

Abstract
Transcription-coupled repair (TCR) plays an important role in removing DNA damage from actively transcribed genes. It has been speculated that TCR is the most important mechanism for repairing DNA damage in non-dividing cells such as neurons. Therefore, abnormal TCR may contribute to the development of many age-related and neurodegenerative diseases. However, the molecular mechanism of TCR is not well understood. Oligonucleotide DNA triplex formation provides an ideal system to dissect the molecular mechanism of TCR since triplexes can be formed in a sequence-specific manner to inhibit transcription of target genes. We have recently studied the molecular mechanism of triplex-forming oligonucleotide (TFO)-mediated TCR in HeLa nuclear extracts. Using plasmid constructs we demonstrate that the level of TFO-mediated DNA repair activity is directly correlated with the level of transcription of the plasmid in HeLa nuclear extracts. TFO-mediated DNA repair activity was further linked with transcription since the presence of rNTPs in the reaction was essential for AG30-mediated DNA repair activity in HeLa nuclear extracts. The involvement of individual components, including TFIID, TFIIH, RNA polymerase II and xeroderma pigmentosum group A (XPA), in the triplex-mediated TCR process was demonstrated in HeLa nuclear extracts using immunodepletion assays. Importantly, our studies also demonstrated that XPC, a component involved in global genome DNA repair, is involved in the AG30-mediated DNA repair process. The results obtained in this study provide an important new understanding of the molecular mechanisms involved in the TCR process in mammalian cells.
INTRODUCTION
In mammalian cells genomic DNA is constantly damaged by both endogenous and environmental factors. This DNA damage needs to be repaired to maintain genetic fidelity and stability. Many DNA repair systems have evolved to remove DNA damage from the genomes of living organisms. Based on the transcriptional activity of the DNA targets, DNA repair can be distinguished as transcription-coupled repair (TCR) or global genome repair (GGR) (1). TCR is used for rapid removal of DNA damage in highly transcribed genes, while GGR is used to remove DNA damage from untranscribed DNA regions at a much slower rate. It has been suggested that TCR plays an important role in repairing DNA damage in non-dividing cells such as neurons since a low level of GGR was detected in neurons (2). Therefore, abnormal TCR may contribute to the development of many age-related diseases (3). At present two genes, CSA (4) and CSB (5), have been shown to promote TCR (5–10). Other components, such as TFIIH, RNA polymerase II (Pol II), BRCA1, hMLH1 and HMLH2, have also been implicated in the TCR process (11–18). In the case of GGR the XPC (19) and XPE genes (20) were found to be involved in this DNA repair process (21–23). However, the molecular mechanisms involved in TCR are not well understood and the correlation between TCR and GGR has not been established.
Oligonucleotides can be designed to bind to homopurine/homopyrimidine sequences of double-stranded DNA targets in a sequence-specific manner to form Hoogsteen triple helix structures (24,25). Triplexes have been used to inhibit transcription of many genes both in vitro and in vivo (26–33). In our previous studies DNA triplexes were found to lead to targeted mutagenesis (34). This triplex-mediated targeted mutagenesis is thought to result from triplex-mediated transcription inhibition and TCR, since the structure of the triplexes does not cause any chemical alterations in the DNA template. However, the molecular basis of triplex-mediated TCR and targeted mutagenesis has not been established.
In this study, using triplex-forming oligonucleotides (TFOs) as transcription inhibition agents, the molecular mechanism of triplex-mediated TCR has been studied in HeLa nuclear extracts. Using plasmid constructs we demonstrate that binding of TFOs induces a much stronger DNA repair activity in promoter-containing plasmids than in promoterless plasmids. This DNA repair activity has been further linked with transcription by the observation that the presence of rNTPs in the reaction was essential for TFO-mediated DNA repair activity. Individual components involved in the triplex-mediated DNA repair process have been determined. Most importantly, our studies suggest that XPC protein, which is involved in GGR, is also involved in the TFO-mediated TCR process. The results obtained in this study provide important data concerning the molecular mechanism involved in triplex-mediated TCR.
MATERIALS AND METHODS
Oligonucleotides and plasmids
The oligonucleotides used in this study are listed in Table 1 and were synthesized by either Oligos Etc. Inc. (Wilsonville, OR) or the W.M. Keck Biotechnology Resource Center at Yale University.
Table 1. Oligonucleotides used in the study.
| Oligonucleotide |
Sequence |
| AG30 | 5′-AGGAAGGGGGGGGTGGTGGGGGAGGGGGAG-3′ |
| CT30 | 5′-CTCTCTCTCTCTCTCTCTCTCTCTCTCTCT-3′ |
| RT primer | 5′-GCCTTATGCAGTTGCTCTCC-3′ |
Plasmids pUSAG15 and pUSAG16 were constructed using a standard DNA cloning protocol (35). Briefly, plasmid pUSAG15 was constructed by inserting a 177 bp DNA fragment containing a 30 bp homopurine/homopyrimidine triplex-binding sequence (36) into the HindIII site of pGL3-Control, a SV40 promoter-driven luciferase reporter gene expression vector (Promega Corp., Madison, WI) (Fig. 1, top). Plasmid pUSAG16 was constructed by inserting the 177 bp DNA fragment into the HindIII site of pGL3-Basic, a promoterless luciferase gene plasmid (Promega Corp.) (Fig. 1, bottom).
Figure 1.

Structure of plasmids pUSAG15 and pUSAG16. pUSAG15 was constructed by cloning a 177 bp supF gene DNA fragment containing a 30 bp homopurine triplex-binding site from pSupFG1 into the HindIII site of vector pGL3-Basic. pUSAG16 was constructed by inserting the 177 bp supF gene DNA fragment into the HindIII site of vector pGL3-Control. TBS, triplex-binding sequence; H, restriction enzyme HindIII digestion site; B, restriction enzyme BamHI digestion site.
Cell lines and preparation of nuclear extracts
Normal human fibroblasts (NF) (GM00637) were obtained from the NIGMS Human Genetic Cell Repository (Camden, NJ) and were maintained in MEM supplemented with 10% fetal calf serum and 2× essential amino acids, non-essential amino acids and vitamins. Xeroderma pigmentosum group A (XPA) (GM02250) cells were obtained from the NIGMS and maintained in RPMI 1640 medium supplemented with 15% heat-inactivated fetal calf serum. Xeroderma pigmentosum group C (XPC) (GM02249) cells were obtained from the NIGMS and maintained in RPMI 1640 medium supplemented with 15% heat-inactivated fetal calf serum. Cockayne’s syndrome group A (CSA) (GM01857) cells were obtained from the NIGMS and maintained in RPMI 1640 medium supplemented with 15% heat-inactivated fetal calf serum. All cells were maintained in a tissue culture incubator supplemented with 5% CO2.
The nuclear extracts were prepared as described (37). Briefly, cells were harvested from the cell culture medium by centrifugation (4°C) and washed twice with phosphate-buffered saline (PBS). The cells were suspended in 5 packed cell pellet vol of buffer A [10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl and 0.5 mM dithiothreitol (DTT)] and incubated on ice for 10 min. The cells were collected by centrifugation and resuspended in 2 packed cell pellet vol of buffer A. The cells were lysed by 20 strokes of a Kontes glass Dounce homogenizer (B type pestle). The homogenate was centrifuged for 10 min at 25 000 g (4°C) to pellet nuclei. The nuclei were resuspended in 3 ml of buffer C [20 mM HEPES pH 7.9, 25% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride (PMSF) and 0.5 mM DTT] per 109 cells and were lysed by 20 strokes of a Kontes glass Dounce homogenizer (B type pestle). The resulting suspension was stirred gently for 30 min at 4°C and centrifuged for 30 min at 25 000 g. The clear supernatant was dialyzed with 50 vol of buffer D (20 mM HEPES pH 7.9, 20% glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM PMSF and 0.5 mM DTT) at 4°C for 6 h. The dialysate was centrifuged at 25 000 g for 20 min and the supernatant was frozen as aliquots in liquid nitrogen and stored at –80°C.
In vitro DNA repair synthesis assay
The in vitro DNA repair synthesis assay was performed as described (34) using plasmid pUSAG15 or pUSAG16. The plasmid DNA was digested with restriction enzyme XhoI at 37°C for 2 h and then analyzed by agarose gel electrophoresis using a 1% gel. The DNA was visualized under UV after ethidium bromide staining and incorporation of [α-32P]dCTP was visualized by autoradiography and quantificated using a Bio-Rad G250 phosphorimager (Bio-Rad, Hercules, CA).
Reverse transcription assay
RNA was synthesized from pUSAG15 and pUSAG16 plasmid DNA via an in vitro transcription reaction. The nascent RNA was purified from the reactants, precipitated with ethanol and dissolved in 5 µl of RNase-free H2O. The reverse transcription experiment was performed in 25 µl containing 5 µl of RNA, 1× AMV buffer, 1 pmol end-labeled RT primer (which binds to the luciferase mRNA 5′ coding sequence), 200 µM dATP, dCTP, dGTP and dTTP, 10 U AMV reverse transcriptase and 10 U RNase inhibitor. The reactants were preincubated at 95°C for 5 min and then cooled to 42°C before addition of AMV reverse transcriptase and RNase inhibitor. The reactants were incubated at 42°C for 60 min and terminated by addition of 12.5 µl of stop solution (Gibco BRL Cycle Sequencing Kit). The reactants were denatured at 95°C for 5 min and then analyzed by DNA sequencing gel electrophoresis using a 6% gel. Synthesis of specific RNA transcripts was determined by autoradiography and quantitated using a Bio-Rad G250 phosphorimager.
Immunodepletion of individual transcription components from HeLa nuclear extracts
Protein A-immobilized agarose beads (Sigma, St Louis, MO) were incubated with antibodies against individual components, including TFIID (SI-1), TFIIH (Q-19), RNA Pol II (N-20) and XPA (FL-273) (Santa Cruz Technologies, Santa Cruz, CA) at 4°C for 1 h. The antibody-bound agarose beads were collected by centrifugation at 4°C for 10 min and washed with PBS twice. The HeLa nuclear extracts (Life Technologies, Gaithersburg, MD) were incubated with the antibody-bound agarose beads at 4°C for 2 h and the supernatant was saved after centrifugation at 4°C for 10 min. The efficiency of immunodepletion for individual transcription factors was confirmed by western blot analysis. The immunodepleted HeLa nuclear extracts were used for DNA repair assays.
Purification of XPA and XPC proteins
Both XPA and XPC proteins were purified by an immunoprecipitation procedure. Briefly, both XPA and XPC antibodies were incubated with protein A-immobilized agarose beads at 4°C for 1 h. The antibody-bound agarose beads were collected by centrifugation at 4°C for 10 min and washed twice with PBS. The HeLa nuclear extracts were incubated with the antibody-bound beads at 4°C for 2 h and the beads were collected by centrifugation. The beads were washed twice in PBS. The proteins were eluted from the beads by resuspending the beads in triethanolamine solution (50 mM triethanolamine pH 11.5, 0.1% Triton X-100 and 0.15 M NaCl) for 2 min and centrifugation at 4°C for 5 min. The supernatants were adjusted to pH ∼7.5 and desalted using a Centricon 10 device (Millipore, MA). The proteins were resuspended in 20% glycerol and stored at –20°C.
RESULTS
Detection of oligo AG30 triplex-mediated TCR in HeLa nuclear extracts
To determine whether triplex structures can stimulate DNA repair synthesis in HeLa nuclear extracts, both pUSAG15, which carries a SV40 promoter-derived luciferase gene with a 30 bp triplex-binding sequence, and pUSAG16, which carries a promoterless luciferase gene with the same triplex-binding sequence, were constructed (Fig. 1). Both plasmids were incubated with oligo AG30, a TFO that has been used previously and exhibits strong triplex-binding affinity (34), to form a 30 bp triplex structure at the homopurine/homopyrimidine sequence of the plasmids. The plasmid–AG30 complex was incubated in HeLa nuclear extracts supplemented with rNTPs, dATP, dGTP, dTTP and [α-32P]dCTP at 30°C for 2 h to allow transcription and DNA repair of the plasmid. Repair of DNA lesions in the plasmid would result in DNA repair synthesis and incorporation of [α-32P]dCTP (Fig. 2). As negative controls both plasmids were preincubated with oligo Mix30 (a control oligo that does not form stable triplexes with the plasmids) and were used as substrates for DNA repair assays. No significant DNA repair synthesis activity was detected with plasmid pUSAG15 pretreated with oligo Mix30 (Fig. 2, lane 1). However, a very strong DNA repair synthesis signal was detected with plasmid pUSAG15 DNA preincubated with TFO AG30 (Fig. 2, lane 2). A similar DNA repair pattern was also observed with plasmid pUSAG16 DNA in a parallel experiment (Fig. 2, lane 5 versus lane 4). However, the level of [α-32P]dCTP incorporation into AG30-treated pUSAG16 DNA was much less than that into AG30-treated pUSAG15 DNA. Phosphorimaging analysis indicated that the amount of [α-32P]dCTP incorporation into AG30-treated pUSAG16 DNA was only 26% of that into AG30-treated pUSAG15 DNA in HeLa nuclear extracts (mean data from five independent experiments) (Fig. 2C).
Figure 2.
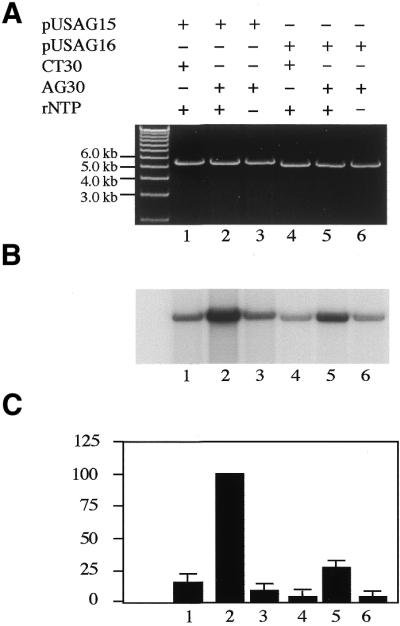
TFO AG30-mediated DNA repair synthesis of pUSAG15 and pUSAG16 in HeLa cell nuclear extracts. The supercoiled plasmid DNA was preincubated with oligonucleotide at 37°C for 2 h in triplex-binding buffer (10 mM Tris pH 7.5, 1 mM spermidine, 20 mM MgCl2) for triplex formation. Then the plasmid DNA (1 µg) was incubated with HeLa nuclear extracts supplemented with dATP, dGTP, dTTP and [α-32P]dCTP at 30°C for 2 h. The plasmid DNAs were linearized with XhoI restriction enzyme and analyzed by agarose gel electrophoresis using a 0.8% gel. (A) Visualization of the plasmid DNA by ethidium bromide staining. (B) Autoradiogram of the same gel showing labeled nucleotide incorporation indicative of DNA repair synthesis. (C) Quantification of incorporation of [α-32P]dCTP into plasmid DNA. The amount of incorporated [α-32P]dCTP in pUSAG15–AG30 was taken as 100%. Incorporation of [α-32P]dCTP in other reactions was calculated as a percentage of pUSAG15–AG30. The data are means obtained from four individual experiments.
To determine whether the elevated levels of DNA repair activity observed with pUSAG15 and pUSAG16 DNA resulted from different levels of transcription in both plasmids an in vitro transcription assay was performed (Fig. 3A). Indeed, the level of transcription of pUSAG15 was much higher than that of pUSAG16 in HeLa nuclear extracts (Fig. 3A, lane 1 versus lane 2). The reverse transcription results indicated that specific RNA transcripts initiated from the SV40 promoter were detected only in plasmid pUSAG15 DNA in HeLa nuclear extract (Fig. 3B, lane 2 versus lane 3). All these results suggest that the difference in TFO AG30-mediated DNA repair activity with pUSAG15 and pUSAG16 was correlated with different levels of transcription of the plasmids in HeLa nuclear extracts and oligo AG30 triplex-mediated TCR.
Figure 3.
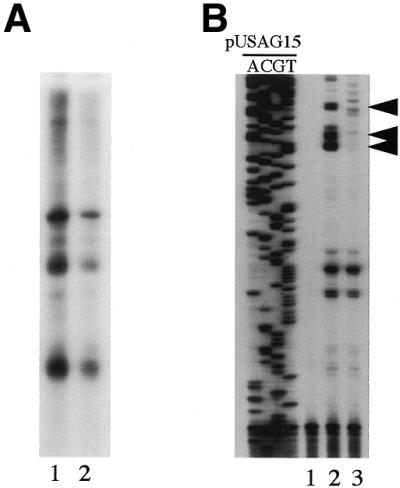
Detection of transcription of both pUSAG15 and pUSAG16 in HeLa nuclear extract. The in vitro transcription reactions with pUSAG15 and pUSAG16 were performed in HeLa nuclear extracts. (A) Detection of RNA transcripts synthesized from pUSAG15 and pUSAG16 plasmid DNA in HeLa nuclear extract. Lane 1, pUSAG15; lane 2, pUSAG16. (B) Reverse transcription assay to determine the initiation sites of RNA transcripts synthesized from pUSAG15 and pUSAG16 plasmid DNA in HeLa nuclear extract. Lane 1, no DNA control; lane 2, pUSAG15; lane 3, pUSAG16 (the arrows indicate specific RNA transcripts initiated from the SV40 promoter).
Most of the DNA repair reactions described above were performed under conditions that favored in vitro transcription to enhance the transcription event. To determine whether transcription enhances triplex-mediated DNA repair activity, oligo AG30-mediated DNA repair was also investigated in HeLa nuclear extracts in which transcription was inhibited. In these assays transcription was inhibited by removal of rNTPs (except ATP) from the reactants, since the absence of rNTPs has been shown to inhibit transcription. The absence of rNTPs in the reactants greatly reduced the TFO AG30-mediated DNA repair activity of both pUSAG15 and pUSAG16 DNA to the background level (Fig. 2, lane 3 versus lane 2 and lane 6 versus lane 5). Since the rNTPs provided in the reactants were used only for transcription, the absence of rNTPs should only lead to transcription inhibition. These results strongly suggest that TFO AG30-mediated DNA repair activity in both pUSAG15 and pUSAG16 DNA directly results from AG30 triplex-mediated transcription inhibition and transcription inhibition-mediated TCR.
The requirement for individual transcription components in the AG30 triplex-mediated DNA repair process
To determine the role of individual transcription components in the triplex-mediated TCR process TFO AG30-mediated TCR was studied in immunodepleted HeLa nuclear extracts (Fig. 4). Transcription components, including TFIID, TFIIH and RNA Pol II, as well as the nucleotide excision repair (NER) component XPA, were individually immunodepleted from HeLa nuclear extracts and depletion efficiency was confirmed by western blot hybridization assay (data not shown). The immunodepleted HeLa nuclear extracts were then used to study TFO AG30-mediated DNA repair of pUSAG15. Depletion of any of these individual components resulted in reduced levels of TFO AG30-mediated DNA repair activity with pUSAG15 in HeLa nuclear extracts. Because of the roles of TFIID, TFIIH and Pol II in transcription, this result suggests that transcription was indeed essential for TFO AG30-mediated DNA repair activity, and depletion of individual transcription components resulted in diminished AG30 triplex-mediated DNA repair activity with pUSAG15 in HeLa nuclear extracts. Depletion of XPA from HeLa nuclear extracts also led to a diminished level of AG30 triplex-mediated DNA repair activity with pUSAG15. This result indicated that the NER pathway is also involved in AG30 triplex-mediated TCR. This result is consistent with our previous targeted mutagenesis studies (34).
Figure 4.
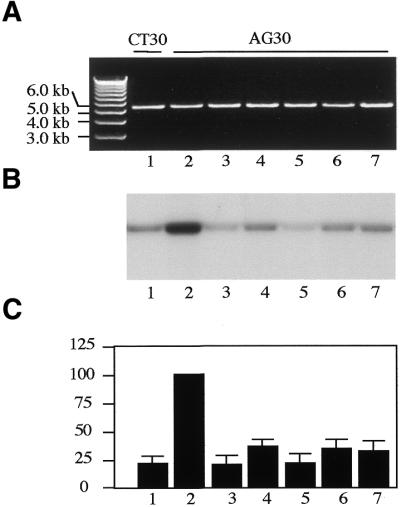
Involvement of individual components in TFO AG30 triplex-mediated DNA repair synthesis of pUSAG15. Individual components were immunodepleted from HeLa nuclear extracts using antibodies against specific components. (A) Visualization of plasmid DNA by ethidium bromide staining. (B) Autoradiogram of the same gel showing labeled nucleotide incorporation indicative of DNA repair synthesis. (C) Quantification of incorporated [α-32P]dCTP in plasmid DNA. Lane 1, CT30 + HeLa nuclear extract; lane 2, AG30 + HeLa nuclear extract; lane 3, AG30 + XPA-depleted HeLa nuclear extract; lane 4, AG30 + TFIID-depleted HeLa nuclear extract; lane 5, AG30 + TFIIH-depleted HeLa nuclear extract; lane 6, AG30 + Pol II-depleted HeLa nuclear extract; lane 7, AG30 + rNTP-lacking HeLa nuclear extract. The data are means obtained from four individual experiments.
Involvement of the TCR pathway and XPC protein in the oligo AG30 triplex-mediated DNA repair process
The results obtained from DNA repair experiments indicated that TCR is the mechanism for overcoming AG30 triplex-mediated transcription blockage. However, the involvement of TCR in the triplex-mediated DNA repair process has not been directly tested. The role of GGR, which is thought to be the mechanism for removing DNA damage from non-transcribed DNA in living cells, in the triplex-mediated DNA repair process is also unclear. To address these questions we studied oligo AG30 triplex-mediated DNA repair in nuclear extracts prepared from XPC cells, which have a defective GGR pathway, and CSA cells, which have a defective TCR pathway (Fig. 5). As controls, AG30 triplex-mediated DNA repair was studied in nuclear extracts prepared from XPA cells, which have defective NER, and nuclear extracts prepared from human NF, which have normal DNA repair. TFO AG30 triplex-mediated DNA repair of pUSAG15 was found to be defective in XPA, XPC and CSA nuclear extracts (Fig. 5, lanes 3, 5 and 9). However, when nuclear extracts of XPA and XPC cells were supplemented with purified XPA and XPC proteins, respectively, AG30-mediated TCR activity of pUSAG15 was detected (Fig. 5, lanes 4 and 6). In contrast, when purified XPA protein was added to XPC nuclear extract and purified XPC protein was added to XPA nuclear extract, TFO AG30-mediated DNA repair activity was not detected (Fig.5, lanes 7 and 8). In a complementary experiment in which both XPC and CSA nuclear extracts were provided, TFO AG30 triplex-mediated TCR activity was partially restored (Fig. 5, lane 10 versus lanes 5 and 9). As a positive control, AG30 triplex-mediated DNA repair activity with pUSAG15 was detected in nuclear extracts prepared from NF as expected (Fig. 5, lane 2 versus lane 1). These results suggest that the XPA, XPC and CSA proteins are required for AG30 triplex-mediated TCR.
Figure 5.

TFO AG30 triplex-mediated DNA repair of pUSAG15 in nuclear extracts prepared from XPA, XPC and CSA cells. Lane 1, CT30 + NF nuclear extract (NF); lane 2, AG30 + NF nuclear extract (NF); lane 3, AG30 + XPA nuclear extract (XPAN); lane 4, AG30 + XPAN + purified XPA protein (XPAP); lane 5, AG30 + XPC nuclear extract (XPCN); lane 6, AG30 + XPCN + purified XPC protein (XPCP); lane 7, AG30 + XPAN + XPCP; lane 8, AG30 + XPCN + XPAP; lane 9, AG30 + CSA nuclear extract (CSAN); lane 10, AG30 + CSAN + XPCN.
DISCUSSION
TCR plays a very important role in removing DNA damage from highly transcribed DNA regions of living cells and abnormal TCR may contribute to the development of many human diseases, including cancer, age-related diseases and neurodegenerative diseases; however, the molecular mechanism of TCR is not well understood. In this study, using oligonucleotide triplex formation to inhibit transcription, we have investigated TFO AG30 triplex-mediated DNA repair in HeLa cell nuclear extracts. Using plasmid constructs we have demonstrated that a TFO AG30 triplex stimulated much higher DNA repair activity in plasmids containing a mammalian promoter than plasmids lacking mammalian promoters in HeLa nuclear extracts. Our in vitro transcription assay also demonstrated that the level of transcription was much higher in the promoter-containing plasmid than the promoter-lacking plasmid in HeLa nuclear extracts. Therefore, the levels of TFO AG30-mediated DNA repair activity were directly correlated with the levels of transcription of the plasmids. This result is consistent with our previous observation that TFOs with stronger transcription inhibition induced stronger DNA repair activity and a higher mutation frequency than TFOs with weaker transcription inhibition (34). This result suggests that HeLa nuclear extract provides a good in vitro system to study the molecular mechanism of TCR. Oligonucleotide triplex structures provide an ideal system to inhibit the transcription of target genes in a sequence-specific manner and promote TCR.
Triplex-mediated DNA repair activity has been further linked with transcription by the transcription inhibition results. We hypothesized that triplex-mediated DNA repair was caused by triplex-mediated transcription inhibition and TCR. Therefore, most DNA repair assays were performed under conditions that favored transcription of the plasmid, by supplementing the reactants with rNTPs. However, when rNTPs were omitted from the reaction TFO AG30-mediated DNA repair activity in HeLa nuclear extracts was reduced to a level similar to the background. Immunodepletion of RNA Pol II activity in HeLa nuclear extracts also greatly reduced TFO AG30-mediated DNA repair activity in the extracts. Since both rNTPs and RNA Pol II are used in transcription, omission of rNTPs or depletion of RNA Pol II should only lead to transcription inhibition. Therefore, the observed reduced levels of DNA repair activity with the plasmid DNA strongly suggest that transcription is essential for TFO AG30-mediated DNA repair activity in HeLa nuclear extracts. Collectively, these results indicate that TFO AG30-mediated DNA repair activity resulted from TFO AG30 triplex-mediated transcription inhibition and TCR in the plasmid.
The involvement of individual transcription components in TFO AG30 triplex-mediated DNA repair has been determined in this study. Using antibodies against specific components, transcription factors TFIID, TFIIH, RNA Pol II and the NER component XPA were individually immunodepleted from HeLa nuclear extracts. Our TFO AG30-mediated DNA repair assay indicates that depletion of any of these individual components from HeLa nuclear extracts resulted in diminished or reduced levels of TFO AG30-mediated DNA repair activity in the plasmid DNA. The requirement for TFIIH in TFO AG30 triplex-mediated TCR is in agreement with the published data that TFIIH is required for TCR (14). Our studies show that the transcription factor TFIID is required for TFO AG30 triplex-mediated TCR. This result was expected, since studies have demonstrated that recognition and binding of TATA-binding protein (TBP), a component of TFIID, to promoter regions of target genes is the initial step of RNA Pol II-related transcription. Therefore, depletion of TFIID from HeLa nuclear extracts is expected to completely abolish RNA Pol II-related transcription. RNA Pol II is the polymerase used in synthesis of mRNA, therefore, depletion of RNA Pol II from HeLa nuclear extracts is expected to inhibit transcription and reduce TFO AG30 triplex-mediated TCR activity in the plasmid. Depletion of XPA from HeLa nuclear extracts resulted in diminished TFO AG30 triplex-mediated DNA repair activity. This result is consistent with our previous observation that the NER pathway is required for triplex-mediated targeted mutagenesis (34) and it provides direct evidence for the involvement of NER in triplex-mediated DNA repair and targeted mutagenesis.
Low levels of DNA repair activity were detected in TFO AG30-bound pUSAG16 plasmid DNA in HeLa nuclear extracts. This result was unexpected, since the plasmid lacks a normal mammalian promoter and no transcription should occur in the plasmid. However, our in vitro transcription assay detected a low level of transcription of plasmid pUSAG16 in HeLa nuclear extracts. The reverse transcription result for pUSAG16 also indicated that a certain level of non-specific transcription occurs with pUSAG16 in the HeLa nuclear extract system. Therefore, the low level of TFO AG30 triplex-mediated DNA repair with plasmid pUSAG16 DNA may result from low levels of transcription of the plasmid. However, we could not exclude the possibility that the low level of DNA repair activity detected with pUSAG16 resulted from GGR activity, since the GGR pathway also exists in HeLa nuclear extracts. The GGR enzymes may recognize the AG30 triplex structure in pUSAG16 and lead to repair of the plasmid. However, a slower repair rate of GGR may lead to a much lower level of DNA repair activity with plasmid pUSAG16.
The absence of TFO AG30 triplex-mediated DNA repair activity in CSA cells indicates the direct involvement of TCR in the TFO AG30-mediated DNA repair process. This result is consistent with our previous observation that the TCR pathway is required for triplex-mediated targeted mutagenesis (34). The absence of TFO AG30 triplex-mediated DNA repair activity in XPC cell nuclear extracts suggests that XPC protein is also involved in triplex-mediated TCR. This result was not predicted since XPC protein was identified as a component of GGR (21). However, recent studies have demonstrated that XPC protein possesses a much higher binding affinity for damaged DNA than either the XPA or RPA proteins (38). Other studies demonstrated that XPC protein serves as an initiator of the GGR pathway (21). It is possible that XPC protein may serve as a universal DNA damage recognition protein that recognizes and binds to a broad range of DNA damage with high affinity in living cells regardless of the nature of transcription of the DNA damage sites. The CSA/CSB proteins may help to recruit DNA repair enzymes quickly to DNA damage sites of transcribed DNA regions, leading to more rapid DNA damage repair in highly transcribed DNA regions. Unfortunately, very little is known about the interaction between XPC and other proteins in DNA repair, especially in TCR, except that it has been shown that XPC protein interacts with HHR23B and HHR23A in vivo (39). It will be important to determine the interaction between XPC and other components in the DNA repair process, especially in the TCR process.
Although a wealth of data has suggested the existence of intracellular triplex structures in living cells (40–48), very little is known about the role of intracellular triplexes in causing DNA repair and mutagenesis. In our previous studies triplexes were demonstrated to lead to targeted mutagenesis (34). Therefore, it is possible that intracellular triplexes may lead to genetic instability and contribute to the development of many human diseases, such as cancer, age-related diseases and neurodegenerative diseases. The work presented in this study provides an important understanding of the molecular mechanism of triplex-mediated TCR and genetic instability. The knowledge obtained from this study may also help provide a molecular basis for the development of many human diseases.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Peter M.Glazer for his helpful discussions and suggestions and Drs R.Balczon and M.Bhatnager for their helpful discussions and critical readings. This work was supported by NIH grant R01 ES09699 to G.W.
References
- 1.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC.
- 2.Nouspikel T. and Hanawalt,P.C. (2000) A common mutational pattern in Cockayne syndrome patients from xeroderma pigmentosum group G: implications for a second XPG function. Mol. Cell. Biol., 20, 1562–1570. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Finch C.E. and Goodman,M.F. (1997) Relevance of ‘adaptive’ mutations arising in non-dividing cells of microorganisms to age-related changes in mutant phenotypes of neurons. Trends Neurosci., 20, 501–507. [DOI] [PubMed] [Google Scholar]
- 4.Henning K.A., Li,L., Lyer,N., McDaniel,L.D., Reagan,M.S., Legerski,R., Schultz,R.A., Stefanini,M., Lehmann,A.R., Mayne,L.V. and Friedberg,E.C. (1995) The Cockayne Syndrome Group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell, 82, 555–564. [DOI] [PubMed] [Google Scholar]
- 5.Troelstra C., van Gool,A., de Wit,J., Vermeulen,W., Bootsma,D. and Hoeijmakers,H.J. (1992) ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell, 71, 939–953. [DOI] [PubMed] [Google Scholar]
- 6.Leadon S.A. and Cooper,P.K. (1993) Preferential repair of ionizing radiation-induced damage in the transcribed strand of an active human gene is defective in Cockayne syndrome. Proc. Natl Acad. Sci. USA, 90, 10499–10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Gool A.J., Citterio,E., Rademakers,S., van Os,R., Vermeulen,W., Constantinou,A., Egly,J.M., Bootsma,D. and Hoeijmakers,J.H. (1997) The Cockayne syndrome B protein, involved in transcription-coupled DNA repair, resides in an RNA polymerase II-containing complex. EMBO J., 16, 5955–5965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Gool A.J., van der Horst,G.T., Citteri,E. and Hoeijmakers,J.H. (1997) Cockayne syndrome: defective repair of transcription? EMBO J., 16, 4155–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tu Y., Bates,S. and Pfeifer,G.P. (1998) The transcription-repair coupling factor CSA is required for efficient repair only during the elongation stages of RNA polymerase II transcription. Mutat. Res., 400, 143–151. [DOI] [PubMed] [Google Scholar]
- 10.Cooper P.K., Nouspikel,T., Clarkson,S.G. and Leadon,S.A. (1997) Defective transcription-coupled repair of oxidative base damage in Cockayne Syndrome patients from XP group G. Science, 275, 990–993. [DOI] [PubMed] [Google Scholar]
- 11.Gowen L.C., Avrutskaya,A.V., Latour,A.M., Koller,B.H. and Leadon,S.A. (1998) BRCA1 required for transcription-coupled repair of oxidative DNA damage. Science, 281, 1009–1012. [DOI] [PubMed] [Google Scholar]
- 12.Leadon S.A. and Lawrence,D.A. (1991) Preferential repair of DNA damage on the transcribed strand of the human metallothionein genes requires RNA polymerase II. Mutat. Res., 255, 67–78. [DOI] [PubMed] [Google Scholar]
- 13.Leadon S.A. and Lawrence,D.A. (1992) Strand-selective repair of DNA damage in the yeast GAL7 gene requires RNA polymerase II. J. Biol. Chem., 267, 23175–23182. [PubMed] [Google Scholar]
- 14.Park C.H., Mu,D., Reardon,J.T. and Sancar,A. (1995) The general transcription-repair factor TFIIH is recruited to the excision repair complex by the XPA protein independent of the TFIIE transcription factor. J. Biol. Chem., 270, 4896–4902. [DOI] [PubMed] [Google Scholar]
- 15.Donahue B.A., Fuchs,R.P.P., Reines,D. and Hanawalt,P.C. (1996) Effects of aminofluorene and acetylaminofluorene DNA adducts on transcriptional elongation by RNA polymerase II. J. Biol. Chem., 271, 10588–10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selby C.P. and Sancar,A. (1997) Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem., 272, 1885–1890. [DOI] [PubMed] [Google Scholar]
- 17.Selby C.P., Drapkin,R., Reinberg,D. and Sancar,A. (1997) RNA polymerase II stalled at a thymine dimer: footprint and effect on excision repair. Nucleic Acids Res., 25, 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leadon S.A. and Avrutskaya,A.V. (1997) Differential involvement of the human mismatch repair proteins, hMLH1 and hMSH2, in transcription-coupled repair. Cancer Res., 57, 3784–3791. [PubMed] [Google Scholar]
- 19.Legerski R. and Peterson,C. (1992) Expression cloning of a human DNA repair gene involved in xeroderma pigmentosum group C. Nature, 359, 70–73. [DOI] [PubMed] [Google Scholar]
- 20.Hwang B.J., Liao,J.C. and Chu,G. (1996) Isolation of a cDNA encoding a UV-damaged DNA binding factor defective in xeroderma pigmentosum group E cells. Mutat. Res., 362, 105–117. [DOI] [PubMed] [Google Scholar]
- 21.Sugasawa K., Ng,J.M., Masutani,C., Iwai,S., van der Spek,P.J., Eker,A.P., Hanaoka,F., Bootsma,D. and Hoeijmakers,J.H. (1998) Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell, 2, 223–232. [DOI] [PubMed] [Google Scholar]
- 22.Shivji M.K.K., Eker,A.P.M. and Wood,R.D. (1994) DNA repair defect in xeroderma pigmentosum group C and complementing factor from HeLa cells. J. Biol. Chem., 269, 22749–22757. [PubMed] [Google Scholar]
- 23.Hwang B.J., Ford,J.M., Hanawalt,P.C. and Chu,G. (1999) Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl Acad. Sci. USA, 96, 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun J.-S. and Helene,C. (1993) Oligonucleotide-directed triple-helix formation. Curr. Opin. Struct. Biol., 3, 345–356. [DOI] [PubMed] [Google Scholar]
- 25.Helene C. (1991) The anti-gene strategy: control of gene expression by triplex-forming oligonucleotides. Anticancer Drug Des., 6, 569–584. [PubMed] [Google Scholar]
- 26.Duval-Valentin G., Thuong,N.T. and Helene,C. (1992) Specific inhibition of transcription by triple helix-forming oligonucleotides. Proc. Natl Acad. Sci. USA, 89, 504–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim H.-G. and Miller,D.M. (1995) Inhibition of in vitro transcription by a triplex-forming oligonucleotide targeted to human c-myc P2 promoter. Biochemistry, 34, 8165–8171. [DOI] [PubMed] [Google Scholar]
- 28.Porumb H., Gousset,H., Letellier,R., Salle,V., Briane,D., Vassy,J., Amor-Gueret,M., Israel,L. and Taillandier,E. (1996) Temporary ex vivo inhibition of the expression of the human oncogene HER2 (NEU) by a triple helix-forming oligonucleotide. Cancer Res., 56, 515–522. [PubMed] [Google Scholar]
- 29.Postel E.H., Flint,S.J., Kessler,D.J. and Hogan,M.E. (1991) Evidence that a triplex-forming oligonucleotide binds to the c-myc promoter in HeLa cells, thereby reducing c-myc mRNA levels. Proc. Natl Acad. Sci. USA, 88, 8227–8231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tu G.C., Cao,Q.N. and Israel,Y. (1995) Inhibition of gene expression by triple helix formation in hepatoma cells. J. Biol. Chem., 270, 28402–28407. [DOI] [PubMed] [Google Scholar]
- 31.Kochetkova M. and Shannon,M.F. (1996) DNA triplex formation selectively inhibits granulocyte-macrophage colony-stimulating factor gene expression in human T cells. J. Biol. Chem., 271, 14438–14444. [DOI] [PubMed] [Google Scholar]
- 32.Kochetkova M., Iversen,P.O., Lopez,A.F. and Shannon,M.F. (1997) Deoxyribonucleic acid triplex formation inhibits granulocyte macrophage colony-stimulating factor gene expression and suppresses growth in juvenile myelomonocytic leukemic cells. J. Clin. Invest., 99, 3000–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faria M., Wood,C.D., Nelson,J.S., Winter,A., White,M.R.H., Helene,C. and Giovannangeli,C. (2000) Targeted inhibition of transcription elongation in cells mediated by triplex-forming oligonucleotides. Proc. Natl Acad. Sci. USA, 97, 3862–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang G., Seidman,M.M. and Glazer,P.M. (1996) Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science, 271, 802–805. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 36.Wang G., Levy,D.D., Seidman,M.M. and Glazer,P.M. (1995) Targeted mutagenesis in mammalian cells mediated by intracellular triple helix formation. Mol. Cell. Biol., 15, 1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wakasugi M. and Sancar,A. (1999) Order of assembly of human DNA repair excision nuclease. J. Biol. Chem., 274, 18759–18768. [DOI] [PubMed] [Google Scholar]
- 39.Li L., Lu,X., Peterson,C. and Legerski,R. (1997) XPC interacts with both HHR23B and HHR23A in vivo. Mutat. Res., 383, 197–203. [DOI] [PubMed] [Google Scholar]
- 40.Agazie Y.M., Lee,J.S. and Burkholder,G.D. (1994) Characterization of a new monoclonal antibody to triplex DNA and immunofluorescent staining of mammalian chromosomes. J. Biol. Chem., 269, 7019–7023. [PubMed] [Google Scholar]
- 41.Agazie Y.M., Burkholder,G.D. and Lee,J.S. (1996) Triplex DNA in the nucleus: direct binding of triplex-specific antibodies and their effect on transcription, replication and cell growth. Biochem. J., 316, 461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guieysse A.L., Praseuth,D. and Helene,C. (1997) Identification of a triplex DNA-binding protein from human cells. J. Mol. Biol., 267, 289–298. [DOI] [PubMed] [Google Scholar]
- 43.Hampel K.J. and Lee,J.S. (1993) Two-dimensional pulsed-field gel electrophoresis of yeast chromosomes: evidence for triplex-mediated DNA condensation. Biochem. Cell Biol., 71, 190–196. [DOI] [PubMed] [Google Scholar]
- 44.Kiyama R. and Camerini-Otero,R.D. (1991) A triplex DNA-binding protein from human cells: purification and characterization. Proc. Natl Acad. Sci. USA, 88, 10450–10454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee J.S., Burkholder,G.D., Latimer,L.J., Haug,B.L. and Braun,R.P. (1987) A monoclonal antibody to triplex DNA binds to eucaryotic chromosomes. Nucleic Acids Res., 15, 1047–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mikhailov V.S. and Bogenhagen,D.F. (1996) Termination within oligo(dT) tracts in template DNA by DNA polymerase γ occurs with formation of a DNA triplex structure and is relieved by mitochondrial single-stranded DNA-binding protein. J. Biol. Chem., 271, 30774–30780. [DOI] [PubMed] [Google Scholar]
- 47.Thomas T.J., Seibold,J.R., Adams,L.E. and Hess,E.V. (1995) Suppression of c-myc oncogene expression by a polyamine-complexed triplex forming oligonucleotide in MCF-7 breast cancer cells. Biochem. J., 311, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Veselkov A.G., Malkov,V.A., Frank-Kamenetskll,M.D. and Dobrynin,V.N. (1993) Triplex model of chromosome ends. Nature, 364, 496. [DOI] [PubMed] [Google Scholar]