars represent fed values. #P < 0.05, #P < 0.01, #P < 0.001 versus regular chow fed control ASO treated rats. *P < 0.05, **P < 0.01, ***P < 0.001 versus high-fructose fed control ASO treated rats.
It has been reported that rat SREBP-1c promoter contains binding sites for SREBP (Cagen et al., 2005) and LXR (Dif et al., 2006) which are both co-activated by PGC-1β (Lin et al., 2005). We next examined whether the binding of SREBP-1 or LXR to the SREBP-1c promoter was affected by PGC-1β ASO treatment. Formaldehyde treatment of fresh liver samples was used to cross-link the transcription factors with the chromosomal DNA. Chromatin immunoprecipitation (ChIP) assays were performed using anti-SREBP-1 (H-160) or anti-LXRα/β (S-20) antibodies to immunoprecipitate fragments of DNA where there was interaction. Figure 3E shows the amplification by PCR (33 cycles) of a specific portion of the proximal promoter of the SREBP-1c gene. we quantified the PCR products using real-time PCR and confirmed that the ampification of the negative control using normal rabbit IgG was less than 0.05% of that the input sample (data not shown). Moreover we also confirmed that there was no difference in LXR mRNA and protein expression among all four groups (Supplemental table1 and data not shown). When rats were refed with a high-fructose diet, there was a marked increase in the association of both SREBP-1 and LXRα/β with the SREBP-1c promoter. PGC-1β ASO decreased both bindings. These data were consistent with SREBP-1c mRNA (Figure 3A) and protein (Figure 3C) expressions.
Thus, the ability of fructose to activate a program of lipogenesis via SREBP-1 was inhibited by PGC-1β ASO.
While PGC-1β ASO decreased liver and plasma triglyceride concentrations in fructose-fed rats, plasma cholesterol concentrations were increased in both diet groups (Table). We also measured the expression of key genes that regulate cholesterol metabolism. Neither high-fructose feeding nor PGC-1β ASO treatment increased expression of SREBP-2 or downstream targets such as HMG-CoA reductase (HMGCoAR) and LDL-receptor (Supplemental table 1). LXRα, a nuclear receptor that balances cholesterol and bile metabolism (Kalaany et al., 2005), is also coactivated by PGC-1s (Lin et al., 2005). Although LXRα expression was unchanged by either diet or ASO treatment, (Supplemental table 1), target gene, cholesterol-7α-hydroxylase (CYP7A1) protein expression was significantly decreased in all PGC-1β ASO treated groups (Figure 3F). CYP7A1 converts cholesterol into 7α-hydroxycholesterol and responsible for the rate-limiting step in the classical bile acid synthesis pathway. Thus, CYP7A1 mediates clearance of cholesterol from the circulation and it is possible that the decrease in CYP7A1 protein expression is responsible for the observed increased concentrations of cholesterol in the PGC-1β ASO treated rats. Some other target genes of LXR (SR-B1, ABCG8) were had decreased transcription by PGC-1β ASO treatment (supplemental table 1), however there was no difference in protein expression (data not shown).
PGC-1β ASO protects high-fructose fed but not regular chow fed rats from hepatic insulin resistance
PGC-1β ASO lowered fasting plasma glucose and insulin concentration in high fructose-fed rats, suggesting improved insulin sensitivity. We performed hyperinsulinemic-euglycemic clamps in order to further understand the mechanisms of improved insulin sensitivity. The basal rate of hepatic glucose production (HGP) was significantly increased in high-fructose fed rats compared with regular chow fed rats (Figure 4A). In fructose fed rats, PGC-1β ASO treatment decreased HGP, along with plasma glucose and insulin concentrations.
Figure 4. PGC-1β ASO treatment results in improved fructose induced hepatic insulin resistance but also induces hepatic insulin resistance in regular chow fed rats.
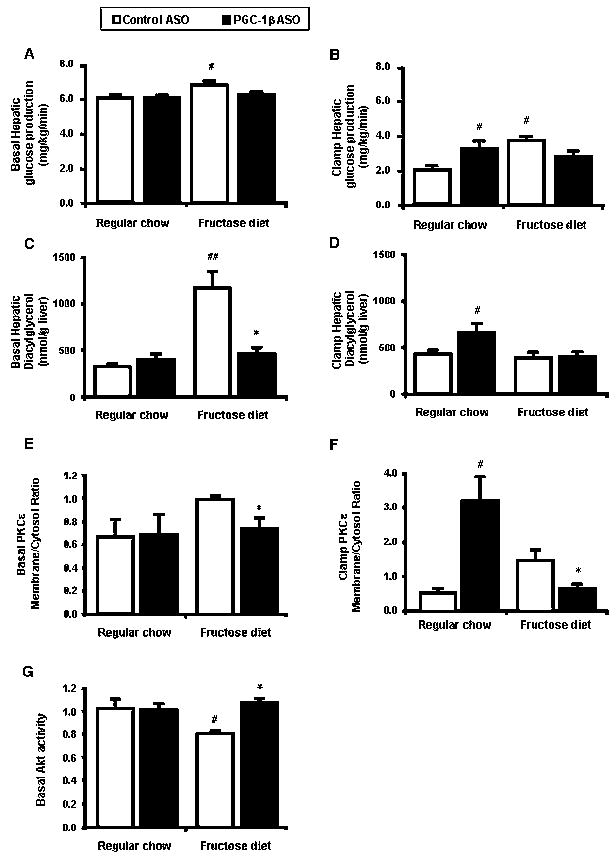
Rate of basal (A) and clamp (B) endogenous glucose production. Hepatic total diacylglycerol obtained from basal (C) and clamp (D) liver. Representative Western blots showing basal (E) and clamp (F) membrane to cytosol fractions ratio for PKCε. The relative densities of the bands in the membrane fraction were compared with the cytosol fraction to derive a quantifiable measure of activation. (G) Basal Akt activity in liver. Data are means ± SE. (n=6-20 rats per treatment group) #P < 0.05 versus regular chow fed control ASO treated rats. *P < 0.05 versus high-fructose fed control ASO treated rats.
Under hyperinsulinemic-euglycemic conditions, plasma insulin concentrations were raised to approximately 80 mU/l while plasma glucose levels were maintained at approximately 5.5 mM in all groups with a variable infusion of glucose (data not shown). Under these conditions, HGP was significantly higher in high-fructose fed rats (Figure 4B). PGC-1β ASO treatment improved the ability of insulin to suppress HGP in high-fructose fed rats (Figure 4B).
We measured hepatic diacylglycerol (DAG) which has previously been associated with insulin resistance (Samuel et al., 2004). In the basal state, DAG concentration was markedly increased in high-fructose fed rats (Figure 4C). PGC-1β ASO treatment decreased DAG concentrations 60% (Figure 4C). In the past, we have shown that DAG accumulation leads to PKCε activation (Samuel et al., 2007). The decreased DAG concentrations seen with PGC-1β ASO were associated with decreased PKCε membrane to cytosolic ratio (Figure 4E) and increased hepatic Akt activity (Figure 4G).
In contrast with fructose feeding, HGP was paradoxically higher in the regular chow fed PGC-1β ASO treated rats (Figure 4B). Hepatic DAG concentration was significantly increased by ~2-fold in regular chow fed PGC-1β ASO treated rats after insulin stimulation (Figure 4D). PKCε membrane to cytosolic ratio was increased over 6 times in regular chow fed PGC-1β ASO treated rats after insulin stimulation (Figure 4F).
The unexpected hepatic insulin resistance seen in the regular chow groups is consistent with previous studies in mice lacking exon 3 to 4 of the PGC-1β gene (PGC-1 β E3,4-/E3,4- mice ) (Vianna et al., 2006). In these mice, defects in mitochondrial oxidative capacity were implicated in the development of insulin resistance. PGC-1β have been reported to coactivate both PPAR-α (Lin et al., 2003) which plays a key role in control of fatty acid oxidation and nuclear respiratory factor (NRF)-1 (Wu et al., 1999) which regulates proteins required for mitochondrial function (Scarpulla, 2002). PGC-1β ASO significantly decreased PPARα mRNA expression and its target genes LCAD, MCAD and CPT1a in both regular chow fed and high-fructose fed rats by ~30% (Figure 5A and 5B). NRF-1 mRNA expression was significantly decreased by 40% (Figure 5C). Consistent with this reduction, mitochondrial copy numbers were also significantly reduced by 30% in PGC-1β ASO treated groups (Fugure 5D). Additionally, PGC-1β ASO decreased expressions of ATPase synthase subunit 6 (ATP6), a mitochondrial encoded gene and cytochrome C, a nuclear encoded mitochondrial gene (Figure 5E).
Figure 5.
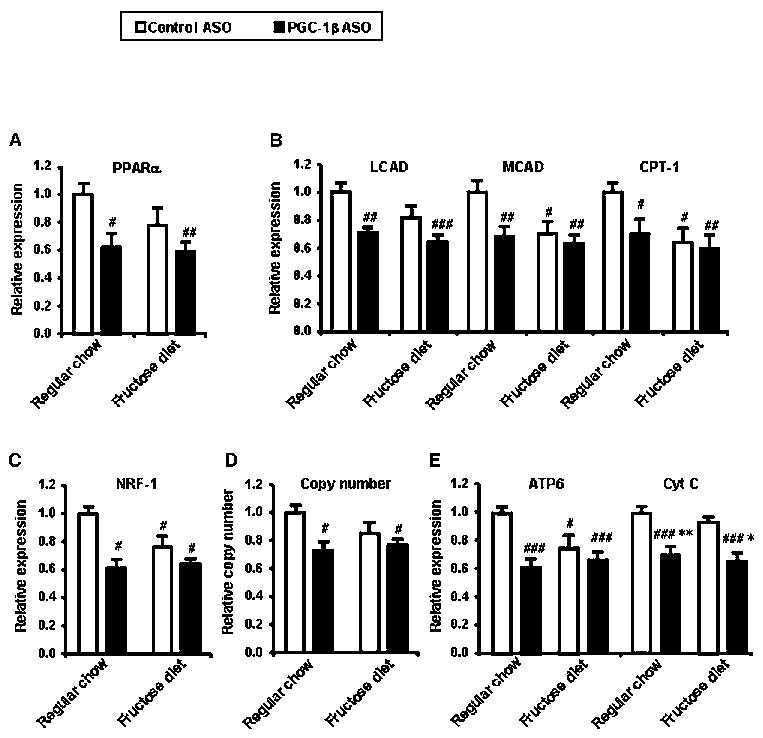
Liver gene expression of PPARα (A) and target enzymes in fatty acid oxidation (B). Liver gene expression of NRF-1 (C) and target enzymes in mitochondrial oxidative phosphorylation (E). mtDNA copy number is determined by real-time quantitative PCR using primers for mitochondrial encoded gene (ATP6) and nuclear encoded gene (β-actin) (D). mtDNA copy number is calculated as the ratio of ATP6 to β-actin. Data are means ± SE. (n=6-8 rats per treatment group) #P < 0.05, #P < 0.01, #P < 0.001 versus regular chow fed control ASO treated rats. *P < 0.05, **P < 0.01 versus high-fructose fed control ASO treated rats.
In order to determine whether PGC-1β ASO has any affect to other diet-induced insulin resistance model, we tested PGC-1β ASO on rats fed a high fat diet. Though both fasting plasma glucose and insulin concentrations were significantly decreased in PGC-1β ASO treated high-fat fed rats (Supplemental figure A and B), there was no difference in HGP both under basal and insulin-stimulated conditions (Supplemental figure C). In addition, the degree of induction of SREBP-1c with re-feeding was less than that observed compared to high- fructose fed rats. PGC-1β ASO did not decrease SREBP-1c mRNA expression (Supplemental figure E). The unchanged expression of SREBP-1c was reflected by unchanged DAG accumulation in liver (Supplemental figure E).
PGC-1β ASO treatment improves adipose insulin action in high-fructose fed rats
Insulin-stimulated whole-body glucose disposal was decreased by ~50% in the high-fructose fed rats compared to the regular chow fed rats (Figure 6A). PGC-1β ASO rescued the fructose induced insulin resistance as shown by the 90% increase in insulin-stimulated whole-body glucose uptake.
Figure 6. PGC-1β ASO treatment also results in improved peripheral insulin sensitivity due to an increase in white adipose tissue glucose uptake in high-fructose fed rats.
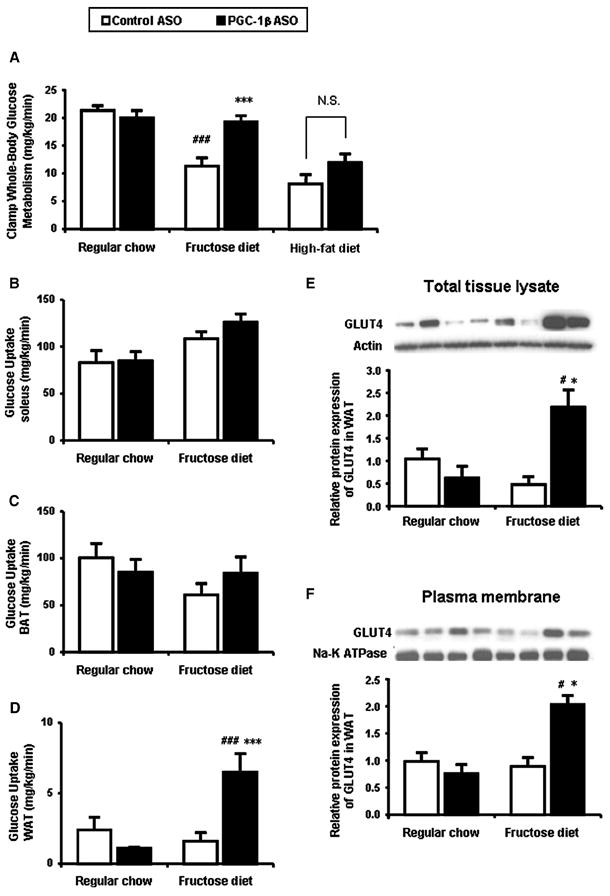
(A) Rate of insulin-stimulated whole-body glucose uptake. (B) Soleus 14C-2-deoxyglucose (2DG) uptake. (C) Brown adipose tissue (BAT) 2DG uptake. (D) Epididymal white adipose tissue (WAT) 2DG uptake. Western blotting of extract from total tissue (E) and plasma membrane fraction (F) using the GLUT4-specific mouse monoclonal antibody (1F8). Two separate samples are shown for each group fraction. Data are means ± SE. (n=6-20 rats per treatment group) #P < 0.001 versus regular chow fed control ASO treated rats; *P < 0.05, ***P < 0.001 versus high-fructose fed control ASO treated rats.
In order to determine the organs response to the increased insulin stimulated glucose uptake seen in high-fructose fed PGC-1β ASO treated rats, we assessed 14C-2 deoxyglucose uptake in muscle and adpose tissue. There was no significant increase in either muscle or BAT 14C-2 deoxyglucose uptake (Figure 6B and 6C). However, in high-fructose fed PGC-1β ASO treated rats, glucose uptake in the epididymal WAT was increased nearly 4-fold compared with that in the other 3 groups (Figure 6D). The augmented adipose insulin sensitivity is corroborated by a decreased plasma fatty acid concentration during the hyperinsulinemic-euglycemic clamps (data not shown). To further explore the mechanism for the increased WAT glucose uptake, we first examined the gene expression of PPAR-γ and its target genes in epididymal WAT. Surprisingly, the expression of PPAR-γ was decreased by 50% and adipocyte protein 2 (aP2), adiponectin and acyl-CoA synthetases (ACS) were decreased in PGC-1β ASO treated groups (Supplemental table 2). PGC-1β also induces mRNA expression of GLUT4 in cultures of primary rat skeletal muscle cells (Mortensen et al., 2006). In the present study, GLUT4 mRNA in WAT was decreased by 50% in PGC-1β ASO treated groups (Supplemental table 2). In contrast, the protein expression in both total tissue lysate and plasma membrane was increased by ~2-fold in only PGC-1β ASO treated high-fructose fed rats (Figure 6E and 6F), consistent with the result of 2DG uptake in WAT. There were no differences in the expression of other glucose transporters (GLUT1 and GLUT5 (known as a fructose transporter) (data not shown).
To investigate the cause of this dissociation between GLUT4 protein and mRNA expression, we evaluated the expression of proteins that regulate GLUT4 trafficking and stability. However, there were no results consistent with the GLUT4 protein expression, although many of these genes were decreased by the PGC-1β ASO treatment (Supplemental table 2). Furthermore, one report suggests that GLUT4 can be degraded by calpain-2 (Otani, 2004). But calpain 1/2 activity was similar across all groups (data not shown).
Interestingly, this amelioration of diet induced insulin resistance by PGC-1β ASO was not seen in high fat fed rats (Figure 6A). Consistent with this result, GLUT4 protein expression in WAT was not increased in high-fat fed rats (Supplemental Figure S1F).
Discussion
It is becoming increasingly clear that PGC-1β plays an important role as a regulator of both carbohydrate and lipid metabolism. In the present study we evaluated the metabolic effects of PGC-1β knockdown in rats with fructose-induced insulin resistance. Here we show for the first time that hepatic de novo lipogenesis and hepatic triglyceride synthesis induced by fructose are both decreased by PGC-1 β ASO treatment. Furthermore we show that knockdown of PGC-1β prevents fructose-induced hypertriglyceridemia and hepatic and peripheral insulin resistance.
PGC-1 β has been reported to coactivate its partners through augmentation of their transcriptional activity (Lin et al., 2005). Though initially described to pair with PPARγ, the number of partners for PGC-1 transcription factors is rapidly growing. One of its partners is SREBP-1c. The transcriptional control of this key regulator of lipogensis is complex. It has been reported that rat and human SREBP-1c promoter contains binding sites for both SREBP itself (Cagen et al., 2005) and LXR (Dif et al., 2006) which are both regulated by PGC-1β (Lin et al., 2005).
The ChIP assay data showed a marked increase in the association of both SREBP-1 and LXRα/β with the SREBP-1c promoter on a high-fructose diet. The bindings were both decreased by PGC-1β ASO treatment. Undoubtedly, the binding of SREBP-1 to SREBP-1c promoter might be reflected with the amount of SREBP-1 total protein, but the binding of LXR to SREBP-1c promoter was also decreased by PGC-1β ASO treatment. LXR protein was unchanged between all groups implying the decreased binding to the SREBP-1c promoter was due to the decreased coactivation with PGC-1β. Thus, this data suggests that PGC-1β ASO treatment reduces SREBP-1c expression by decreased co-activation of LXR and SREBP.
Consistent with the reduction of SREBP-1 expression, a target lipogenic gene of SREBP-1, FAS was down-regulated in high-fructose fed PGC-1β ASO treated rats. This likely accounted for the observed decreased in hepatic de novo lipogenesis, hepatic triglyceride content and reductions in hypertriglyceridemia in the high-fructose fed PGC-1β ASO treated rats. Supporting these observations, we found that in vitro hepatic triglyceride synthesis was significantly inhibited in primary rat hepatocytes by PGC-1β ASO treatment.
Knockdown of PGC-1β in liver protected rats from fructose-induced hepatic insulin resistance. This protection from fructose-induced hepatic insulin resistance could mostly be attributed to reduction in hepatic lipogenesis resulting in reduced hepatic diacylglycerol content and decreased PKCε activation. These data were also reflected in Akt activity.
In marked contrast to these findings, the effects of PGC-1β ASO on the liver differed in the regular chow and high fructose-fed rats. In regular chow fed rats PGC-1β ASO induced slight, but significant reductions in insulin suppression of hepatic glucose production. These reductions in insulin responsiveness could be attributed to decreased mitochondrial fatty acid oxidation resulting in increased diacylglycerol content and increased PKCε activation resulting in decreased insulin signaling (Samuel, 2007). PGC-1s have been reported to coactivate both PPAR-α and NRF-1. PPARα plays a key role in the transcriptional control of genes encoding mitochondrial fatty acid oxidation enzymes such as LCAD and MCAD. NRFs regulate expression of mitochondrial transcription factor A (Tfam), a nuclear-encoded transcription factor essential for replication, maintenance, and transcription of mitochondrial DNA. NRFs also control the expression of nuclear genes encoding respiratory chain subunits and other proteins required for mitochondrial function (Scarpulla, 2002). PGC-1β ASO decreased expression of genes encoding fatty acid oxidation and oxidative phosphorylation as well as mitochondrial copy number, suggesting that PGC-1β is required in liver for normal expression of genes encoding fatty acid oxidation and oxidative phosphorylation. These results are consistent with previous studies in PGC-1 β E3,4-/E3,4- mice (Vianna et al., 2006) and LCAD knockout mice, which both develop hepatic steatosis and hepatic insulin resistance due to decreased hepatic fat oxidation resulting in increased hepatic diacylglycerol content and increased PKCε activation (Zhang et al., 2007).
PGC-1β ASO also prevented fructose-induced insulin resistance in peripheral tissue. This preservation of insulin responsiveness could be attributed largely to a threefold increase in insulin stimulated WAT glucose uptake. Since the PGC family members are known coactivators of PPARs, we examined the expression of PPAR-γ and its target genes. The expression of PPARγ itself, as well as key target genes were all reduced with PGC-1β ASO treatment and could not explain the observed improvements in insulin action.
We next investigated GLUT4 expression and found that the GLUT4 protein expression in WAT was increased by more than fourfold in the high-fructose fed PGC-1β ASO treated rats. This was compatible with our findings in the glucose uptake, although the mRNA expression of GLUT4 was surprisingly decreased by PGC-1β ASO treatment. A similar dissociation between GLUT4 protein and mRNA expression has been reported by Otani et al. (Otani et al., 2004). We believe this result may reflect enhanced stability of GLUT4 protein, leading to its accumulation. However, in the present study, we did not find any explanations in the gene expression of GLUT4 trafficking proteins. Further studies are required to explain why the increases of 2DG uptake and GLUT4 protein expression were seen in only high-fructose fed PGC-1β ASO treated rats.
We tested PGC-1β ASO on rats fed a high fat diet to understand if the protection from diet induced insulin resistance was specific for high-fructose diet. PGC-1β ASO failed to ameliorate both hepatic and peripheral insulin resistance in high fat fed rats. The degree of SREBP-1 induction with high-fat diet was less than that with high-fructose diet, and PGC-1β ASO did not affect to the SREBP-1 mRNA expression on high fat diet. Consequently, the unaffected lipogenesis resulted in similar hepatic lipid content between control ASO and PGC-1β ASO group. These findings raise the possibility that fructose can directly induces the transcriptional activity of SREBP-1 via PGC-1β.
Interestingly we also found that PGC-1β ASO treatment increased plasma cholesterol concentrations. We did not observe any increases in the expression of SREBP-2, a key transcriptional regulator of cholesterol biosynthesis or in the SREBP2 responsive genes HMGCoAR or LDL-R following PGC-1β ASO treatment. These data suggest that cholesterol biosynthesis in liver and LDL uptake from plasma by liver were unlikely to be responsible for the observed increases in plasma cholesterol in the PGC-1β ASO treated groups. Consistent with these observations we found that reduction in PGC-1β expression in primary rat hepatocytes had no effect on the incorporation rate of 14C acetic acid into sterol. We next investigated the effects of PGC-1β knockdown on the expression of LXRα and its target genes. LXRα has been shown to be an important regulator of cholesterol (Kalaany et al., 2005) and is co-activated by PGC-1s (Lin et al., 2005). Although we found that LXRα expression was not different in the four groups, hepatic CYP7A1 expression was decreased in both PGC-1β ASO treated groups. Since CYP7A1 is responsible for the rate-controlling step in the bile acid synthesis pathway, it is likely that this reduction in CYP7A1 may explain the increased plasma cholesterol concentrations in the PGC-1β ASO treated rats.
In conclusion these data support an important role for PGC-1 β in the pathogenesis of fructose-induced hypertriglyceridemia and insulin resistance. Furthermore, given recent studies suggesting an important role for increased hepatic de novo lipogenesis in the pathogenesis of hypertriglyceridemia and NAFLD associated with the metabolic syndrome (Petersen et al., 2007) these data suggest that PGC-1 β inhibition may be a novel therapeutic target for treatment of this condition.
EXPERIMENTAL PROCEDURES
Animals
All rats were maintained in accordance with the Institutional Animal Care and Use Committee of Yale University School of Medicine. Healthy male Sprague-Dawley rats weighing ~200 g were obtained from Charles River Laboratories and acclimated for 1 week after arrival before initiation of the experiment. Rats received water ad libitum and were maintained on a 12:12 h light/dark cycle (lights on at 6:00 a.m.). They received either regular rodent chow (60% carbohydrate, 10% fat, 30% protein calories) , a high fructose diet (66.8% carbohydrate, 13% fat, 20.2% protein calories, TD.89247; Harlan Teklad, WI) , or a high fat diet (24.0% carbohydrate, 54.8% fat, 21.2% protein calories, TD.93075; Harlan Teklad, WI) three times per week at a weight of 225g per week for 4 weeks. Intraperitoneal (i.p.) ASO therapy was initiated 3 days after commencing the high fructose diet. All ASOs (control and PGC-1 β) were prepared in normal saline, and the solutions were sterilized through a 0.2-μm filter. Rats were dosed with ASO solutions twice per week via i.p. injection at a dose of 50 mg/kg per week for 4 weeks. During the treatment period, body weight was measured weekly. After 14–20 days of ASO treatment, rats underwent the placement of jugular venous and carotid artery catheters. They recovered their presurgical weights by 5–7 days after the operation.
Selection of Rat PGC-1 β ASOs
To identify rat PGC-1 β ASO inhibitors, rapid throughput screens were performed in vitro as described previously (Qu et al., 1999). In brief, 80 ASOs were designed to the rat PGC-1 β mRNAs sequences and initial screens identified several potent and specific ASOs, all of which targeted a binding site within the coding region of the PGC-1 β mRNAs. After extensive dose response characterization, the most potent ASO from the screen was chosen: ISIS-384891, with the following sequence: 5-CTGGAAGTCCTGGGAGACAC-3. The control ASO, ISIS-141923, has the following sequence, 5-CCTTCCCTGAAGGTTCCTCC-3, and does not have perfect complementarily to any known gene in public data bases.
Hyperinsulinemic-euglycemic clamp studies
After 4 weeks of high-fructose feeding, rats were fasted overnight. The following morning, the clamp study began with a prime (1 mg/kg for 8 minutes) of [6,6-2H]glucose followed by a continuous infusion at a rate of 0.1 mg/kg per minute for 2 hours to assess the basal glucose turnover. After the basal period, the hyperinsulinemiceuglycemic clamping was conducted for 140 minutes with a primed/continuous infusion of human insulin (400 mU/kg prime for 5 minutes, 4 mU/kg per minute infusion) (Novo Nordisk) and a variable infusion of 20% dextrose to maintain euglycemia (approximately 100 mg/dl). The 20% glucose was enriched with [6,6-2H]glucose to approximately 2.5% to match the enrichment in the plasma achieved after the basal period. A 30 μCi bolus of 2-deoxy-d-[1-14C] glucose (PerkinElmer) was injected 120 minutes into the clamp to estimate the rate of insulin-stimulated tissue glucose uptake. At the end of the clamping, rats were anesthetized with pentobarbital sodium injection (150 mg/kg), and all tissues were taken within 4 minutes, frozen immediately with the use of liquid N2–cooled aluminum tongs, and stored at −80°C for subsequent analysis.
Biochemical analysis and calculations
Plasma glucose was analyzed during the clamping with the use of 10 μl plasma by a glucose oxidase method on a Beckman Glucose Analyzer II (Beckman Coulter). Plasma insulin and adiponectin were measured by RIA using kits from Linco. Plasma leptin was measured using the LINCOplex Assay system (Linco). Plasma RBP4 was measured using EIA kit from ALPCO.
Plasma fatty acid concentrations were determined using an acyl-CoA oxidase–based colorimetric kit (Wako). To determine the enrichment of [6, 6-2H] glucose in plasma, samples were deproteinized with 5 volumes of 100% methanol, dried, and derivatized with 1:1 acetic anhydride/pyridine to produce the pentacetate derivative of glucose. The atom percentage enrichment of glucoseM+6 was then measured by gas chromatographic/mass spectrometric analysis using a Hewlett-Packard 5890 gas chromatograph interfaced to a Hewlett-Packard 5971A mass-selective detector operating in the electron ionization mode (Hundal et al., 2002). GlucoseM+2 enrichment was determined from the m/z ratio 202:200. Rates of basal and insulin-stimulated whole-body glucose turnover were determined as the ratio of the rate of [6, 6-2H] glucose infusion (mg/kg per min) to the atom percentage excess glucoseM+2 (%) in the plasma. This rate was corrected by subtraction of the rate of [6, 6-2H] glucose infusion. Hepatic glucose production was determined by subtraction of the glucose infusion rate from the rate of total glucose appearance. For the determination of muscle and WAT 14C-2-deoxyglucose-6-phosphate content, tissue samples were homogenized, and the supernatants were subjected to an ion-exchange column to separate 14C-2-deoxyglucose-6-phosphate from 2-deoxyglucose as previously described (Youn et al., 1993).
Tissue lipid measurement
The DAG extraction and analysis were performed as previously described (Yu et al., 2002). After purification, DAG fractions were dissolved in methanol/H2O (1:1, vol/vol) and subjected to liquid chromatography-tandem mass spectrometry (LC/MS/MS) analysis. A Turbo Ion Spray source was interfaced with an API 3000 Tandem Mass Spectrometer (Applied Biosystems) in conjunction with 2 PerkinElmer Series 200 Micro Pumps and a PerkinElmer Series 200 Autosampler. Total DAG content is expressed as the sum of individual species. Tissue triglyceride was extracted by the method of Bligh and Dyer (Bligh et al., 1959) and measured with the use of a DCL Triglyceride Reagent (Diagnostic Chemicals Ltd.).
Total RNA preparation and RT-PCR analysis
Total RNA was extracted from liver samples using the RNeasy kit (QIAGEN). RNA was reverse-transcribed into cDNA with the use of M-MuLV Reverse Transcriptase (New England Biolabs). The abundance of transcripts was assessed by real-time PCR on an Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems) with a SYBR Green detection system. For each run, samples were run in duplicate for both the gene of interest and actin. The expression data for each gene of interest and actin were normalized for the efficiency of amplification, as determined by a standard curve included on each run (Pfaffl, 2001).
mtDNA content
Total DNA was isolated from 20 mg of each basal liver using a QIAamp DNA Micro Kit (QIAGEN). Fifty nanograms of total DNA was used as a template in 20 μl PCR reaction using an Applied Biosystems 7500 Real-Time PCR System with a SYBR Green detection system against ATPase synthase subunit 6 (mitochondrial encoded gene [ATP6]) and β-actin (nuclear encoded gene). mtDNA copy number was presented as a ratio of ATP6 to β-actin as previously described (Bhat et al., 2004).
Determination of fatty acid and sterol synthesis in transfected rat hepatocytes in vitro
Primary rat hepatocytes were isolated as previously described and plated onto collagen-coated 25-cm2 flasks for 60-mm plates for the synthesis measurement. Hepatocytes were treated with ASO (150 nM) and Lipofectin (Invitrogen Corp.) mixture for 4 hours in serum-free William’s E media (Invitrogen Corp.). ASO and Lipofectin were mixed in a ratio of 3 μg of Lipofectin for every 1 ml of 100 nM ASO concentration. After 4 hours, ASO reaction mixture was replaced with normal maintenance media (William’s E media with 10% FBS and 10 nM insulin). The cells were incubated under normal conditions for 20–24 hours, and then fatty acid and sterol synthesis (incorporation of 14C acetic acid into fatty acid and sterol) were measured as described previously (Yu et al., 2005).
Western blotting
For SREBP-1 quantification, nuclear protein ware extracted using NE-PER Nuclear and cytoplasmic Extraction Reagents (PIERCE). The protein extracts were resolved by SDS-PAGE using 4-12% gradient gel (Invitrogen) and electroblotted onto PVDF membrane (DuPont) with the use of a wet transfer cell (Bio-Rad). The membrane was then blocked for 2 hours at room temperature in PBS-Tween (10 mmol/l NaH2PO4, 80 mmol/l Na2HPO4, 0.145 mol/l NaCl, and 0.1% Tween-20, pH 7.4) containing 5% (wt/vol) nonfat dried milk, washed twice, and then incubated overnight with rabbit anti-peptide antibody against SREBP-1 (H-160; Santa Cruz Biotechnology Inc.) diluted 1:100 in rinsing solution. After further washings, membranes were incubated with HRPconjugated IgG fraction of goat anti-rabbit IgG (Bio-Rad), diluted 1:5,000 in PBS-Tween, for 2 hours. These blots were stripped and reblotted with rabbit anti- retinoblastoma-associated protein (Rb) IgG to normalize for variations in protein loading. Blots were scanned and analyzed with ImageJ (NIH).
A separate group of rats was used to assess the impact of hepatic DAG accumulation and PKCε quantification. These rats were treated exactly as described above and underwent 20 minutes of hyperinsulinemic-euglycemic clamping after a basal infusion. Tissues were harvested in situ immediately at the end of the clamping. For PKCε quantification, plasma membrane and cytosol protein ware extracted using ProteoExtract Native Membrane Protein Extraction kit (Calbiochem). Western blotting was performed as described above. 20 μg of homogenized samples were blotted on PVDF membranes. The membrane was incubated overnight with rabbit anti-peptide antibody against PKCε (1:1000, Santa Cruz Biotechnology Inc.). These blots were stripped and reblotted with plasma membrane marker, mouse sodium potassium ATPase IgG (abcam) and rabbit anti-actin IgG and to normalize for variations in protein loading. The polyclonal PGC-1β antibodies were a kind gift provided by Dr. Spiegelman, Harvard Medical School and purchased from NOVUS.
Chromatin Immunoprecipitaion (ChIP) assay
ChIP assays were performed according to the procedure described previously (Gosmain et al., 2005), (Deng et al., 2007) with minor modifications using ChampionChIP One-day Kit (SABiosciences). Fresh liver samples (300 mg) were minced and treated for 15 min with 10 ml formaldehyde (1% final concentration in Dulbecco’s modified Eagle’s medium (DMEM) at room temperature. Cross-linking was stopped by the addition of glycine to a final concentration of 125 mM. Chromatin extracts was prepared from tissue homogenates and further fragmented by sonication. The sonicated chromatin was first precleared for 1 h with protein G Agarose / Salmon Sperm DNA beads (Millipore). After centrifugation, supernatants were incubated overnight at 4°C with 4 μg of an anti-SREBP-1 antibody (H-160; Santa-Cruz Biotechnology), 4 μg of anti-LXRα/β antibody (S-20; Santa-Cruz Biotechnology), or normal-rabbit IgG. The immunoprecipitated DNA/protein complex was bound to protein A-Sepharose beads during 1 h at 4°C and washed in a low-salt buffer, high-salt buffer, LiCl buffer, and Tris-EDTA buffer. Proteins were eliminated using proteinase K for 30 min at 45°C. The DNA was purified and used as a template for PCR. The sets of PCR primers used for the analysis of the rat SREBP-1c proximal promoter were 5-TGGTTGCCTGTGCGGCAG-3 (SREBP1c-Pro-S) and 5-TCAGGCCCCGCCAGGCTTTAA-3 (SREBP1c-Pro-AS). Real-time PCR analyses were performed using Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems). The PCR amplification products were analyzed on ethidium bromide-stained 2% agarose gels.
Akt activity
Liver Akt activity was measured using a commercially available kit (Assay Designs).
Calpain activity
Calpain activity in WAT was determined using a commercially available kit (Calbiochem).
Statistics
Values are expressed as mean ± SEM. The significance of the differences in mean values among different treatment groups was evaluated by two-tailed Student’s t tests or 1-way ANOVA followed by post hoc analysis using the Tukey’s Honestly Significant Differences (HSD) test. P values less than 0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank Aida Grossmann, Yanna Kosover, Todd May, Mario Kahn, Rebecca Pongratz, Gary Cline, Takeshi Yoshizaki and Katsutaro Morino for excellent technical assistance.
This work was supported by grants from the United States Public Health Service (R01 DK-40936 and P30 DK-45735 to G.I. Shulman, K23 RR-17404 to V.T. Samuel, R01 DK-075772 to J.S. Bogan.) and a Distinguished Clinical Scientist Award from the American Diabetes Association. G.I. Shulman is an investigator of the Howard Hughes Medical Institute. V.T. Samuel also receives support from the Veterans Administration Medical Center, West Haven, Connecticut, USA. J.S. Bogan also receives support from the W.M. Keck Foundation.
References
- Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab (Lond) 2005;2:5. doi: 10.1186/1743-7075-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat HK, Epelboym I. Quantitative analysis of total mitochondrial DNA: competitive polymerase chain reaction versus real-time polymerase chain reaction. J Biochem Mol Toxicol. 2004;18:180–186. doi: 10.1002/jbt.20024. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Cagen LM, Deng X, Wilcox HG, Park EA, Raghow R, Elam MB. Insulin activates the rat sterol-regulatory-element-binding protein 1c (SREBP-1c) promoter through the combinatorial actions of SREBP, LXR, Sp-1 and NF-Y cis-acting elements. Biochem J. 2005;385:207–216. doi: 10.1042/BJ20040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CS, Savage DB, Kulkarni A, Yu XX, Liu ZX, Morino K, Kim S, Distefano A, Samuel VT, Neschen S, et al. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J Biol Chem. 2007;282:22678–22688. doi: 10.1074/jbc.M704213200. [DOI] [PubMed] [Google Scholar]
- Daly ME, Vale C, Walker M, Alberti KG, Mathers JC. Dietary carbohydrates and insulin sensitivity: a review of the evidence and clinical implications. Am J Clin Nutr. 1997;66:1072–1085. doi: 10.1093/ajcn/66.5.1072. [DOI] [PubMed] [Google Scholar]
- Deng X, Yellaturu C, Cagen L, Wilcox HG, Park EA, Raghow R, Elam MB. Expression of the rat sterol regulatory element-binding protein-1c gene in response to insulin is mediated by increased transactivating capacity of specificity protein 1 (Sp1) J Biol Chem. 2007;282:17517–17529. doi: 10.1074/jbc.M702228200. [DOI] [PubMed] [Google Scholar]
- Dif N, Euthine V, Gonnet E, Laville M, Vidal H, Lefai E. Insulin activates human sterol-regulatory-element-binding protein-1c (SREBP-1c) promoter through SRE motifs. Biochem J. 2006;400:179–188. doi: 10.1042/BJ20060499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ. Fructose, weight gain, and the insulin resistance syndrome. Am J Clin Nutr. 2002;76:911–922. doi: 10.1093/ajcn/76.5.911. [DOI] [PubMed] [Google Scholar]
- Gosmain Y, Dif N, Berbe V, Loizon E, Rieusset J, Vidal H, Lefai E. Regulation of SREBP-1 expression and transcriptional action on HKII and FAS genes during fasting and refeeding in rat tissues. J Lipid Res. 2005;46:697–705. doi: 10.1194/jlr.M400261-JLR200. [DOI] [PubMed] [Google Scholar]
- Havel PJ. Dietary fructose: implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr Rev. 2005;63:133–157. doi: 10.1301/nr.2005.may.133-157. [DOI] [PubMed] [Google Scholar]
- Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, Shulman GI. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest. 2002;109:1321–1326. doi: 10.1172/JCI14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaany NY, Gauthier KC, Zavacki AM, Mammen PP, Kitazume T, Peterson JA, Horton JD, Garry DJ, Bianco AC, Mangelsdorf DJ. LXRs regulate the balance between fat storage and oxidation. Cell Metab. 2005;1:231–244. doi: 10.1016/j.cmet.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta ), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277:1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- Lin J, Tarr PT, Yang R, Rhee J, Puigserver P, Newgard CB, Spiegelman BM. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J Biol Chem. 2003;278:30843–30848. doi: 10.1074/jbc.M303643200. [DOI] [PubMed] [Google Scholar]
- Lin J, Yang R, Tarr PT, Wu PH, Handschin C, Li S, Yang W, Pei L, Uldry M, Tontonoz P, et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell. 2005;120:261–273. doi: 10.1016/j.cell.2004.11.043. [DOI] [PubMed] [Google Scholar]
- Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, Kelly DP, Spiegelman BM. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci U S A. 2001;98:3820–3825. doi: 10.1073/pnas.061035098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen OH, Frandsen L, Schjerling P, Nishimura E, Grunnet N. PGC-1alpha and PGC-1beta have both similar and distinct effects on myofiber switching toward an oxidative phenotype. Am J Physiol Endocrinol Metab. 2006;291:E807–E816. doi: 10.1152/ajpendo.00591.2005. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Nishio Y, Nakamura T, Maegawa H, Kikkawa R, Kashiwagi A. Amelioration of high fructose-induced metabolic derangements by activation of PPARalpha. Am J Physiol Endocrinol Metab. 2002;282:E1180–E1190. doi: 10.1152/ajpendo.00471.2001. [DOI] [PubMed] [Google Scholar]
- Nagata R, Nishio Y, Sekine O, Nagai Y, Maeno Y, Ugi S, Maegawa H, Kashiwagi A. Single nucleotide polymorphism (-468 Gly to A) at the promoter region of SREBP-1c associates with genetic defect of fructose-induced hepatic lipogenesis [corrected] J Biol Chem. 2004;279:29031–29042. doi: 10.1074/jbc.M309449200. [DOI] [PubMed] [Google Scholar]
- Neschen S, Morino K, Hammond LE, Zhang D, Liu ZX, Romanelli AJ, Cline GW, Pongratz RL, Zhang XM, Choi CS, et al. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2005;2:55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Otani K, Han DH, Ford EL, Garcia-Roves PM, Ye H, Horikawa Y, Bell GI, Holloszy JO, Polonsky KS. Calpain system regulates muscle mass and glucose transporter GLUT4 turnover. J Biol Chem. 2004;279:20915–20920. doi: 10.1074/jbc.M400213200. [DOI] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, Cline GW, Befroy D, Zemany L, Kahn BB, et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci U S A. 2007;104:12587–12594. doi: 10.1073/pnas.0705408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Qu X, Seale JP, Donnelly R. Tissue and isoform-selective activation of protein kinase C in insulin-resistant obese Zucker rats - effects of feeding. J Endocrinol. 1999;162:207–214. doi: 10.1677/joe.0.1620207. [DOI] [PubMed] [Google Scholar]
- Rhee J, Inoue Y, Yoon JC, Puigserver P, Fan M, Gonzalez FJ, Spiegelman BM. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc Natl Acad Sci U S A. 2003;100:4012–4017. doi: 10.1073/pnas.0730870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, Kahn M, Zhang XM, Monia BP, Bhanot S, Shulman GI. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest. 2007;117:739–745. doi: 10.1172/JCI30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DB, Choi CS, Samuel VT, Liu ZX, Zhang D, Wang A, Zhang XM, Cline GW, Yu XX, Geisler JG, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. 2006;116:817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta. 2002;1576:1–14. doi: 10.1016/s0167-4781(02)00343-3. [DOI] [PubMed] [Google Scholar]
- Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL. Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest. 1997;99:846–854. doi: 10.1172/JCI119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleder J, Chen YD, Cully MD, Reaven GM. Hyperinsulinemia in fructose-induced hypertriglyceridemia in the rat. Metabolism. 1980;29:303–305. doi: 10.1016/0026-0495(80)90001-3. [DOI] [PubMed] [Google Scholar]
- Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vianna CR, Huntgeburth M, Coppari R, Choi CS, Lin J, Krauss S, Barbatelli G, Tzameli I, Kim YB, Cinti S, et al. Hypomorphic mutation of PGC-1beta causes mitochondrial dysfunction and liver insulin resistance. Cell Metab. 2006;4:453–464. doi: 10.1016/j.cmet.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Youn JH, Buchanan TA. Fasting does not impair insulin-stimulated glucose uptake but alters intracellular glucose metabolism in conscious rats. Diabetes. 1993;42:757–763. doi: 10.2337/diab.42.5.757. [DOI] [PubMed] [Google Scholar]
- Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- Yu XX, Murray SF, Pandey SK, Booten SL, Bao D, Song XZ, Kelly S, Chen S, McKay R, Monia BP, et al. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology. 2005;42:362–371. doi: 10.1002/hep.20783. [DOI] [PubMed] [Google Scholar]
- Zhang D, Liu ZX, Choi CS, Tian L, Kibbey R, Dong J, Cline GW, Wood PA, Shulman GI. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci U S A. 2007;104:17075–17080. doi: 10.1073/pnas.0707060104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.