

SYNOPSIS
Rhomboid proteases are a fascinating class of enzymes that combine a serine protease active site within the core of an integral membrane protein. Despite having key roles in animal cell signaling and microbial pathogenesis, the membrane-immersed nature of these enzymes had long imposed obstacles to elucidating their biochemical mechanisms. But recent multidisciplinary approaches, including eight crystal structures, four computer simulations, and nearly one hundred engineered mutants interrogated in vivo and in vitro, are coalescing into an integrated model for one rhomboid ortholog in particular, bacterial GlpG. The protein creates a central, hydrated microenvironment immersed below the membrane surface to support hydrolysis by its serine protease-like catalytic apparatus. Four conserved architectural elements in particular act as ‘keystones’ to stabilize this structure, while the lateral, membrane-embedded L1 loop functions as a ‘flotation device’ to position the protease tilted in the membrane. Complex interplay between lateral substrate gating by rhomboid, substrate unwinding, and local membrane thinning leads to intramembrane proteolysis of selected target proteins. Although far from complete, studies with GlpG currently offer the best prospect for achieving a thorough and sophisticated understanding of a simplified intramembrane protease.
Keywords: presenilin, site-2 protease, signal peptide peptidase, cell signalling, parasite invasion, pathogenesis
INTRODUCTION
The remarkable synthetic ability of cells relies on a vast orchestra of enzymes that transform simple chemicals from the environment into complex biomolecules and energy that sustain life. As a result, enzymes have been the focus of biochemical investigation for over a century. While most enzymes catalyze metabolic reactions, a subset evolved to function within information-processing circuits, where they are entrusted to regulate cellular processes. To this daunting task enzymes bring key advantages: they act rapidly and with a high degree of specificity. The modern challenge is to understand how the properties of these regulatory enzymes have been tailored to fit the specific needs of the cell.
Proteases cleave proteins by catalyzing hydrolysis of peptide bonds. Because of their speed, selectivity and versatility, proteolytic mechanisms have come to regulate a broad array of cellular processes. It’s often stated that evidence of their importance is encrypted directly in our genomes: up to 5% of a genome is often devoted to encoding proteolytic components [1, 2]. Moreover, serine proteases in particular have served as paradigms for understanding enzyme catalysis through integration of chemical, enzymological and structural investigations for over a century [3–6]. Yet even this well-studied area of biochemistry is not immune to surprise: in the past decade an unforeseen class of proteases was discovered that reside immersed within the cell’s membrane. Without exception, each of the three types of intramembrane proteases were discovered unintentionally, through analyses focused on cell regulation or human disease [7–9]. Although initially thought to be rare, intramembrane proteases are now recognized to be among the most prevalent membrane proteins known, conserved from bacteria to man [10–12].
Rhomboid proteins are the largest family of intramembrane proteases [11]. In contrast to the site-2 protease and γ-secretase/signal peptide peptidase intramembrane protease superfamilies, rhomboid proteins are serine proteases mechanistically, and differ functionally by releasing factors to the outside of the cell, rather than into the cytosol [13]. Originally discovered through genetic analysis of Drosophila embryogenesis, rhomboid proteases initiate Drosophila EGF signaling by releasing transmembrane EGF precursors Spitz, Keren and Gurken from the membrane [9, 14, 15]. Particularly exciting is the recent implication of rhomboid proteases in central processes within human pathogens (recently reviewed in [16]). A rhomboid functions in quorum sensing by a pathogenic bacterium, although the substrate that it activates isn’t the signal itself, but rather a component of a transporter [17]. Malaria and related parasites use their rhomboid enzymes to cleave adhesins in order to dismantle the junction formed between parasite and host during invasion [18–20]. The role of rhomboid in other protozoa is less clear, but may also involve shedding of adhesion molecules, like in a parasitic ameba, for example, during phagocytosis and immune evasion [21].
The immense spectrum of roles that intramembrane proteases are known to play continues to expand, but already warrants a thorough investigation of the underlying enzyme mechanisms. In addition to the general interest in intramembrane proteases, it’s also becoming appreciated that such insights could have useful applications, especially for inhibitor development and its therapeutic potential [16]. Although the biochemical complexity of these enzymes has made achieving a detailed knowledge of their function difficult, recent years have witnessed a dramatic shift in our understanding of intramembrane protease mechanisms. In this regard one enzyme stands out: currently multiple structures, defined reconstitution systems, a myriad of mutants, and molecular dynamics simulations have been achieved for the bacterial rhomboid GlpG. My goal is to integrate the diverse experimental approaches that have recently been applied to study GlpG, scrutinize the picture that is starting to emerge, and evaluate to what extent these properties may be common to rhomboid proteases in general.
THE STARTING LINE: BIOCHEMICAL RECONSTITUTION
The catalytic function of Drosophila rhomboid-1 in signaling was deduced from a combination of cell-based assays, protease inhibitor profiling, and mutagenesis experiments [9]. All of these approaches unified into a model whereby a serine protease-like active site, assembled from transmembrane segments, lies immersed within the membrane. But the experimental support for this model was entirely indirect: intramembrane proteolysis could not be reconstituted outside of a living cell, making biochemical interrogation impossible.
This obstacle took nearly four years to overcome, and ultimately just required a better understanding of rhomboid’s enzymatic properties. The initial confounding issue was whether a single polypeptide alone could catalyze intramembrane proteolysis; γ-secretase, the only intramembrane protease that had been purified in active form, required three other proteins for activity [22, 23]. Ultimately, analysis of dozens of rhomboid orthologs revealed most were active when examined in heterologous cells despite sharing less than 20% sequence identity [24–26] (Figure 1), and displayed common substrate requirements [27]. These studies allowed a more informed approach; simplified prokaryotic rhomboid proteases that share overall enzymatic properties and cleave surrogate substrates made attractive models for recombinant expression and biochemical reconstitution.
Figure 1. Sequence and structural features of the rhomboid transmembrane core.

Top panel lists all residues used in the E. coli GlpG structure analyses (residues 87–276), aligned with the corresponding regions of three other well-studied rhomboid enzymes (rhomboid-1 from Drosophila, human RHBDL2, and B. subtilis YqgP). As such, amino terminal domains of all enzymes, and the seventh TM and carboxy terminal domains of DmRho1, RHBDL2 and YqgP are not shown. Residues involved in nucleophilic catalysis (S201 and H254 for GlpG) are shaded in green, presumed electrophilic catalysis residues (H150 and N154 for GlpG) are shaded in blue, while conserved structural residues are shaded in red. Helical secondary structure is highlighted by common color shading among the sequence alignment (top), cylinders in the topology diagram (lower left), and crystal structure images (lower middle and right). Note the intramembrane location of the catalytic serine (circled with red star), and the protruding L1 loop (boxed lower right). By convention, the ‘back view’ centers on TM5 and TM2, while the ‘front view’ centers on TM1 and the L1 loop.
Despite the inherent complexity of membranes, the reconstitution assays were elegantly simple: proteolytic activity was achieved with recombinantly-expressed and purified rhomboid proteins when the appropriate detergent, substrate and, for some, membrane lipid were provided [28–30]. In fact, rhomboid polypeptides consisting of fewer than 200 residues could catalyze complete intramembrane turnover of a substrate, without the need for protein partners, cofactors, energy, or prosthetic groups [28]. This advance proved that rhomboid proteins themselves were catalytic, and paved the way for including them in the Enzyme Commission database as the only family of intramembrane serine proteases (EC 3.4.21.105). Importantly, this work also provided the long-sought tractable system with which to interrogate their biochemical mechanism, as well as offering validated candidates for structural analysis.
RHOMBOID STRUCTURE: GUIDED TOUR OF A PROTEASE
Structures come into focus
Deducing the atomic structure of a rhomboid protease heralded a renaissance in the intramembrane proteolysis field. Long-standing disbelief in the phenomenon was dispelled, and a new era of pursuing a sophisticated knowledge of rhomboid protease mechanism could begin. The structure of a site-2 family protease has also been determined recently [31], but the current discussion will be focused on rhomboid, since a detailed comparison has been reviewed recently [32].
Within the span of less than a year, eight structures of a bacterial rhomboid ortholog GlpG were published. Five of these were different conformers of GlpG from Escherichia coli [33–35], another two resulted from a DMSO-soaked crystal [36] and a point mutant [37], and another still was of an ortholog from Haemophilus influenzae [38]. All structures were of GlpG in the detergent-solubilized state, and only of the transmembrane core: obtaining diffracting crystals required removal, or in the case of H. influenzae absence, of the cytoplasmic amino terminal domain (Figure 1). Despite the diversity of the eight structures, which were crystallized under varying conditions and in four different laboratories, the structures overlaid with a surprising degree of congruity in all regions but one (discussed later). This remarkable observation inspired some confidence in the notion that the structure is not perturbed to a large extent by its environment, and thus could represent the cellular conformation of GlpG.
The overall shape of GlpG is a compact, helical bundle comprised of six helical transmembrane segments (TMs) and an unusual lateral protrusion (Figure 1). Surprisingly, nearly the entire protein appears to be immersed in the membrane: even the exceptionally long L1 loop, which that connects TMs 1 and 2, emanates laterally to adopt a hairpin orientation that would be partially immersed in the top leaflet of the membrane. This asymmetric shape has not been encountered previously, or since, in membrane protein structure, and the protrusion likely serves as a ‘flotation device’ to position the enzyme in the membrane (discussed later). This property may be of wider significance, because this region of GlpG shares sequence homology with the derlin family of proteins implicated in ER associated degradation [30, 39, 40].
It is worth emphasizing that two structural characteristics in particular ratified the intramembrane serine protease model. The catalytic serine lies submerged ~10A from the presumed surface of the membrane, making the site of catalysis intramembrane (Figure 1). Secondly, visualizing in all structures the catalytic serine hydrogen-bonded to the presumed catalytic histidine base, which is distant from the serine in the primary sequence (Figure 1), provided the final proof for the widely-accepted serine protease model.
The structures coupled with functional analyses also offer new and deeper insights into how rhomboid enzymes are constructed and function.
Architectural features: the four keystones
Sequence analysis of rhomboid proteins identified 20 residues that are conserved in most if not all members (Figure 1). Early experimental analysis suggested catalytic functions for four residues [9]. The other conserved residues were anticipated to play important structural roles, and integrating crystal structures and functional analyses now provides a framework for understanding their roles. While a multitude of local interactions permeate the enzyme, evolutionary conservation highlights four elements that act as architectural keystones to stabilize the structure (Figure 2).
Figure 2. Four keystones of E. coli GlpG rhomboid protease structure.
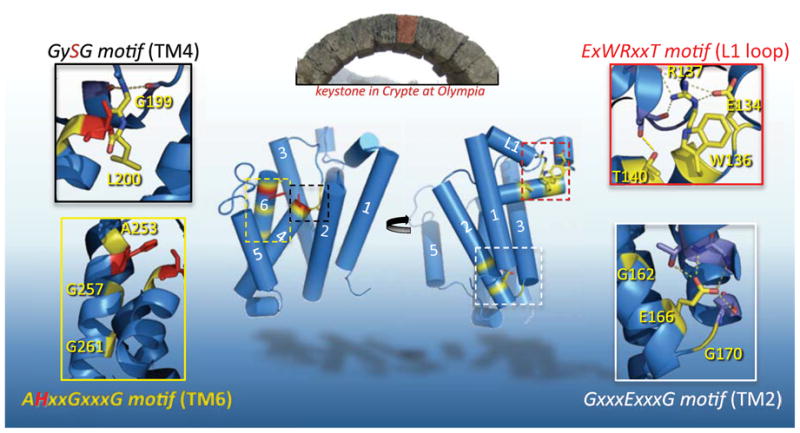
The central stone in an arch is called a keystone, and although arch stability depends on many factors, the keystone occupies a key, central stabilizing position. Pictured in the top inset is the Crypte at Olympia, Greece, with the keystone false-colored red (photo: S. Urban). The back (left) and front (right) views of GlpG are shown. By analogy, ‘keystone’ residues (highlighted in yellow, active site residues in red) of four conserved motifs are depicted in each inset. From top right and moving clockwise: the sidechain of R137 of the ExWRxxT motif reaches upwards to make 5 hydrogen bonds within the L1 loop, two to E134, and three to backbone carbonyls of L121 and A124, while T140 hydrogen bonds to the carbonyl of L123. This array of interactions together stabilizes the lateral hairpin. The E166 sidechain of the GxxxExxxG motif in TM2 makes four hydrogen bonds: three to backbone and sidechain atoms of V96 and T97 in TM1, and one to the hydroxyl of S171 in TM3, bringing the three TMs into an apex on the cytosolic side (bottom). TM2 and the L2 loop bend around G162 and G170, respectively. G199 in the GySG motif of TM4 hydrogen-bonds with the backbone carbonyls of H141 and M144 of the L1 loop, while L200 is buried into the ‘floor’ of the enzyme. AHxxGxxxG motif in TM6 mediates close approach of TM6 with TM3 (via A253), and with TM4 (via G257 and G261). Oxygen atoms are in red, nitrogen atoms are in blue, and dashed lines depict hydrogen bonds throughout.
The first of these keystones is a conserved E/QxWRxxS/T motif situated in the L1 loop, which protrudes laterally to form the hairpin extension. This membrane-embedded conformation is stabilized by an under-belly of hydrophobic residues, which when mutated, greatly decrease enzymatic activity [41]. The residues of the E/QxWRxxS/T motif lie on the bottom face of the hairpin, yet the sidechains of these residues do not point into the membrane, but rather point upwards into the loop to stabilize it (Figure 2). The arginine donates five hydrogen bonds to other residues within the L1 loop, including the preceding glutamate, providing a key stabilizing influence: mutagenesis of the single arginine mutant strongly reduces or abolishes protease activity [37, 41]. The conserved threonine lies one helical turn after the arginine, and also hydrogen bonds with a mainchain carbonyl on the upper half of the L1 loop, providing another stabilizing interaction between the lower and upper helices of the lateral L1 loop hairpin. The role of the tryptophan is less clear, but could be involved in shielding the arginine from the membrane. In support of this hypothesis, mutating the tryptophan to alanine strongly reduces enzyme activity in cell membranes [42], but this mutant has enzymatic activity indistinguishable from wildtype when freed from membranes and assayed in the detergent-solubilized state [41].
A conserved GxxxExxxG sequence is situated on the lower third of TM2 and continues into the cytosolic L2 loop (Figure 2). In the structure, these residues form and stabilize an abrupt turn, because the polypeptide chain needs to cross over to the other side of the membrane-embedded L1 loop. This cross-over allows TM3 to form a V-shape with TM1, cradling the L1 loop in the upper gap between them. The turn is first accomplished by a bend of the TM2 helix around the first glycine residue in the GxxxExxxG motif. The glutamate lies at the bottom of TM2, with its side-chain reaching out to hydrogen-bond with the bottom of TM1 and TM3. The final glycine of the motif forms a bend in the cytosolic turn before TM3. The overall effect is bringing together of helices TM1, 2 and 3 into almost an apex at the cytosolic face of the enzyme’s front half (Figure 2). Mutation of either of the two glycines or the glutamate dramatically reduces enzyme activity (Moin and Urban, unpublished observations), supporting an important role in structure stabilization.
Following TM3, a conserved GySG motif centered on the catalytic serine in TM4 served as a signature motif in rhomboid enzymes [9, 11]. Transmembrane prediction programs were ambiguous on where TM4 starts, with some predicting the catalytic serine to lie at the very top of TM4. This was initially confounding because the active site was anticipated to be intramembrane. The structure confirmed that the serine was indeed recessed into the plane of the membrane, but the serine sits as the topmost residue on TM4, because residues preceding the serine are in an extended loop conformation (Figure 2). The structure also revealed that the conserved residues around the active site serine provide stabilizing points, holding the extended L3 loop in place. The backbone NH group of the first glycine hydrogen bonds to the backbone carboxyl oxygens of H141 and M144 in the L1 loop. Several different residues preceding the catalytic serine are permitted among various rhomboid orthologs, and in GlpG this residue is a leucine that faces downward and, as such, is buried into the ‘floor’ of the enzyme. The overall consequence of this structure is a stabilized, unobstructed cavity above the catalytic serine, which presumably allows entry of water from the outside of the cell, and space for substrate polypeptide.
Lastly, a conserved alanine was noted to precede the catalytic histidine, while a canonical GxxxG transmembrane dimerization motif exists below it on TM6 (AHxxGxxxG). Both of these elements are universally conserved among all rhomboid proteases (Figure 1). The GxxxG motif is a particularly common and strong transmembrane dimerization motif that mediates close association between helices in various membrane proteins [43, 44]. However, it was unclear with what this segment might pair [9]. The crystal structure revealed that the GxxxG motif of TM6 forms a tight association with TM4, bringing together the catalytic serine on TM4 and its base partner histidine on TM6 (Figure 2). Accordingly, even a single valine substitution in the second glycine of the GxxxG abolishes detectable protease activity [41]. Similarly, the alanine preceding the histidine packs into a small hole on TM3, forming a tight association with a neighboring helix on the other side of TM6. In this way, TM6 makes a series of tight contacts with neighboring helices both immediately above and below the catalytic histidine, thus cementing it in place.
The catalytic core
The active site chemistry of soluble serine proteases has been the focus of investigation for over half a century [3, 4]. With defined reconstitution systems and crystal structures now available, rhomboid proteases are starting to enter this exciting phase of analysis. However, our current understanding of rhomboid catalysis is strictly limited to analogy to serine protease paradigms; structures of rhomboid with bound substrate or inhibitors, or chemical probes for interrogating its active site chemistry, have not yet been realized. Nevertheless, soluble serine proteases, although evolutionarily and structurally distinct, serve as an instructive starting point for rationalizing rhomboid catalysis.
Two catalytic elements comprise the active sites of serine proteases: residues that directly sever the C-N amide bond, and neighbouring electrophiles that stabilize the negatively-charged O of the transition state (for a recent review, see [3]). Initial investigation of rhomboid in signalling focused attention on four transmembrane residues in particular as key for protease activity: an asparagine on TM2, a glycine and serine on TM4, and a histidine on TM6 [9]. The role of the serine and histidine were straightforward to understand: the amide bond is initially severed through concerted action by a serine nucleophile and a general base, usually a histidine or lysine. The unsung hero of the reaction is the base since it catalyzes nearly every step in the reaction, while the serine is simply the initial attacking moiety. By analogy, if the serine is the scalpel, the histidine is the surgeon’s hand. The crystal structures revealed the serine interacts with the presumed catalytic histidine (Figure 3). Yet intriguingly the serine is in an unusual position, being the only residue in the entire rhomboid protein that lies in a disallowed region of the Ramachandran plot. While the significance of this, if any, is not clear, it might serve to position the serine for catalysis, by analogy to the ‘nucleophile elbow’ of α/β hydrolases [45].
Figure 3. The two catalytic elements of the GlpG rhomboid protease active site.
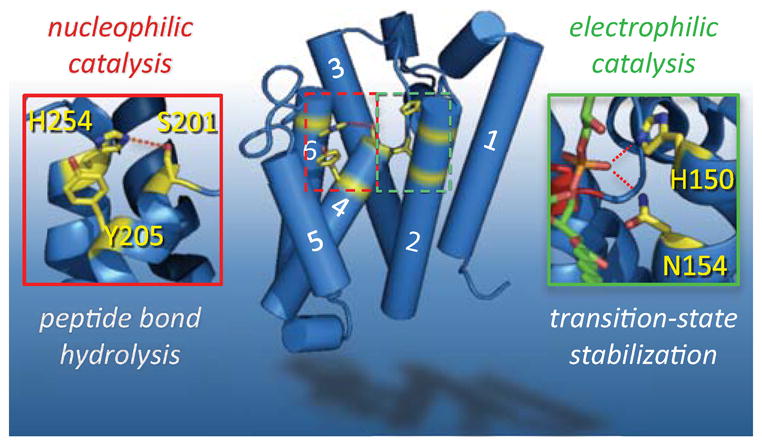
Nucleophilic catalysis (left) that is responsible for severing the peptide bond is carried out by a serine protease-like catalytic dyad: S201 (on TM4) serves as the nucleophile while the catalytic base is H254 (on TM6). H254 hydrogen-bonding to S201 is depicted with a dashed red line. The phenol ring of Y205, emanating from TM4, stacks under the H254 imidazole ring, providing some support for the catalytic base (although a tyrosine could not salt-bridge with the imidazolium ion or alter its pKa, as might occur in some proteases). Electrophilic catalysis (right) is required to stabilize the oxyanion of the tetrahedral intermediate. The residues that form this entity are less defined, but are suggested by interaction with a lipid phosphate that protrudes into the active site of structure 2IRV. The lipid is shown in green, with oxygen atoms in red. Hydrogen bonds between the phosphate oxygen and the sidechains of H150 and N154 (emanating from TM2) are depicted by red dashed lines.
Prior to structure determination, a major point of debate was how water is delivered to the membrane-submerged active site. All rhomboid structures revealed that the catalytic serine, the region to which water must be delivered, lies at the bottom of a funnel-shaped cavity that is ~10 A in diameter at its opening and is lined with hydrophilic groups (Figure 1). This architecture immediately suggested that water accesses the active site from the large opening that faces the periplasm. This has been confirmed in E. coli cells [46]; probe accessibility studies indicate that a ~5 kDa membrane-impermeable probe can modify several transmembrane positions including that of the catalytic serine (although not the histidine). The limitation of cross-linking experiments is that they are a ‘covalent trap’: even if the conformation that is susceptible to cross-linking is rare or short-lived, once cross-linked it becomes permanently labeled. Nevertheless, these studies indicate that the cavity is open at least part of the time, and to surprisingly large molecules.
What has been less apparent is the identity of the second necessary element for serine protease catalysis, the oxyanion-stabilizing pocket. Early analyses of rhomboid function put forward two possibilities [9]. As in chymotrypsin, the backbone NH of a glycine two residues upstream of the catalytic serine might stabilize the oxyanion [3, 4], since its mutation to even alanine abolishes protease activity in vitro and in cells [9]. Alternatively, by analogy to subtilisin [3], in which a conserved asparagine stabilizes the oxyanion, the asparagine in TM2 of rhomboid was noted as an alternative oxyanion-stabilizing entity [9].
The oxyanion transition state is difficult to study directly because of its unstable nature. Instead, stable surrogate compounds are often designed to mimic the tetrahedral intermediate [3], but currently no rhomboid structure has been solved with a transition state inhibitor bound. Nevertheless, one fortuitous structure provides the best alternative currently available: a structure was solved with a phosphate lipid headgroup protruding laterally into the active site [35]. The phosphate shares some properties with the transition state, being both tetrahedral and containing negatively-charged oxygen. In the structure, the phosphate lies within hydrogen bonding distance to the asparagine, and a conserved histidine one helical turn above the asparagine on TM2 (Figure 3). It may also be bonded to the backbone nitrogen of the catalytic serine, but not G199 that lies two residues upstream of the catalytic serine, which instead forms a ‘keystone’ interaction (Figure 2). Therefore, the best candidates for oxyanion-stabilizing elecrophiles are the sidechains of the asparagine and histidine on TM2, both of which are universally conserved. Mutation of either residue reduces catalytic activity in some, but not all, rhomboid enzymes that have been investigated [9, 24, 29, 41, 47].
It is worth noting that the structure also definitively resolved the long-standing question of whether rhomboid proteases use a catalytic triad. The role of the third residue in catalytic triads, and its conspicuous absence in some serine proteases, continues to be a point of heated debate despite four decades of investigation [3, 6, 48–50], and rhomboid was no exception to this inevitability. The only candidate for a histidine-stabilizing residue based on sequence conservation was the asparagine on TM2. Asparagines function within catalytic triads of cysteine proteases and some lipases. But the importance of the asparagine for rhomboid activity was contradictory from the beginning: the asparagine was important for activity of Drosophila rhomboid-1 [9], but not for human RHBDL2 [9]. Subsequent analyses showed it to be required in some rhomboid proteases, but not others, and this did not correlate with whether the enzyme was studied in vivo [9, 24, 29, 51] or in vitro [30]. This mystery had to wait until the structure revealed its position; the asparagine was not bonded to the histidine base, but was potentially in a position to stabilize the oxyanion (Figure 3). A notable minority of serine proteases use catalytic dyads [50].
Completely unexpected was the visualization of a tyrosine residue, one helical turn underneath the catalytic serine, stacking onto the catalytic histidine ring (Figure 3). The tyrosine would thereby serve to orient the histidine ring, which is thought to be one of several postulated functions of the third residue in catalytic triads [3]. Accordingly, a tyrosine (or phenylalanine) at this position is well-conserved among rhomboid proteases (Figure 1), and its mutation reduces protease activity by 4–5 fold both in vitro and in living E. coli cells [41, 42].
It must be stressed that the current rhomboid structures are of a ‘resting’ protease. In the absence of structures with bound substrate, transition-state mimics, and/or inhibitors, it is not possible to delineate the mechanistic details of the catalytic events. In particular, the key question of what triggers abstraction of the proton from the serine by the histidine base remains a mystery, as it does for other serine proteases. In this light, rhomboid proteases present a new and radically different, albeit challenging, context for investigating this central question of enzyme catalysis.
RHOMBOID PROTEASE FUNCTION AND DYNAMICS
The molecular architecture of rhomboid proteins illustrates the enzymatic complexity of intramembrane proteolysis: the hydrated catalytic core is submerged beneath the presumed membrane surface, but is held separate from membrane lipids by protein segments. How does a transmembrane substrate, which normally resides in membrane lipid, gain access to the internal active site of rhomboid?
This problem emphasizes the importance of understanding protein dynamics in rhomboid catalysis, and this to a large extent remains the principal challenge in the field. Recent structure-function enzyme analyses, and computer simulations, have taken the first steps towards outlining these key issues. The answer lies in a complex, and as yet poorly understood, interplay between rhomboid protease gating, substrate unwinding, and membrane lipid dynamics.
Substrate gating: finding a way in
Substrate gating served as a starting point to begin addressing rhomboid protein dynamics: part of the rhomboid protein needs to move to open a path for substrate entry. Addressing how this might occur was initially facilitated by the availability of multiple crystal structures revealing different enzyme conformations, but two contradictory models were put forward (Figure 4). The first structure to be solved revealed a compact molecule with transmembrane segments packing against each other along the perimeter of the enzyme, except between TM1 and TM3. Here, a gap existed between the tops of these segments owing to their V-shaped orientation, but this gap is plugged by insertion of the overlying L1 loop. Because this gap was large enough to accommodate a polypeptide chain, and given its conserved and unusual nature, the L1 loop was assumed to be the lateral substrate gate [34, 52, 53].
Figure 4. Structural and functional analyses of GlpG substrate gating.
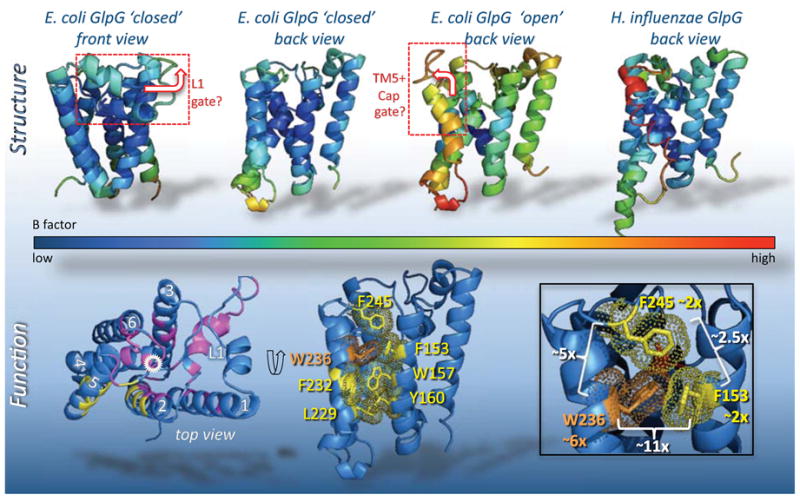
Models for substrate entry posited either lifting away of the L1 loop (boxed in top left), or displacement of TM5 and the overlying Cap (shown already in the open position). In addition to the conformational differences between structures, B factors also serve indirectly to suggest regions of conformational flexibility. The three structures are colored by B factor values (see underlying spectrum). In the first structure (2IC8, leftmost two views), the B factors are relatively low throughout, while in subsequent structures the higher B factors tend to center on TM5 and connecting loops. Bottom panels summarize effects of GlpG mutants on protease activity from a screen to identify gating residues [41]; from an analysis of ~50 mutants engineered in all TMs and outer loops, mutants that increased protease activity only localized in the TM5, TM2 and Cap interface (in yellow). Locations of targeted mutants that did not increase activity are shown in magenta (catalytic serine is circled in white). In the middle lower panel, the interface that prohibits substrate access is shown in the ‘closed’ form, with protruding sidechains and their electron density in yellow or orange (for W236). The lower right panel magnifies the interface formed by W236 (TM5), F245 (Cap) and F153 (TM2). The greatest stimulatory effect of any single mutant within GlpG occurs when W236 (shown in orange) is mutated to alanine, resulting in a ~6-fold (~6x) increase in enzyme activity, compared to an approximate doubling (~2x) when either F153 or F245 are mutated to alanine. White brackets summarize the increase in protease activity relative to wildtype of all pairwise double mutants [42]: note that only the F153A+W236A mutant combination is synergistic, resulting in a ~11-fold increase in protease activity.
Conversely, other structures, resulting from different crystallization conditions, revealed major conformational differences in TM5 and the L5 Cap connected to it, rather than in the L1 loop [33, 35]. The TM5 tilted laterally away from the core by about 35 degrees at its top and the Cap moved upwards, opening a potential path for substrate entry on the side opposite the L1 loop. This ‘open’ conformation was not observed initially because in the first structure TM5 was involved in crystal packing against a neighbouring rhomboid molecule in the asymmetric unit. However, the physiological significance of this ‘open’ conformer was equally unclear, because all structures were solved in the absence of membrane, which would normally apply a lateral pressure on the enzyme that might hold TM5 in place. Moreover, substrate entry from the TM5 direction seemed unlikely because it would place the substrate into the active site in the opposite orientation compared to nearly all serine proteases (discussed later) [38].
Crystal structures only capture stable conformations as snapshots of a dynamic enzyme. Ultimately an enzymatic approach was used to evaluate these alternatives [41]. The rationale relied on the induction that gate opening would be a slow, rate-limiting step; therefore mutating the contacts that stabilize the gate, wherever it might be, in the closed conformation might lead to gate opening more readily and therefore rhomboid variants with higher protease activity. To avoid bias on the one hand, and ‘one-hit wonders’ on the other, ~50 different mutants of rhomboid were engineered that disrupted key interactions within all transmembrane segments and outer loops in multiple ways (Figure 4) [41].
The analysis converged on the interface between TM5 and TM2 that is mediated by interdigitating large, hydrophobic residues as the substrate gate (Figure 4). Mutants in all other parts of the enzyme, especially those that disrupted the stability or interactions with the L1 loop, resulted in decreased enzyme activity [41]. Recent molecular dynamics simulations also support structural roles for the L1 loop, both within the enzyme and how the enzyme sits in the membrane [54]. Conversely, not one but three classes of engineered variants in TM5 displayed dramatically enhanced protease activity: reducing the size of the three residues on the helical face of TM5 that interfaces with TM2, changing pairwise interacting residues on TM5 and TM2 to alanine, or placing a proline on the opposite side of TM5 to kink it outwards but without affecting the TM2:TM5 interface (Figure 4). All of these classes increased enzyme activity compared to the wildtype enzyme, from four-fold for the triple valine mutant on TM5, to a full order of magnitude increase when the upper-most residues on TM5 and TM2 were changed to alanine in a double F153A+W236A mutant [41].
What is equally striking is how a simple perturbation can produce an enzyme with dramatically increased activity: the strongest single activating mutant identified was W236A on TM5 (Figure 4), which by itself increased activity ~6-fold [42]. Other neighbouring residues failed to have such activating effects, including phenylalanine 245 that emanates from the overlying L5 Cap and dips to a position by W236 (Figure 4). Moreover, synergistic effects were observed only when mutations on TM5 were combined with those on TM2, rather than with the L5 Cap phenylalanine (Figure 4) [42]. These observations suggest that upper residues of TM5 have the greatest effect on substrate gating. Remarkably similar stimulatory effects were observed when these gate-open mutants were analyzed and quantified in living E. coli cells, arguing that the gate-open mutations both have the same effect in natural cell membranes [42].
It is important to realize that a defining feature of a gate is that it acts to limit the activity of an enzyme. In this regard, mutations in TM5 are strongly activating while those in the L5 Cap are weak, arguing that TM5 movement, not Cap movement, acts as the rate-limiting gate [41, 42]. This is also consistent with recent molecular dynamics simulations that indicate the L5 Cap to be highly mobile [54]. As such, the L5 Cap may not be able to occlude substrate access as would be required of a gate, and this may explain why mutations in the L5 Cap are only weakly activating. In contrast to the mobility of the loops, the TM segments are much more stable, but notably TM5 is unique as it is the only TM that is not hydrogen bonded, thus appearing to be poised to open. Beyond these observations, it should be noted that the timescale of current simulations is too limited to provide any information on the interchange between various conformers.
While these structure-function enzymatic analyses provide a compelling model, little is known about the gating mechanism in detail. In particular, the order of events in substrate binding, gate opening and substrate access remain entirely unknown. Even the extent of the motions required for substrate access and how far they extend into the membrane are unclear [36]. Motion in the TM5/L5 Cap region is likely to be a continuum, and the extent of gate opening may depend on the requirements of the substrate being cleaved. Nevertheless, direct visualization of a phospholipid headgroup in the GlpG active site in one structure suggests that exposure of the active site to lipid deeper within the membrane may not be that uncommon or unfavourable [35].
Substrate specificity and diversity
The notion of a rhomboid transmembrane segment tilting at its top to clear a path for substrate entry into the active site is also attractive because it fits well with previously-defined substrate requirements [27]. Unlike most soluble proteases, rhomboid proteins display a notable lack of dependence on a particular sequence or single residue in substrates. Instead, rhomboid proteases rely primarily (but not exclusively) on residues that disrupt alpha helical structure. This property implied that transmembrane helix unwinding to facilitate entry into the active site is a defining requirement for rhomboid intramembrane proteolysis [27].
Substrate specificity was first assessed with Drosophila rhomboid-1 and Spitz, the first identified natural enzyme-substrate pair, but these principles were found to be widely-applicable [24, 27, 28]. Chimeras between cleaved and uncleaved proteins revealed that neither the extracellular nor cytosolic regions were required for substrate recognition; specificity relies on only the topmost seven transmembrane residues of Spitz (Figure 5), which alone are sufficient to convert other type I transmembrane proteins into rhomboid substrates. Extensive mutagenesis of the seven-residue substrate region revealed two determinants [27]: there was a requirement for the first two residues to be small and hydrophilic, while distal residues had to have low helical propensity. This helix-breaking glycine-alanine pair in Spitz could be moved, but its placement was constrained and correlated with remaining on the same face of the helix [27]. These general features were ultimately evaluated with a panel of over 15 other rhomboid proteases across evolution (including GlpG) both in vivo and in vitro [24, 25, 27], and in general were found to apply to most rhomboid enzymes, although some rhomboid proteases display specializations [18].
Figure 5. Substrate requirements for rhomboid processing.
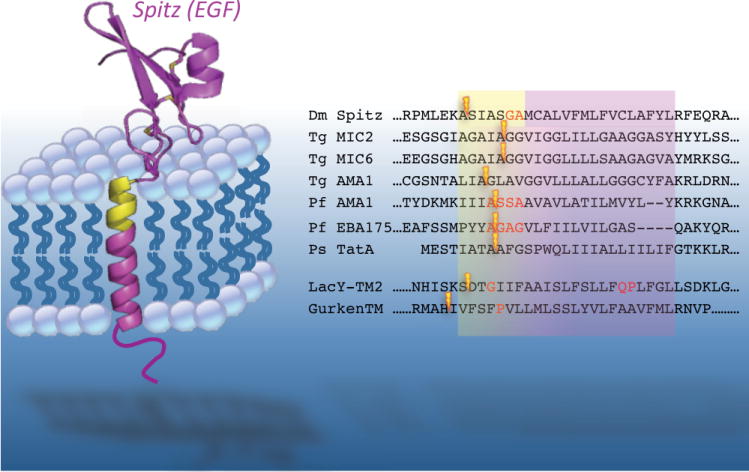
The first identified rhomboid substrate, the Drosophila EGF ligand Spitz, is depicted in the membrane. Note that structural information is available for only the EGF domain. The ‘substrate motif’ in Spitz that is necessary and sufficient for rhomboid cleavage is highlighted in yellow. The right panel shows all of the cleavage sites, depicted with lightning bolts, currently known for different rhomboid protease substrates (excluding the mitochondrial rhomboid substrates). Approximate transmembrane and substrate regions are depicted, while residues experimentally determined to act as helix-breaking are in red font. The cleavage sites for the top seven substrates were determined with natural proteins, while the bottom two are artificial substrates. Note that in natural substrates the cleavage site always follows an alanine residue. Organism designations are: Dm Drosophila, Tg Toxoplasma gondii, Pf Plasmodium falciparum, Ps Providencia stuartii. The artificial substrates are chimeric proteins harbouring the transmembrane segments from the E. coli LacY permease and Drosophila Gurken.
GlpG substrate specificity has recently been interrogated in greater detail using a chimeric substrate harbouring the second transmembrane segment from the E. coli LacY permease [55]. This is not a physiological substrate for any rhomboid, but serves as a useful surrogate because it is cleaved with high efficiency. A notable strength of these studies is that they were conducted with endogenous GlpG, by comparing effects in wildtype cells to the GlpG deleted strain, and confirmed in vitro with purified enzyme. What is particularly remarkable is that this independent approach with a completely different substrate found all of the same features applied: the upper cleavage site required small residues, while the only distal glycine (Figure 5), presumably analagous to the Spitz glycine-alanine, was exquisitely sensitive to mutation: replacing only this glycine with a cysteine abolished cleavage [46].
Interestingly, two additional properties were clarified through these analyses. First, a second helix-breaking motif was discovered further down the helix (Figure 5), because the structure of the LacY permease showed that TM2 bends around a helix-breaking glutamine-proline pair [56]. Mutation of this QP pair in LacY reduced cleavage, while placing it into other substrates enhanced cleavage, explaining why the LacY transmembrane segment is such an efficient substrate for GlpG compared to Spitz or other natural substrates [55]. Like the GA in Spitz, the QP could be moved, but its position was constrained to the same face of the transmembrane helix. This requirement could be partially overcome if the cleavage site was also moved, arguing that it is not a particular face of the helix per se, but rather the placement of the helix break relative to the cleavage site that is important.
Second, properties of the cleavage site in LacY were also examined in greater detail [55]. A genetic screen identified 8 pairs of residues flanking the cleavage site that supported efficient proteolysis. The clear preference was for small residues preceding the scissile bond (P1 position), and negatively-charged residue after the cleavage site (P1’ position). The cleavage site in LacY TM2 lies at the extracellular-transmembrane junction, rather than being embedded within the membrane. Indeed, cysteine residues introduced into this region could be labelled with a membrane-impermeable agent that reacts with thiols [46]. This is consistent with residues essential for rhomboid cleavage being at the top of transmembrane segments (Figure 5), where water is known to permeate the lipid headgroup layer of membranes [57]. Nevertheless, the position of the cleavage site in the substrate probably does not reflect where cleavage occurs relative to the membrane: the complex geometry of the internal active site cavity of GlpG, which lies a lateral distance from the initial point of contact with the substrate, blurs the distinction of where precisely the cleavage is received within the substrate molecule. Indeed, the cleavage site probably represents how far each polypeptide has to travel as it bends into the membrane-recessed active site.
While these studies define general principles underlying rhomboid substrate requirements, the specific details should be treated with caution; all of the analyses were conducted with artificial substrates since the natural substrates of GlpG remain unknown [29, 51]. This artificial context could have unintended differences. For example, all known cleavage sites for rhomboid enzymes analyzed in their natural environment invariantly followed an alanine in the P1 position, and never has a charged residue been found in the P1’ position (Figure 5) [17, 19, 58–60]. Thus, the residues that flank the cleavage site in natural substrates under physiological conditions may have properties that are different from those experienced in current assays. Although not yet complete, all current evidence agrees that substrate dynamics via helix unwinding, in addition to gate opening in rhomboid proteases, is an integral part of intramembrane proteolysis, and underlies its specificity.
Protein dynamics and conformational transitions
Perhaps the most enigmatic aspects of rhomboid function currently center on the general dynamics of the protein. The only study in this area comes from recent molecular dynamics simulations [54]. While in the simulations GlpG is globally stable overall, it’s intrinsically dynamic, even wiggly, compared to other membrane proteins. For example, the catalytic serine moves up and down by several angstroms during the simulations, and multiple hydrogen bonds within the molecule are redistributed. The role of the observed flexibility in catalysis remains unknown, but warrants further scrutiny. Moreover, there appear to be two main hydrogen bond clusters within the molecule, with TM3 being shared by the two. One possible implication of this arrangement is that TM3 could relay structural conformation changes from one part of the molecule to the other. Since the structure with a lipid trapped within the active site served as the subject for the simulations, it remains to be seen whether the observed properties are common to all conformers, and whether the dynamics change when lipid is removed or substrate is bound.
Beyond the general motions within the enzyme, little is known about the conformational changes that accompany protease activity. Current molecular dynamics simulations are not instructive because the limited timeframe is insufficient to observe interchange between different conformations.
MEMBRANE IMMERSION OF A PROTEASE
In addition to being serine proteases, rhomboid proteins are integral membrane proteins with an unusual shape, and this reality raises the challenge of membrane biophysics to understanding enzyme function. Yet rhomboid proteins have been studied as proteases first, and only recently as membrane proteins.
Experimental reconstitution of rhomboid proteins into different phospholipid environments in vitro was found to have profound consequences on activity [28]. What’s particularly intriguing is the diverse response of rhomboid enzymes to different reconstitution lipids: the activity of some rhomboid enzymes is dramatically increased in almost any type of phospholipid, while others display a clear preference for particular lipids, but these preferences are different among rhomboid proteins. In fact, the type of lipid used in reconstitution was found to be the condition that has the greatest influence on protease activity. These experiments indicate that the way in which rhomboid proteases are positioned into the membrane is likely to be an important factor in their enzymatic function, and could potentially constitute a regulatory function [28].
The asymmetric, and unprecedented, shape of detergent-solubilized GlpG made it impossible to predict from structural information alone how it might be positioned in a cellular membrane [34, 37]. Recent molecular dynamics simulations have started to address this complex issue [54]. The simulations were conducted with one structure as a starting point, and in 1-palmytoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine (POPE), a natural component of bacterial membranes, and 1-palmytoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC), which E. coli cannot synthesize. In the simulations, GlpG sits tilted in the membrane (Figure 6). This orientation is the result of many interactions between the enzyme and surrounding lipids, as mediated primarily by hydrogen bonding of residue sidechains to lipid headgroups. The L1 loop is responsible for most of the lipid interactions on the upper surface of the membrane, while interactions to the cytosolic surface of the membrane are mediated primarily by positively-charged residues, which are concentrated on the lower surface of the enzyme. The outcome is GlpG tilting ~12 degrees from the membrane normal in POPE, with the L1 loop acting as a ‘flotation device’ (Figure 6). The tilt is slightly greater in a POPC membrane, owing mainly to altered interactions with the L1 loop residues, but it should be noted that GlpG displays low enzymatic activity when reconstituted into PC lipids [28].
Figure 6. GlpG in a membrane: protease tilt and membrane thinning.
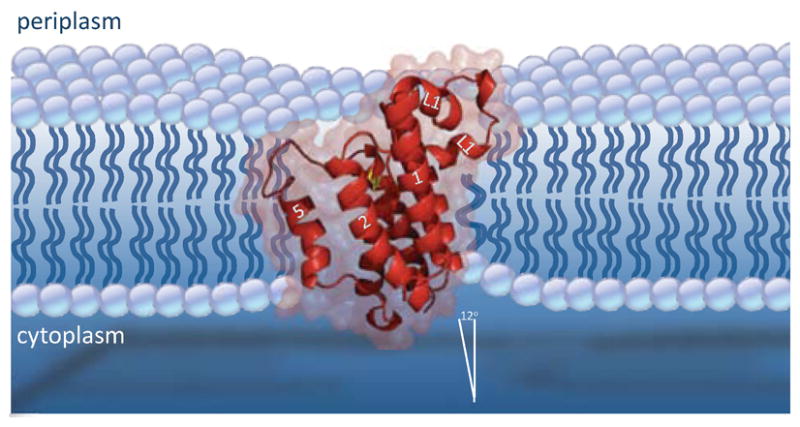
A stylized diagram of GlpG tilt and membrane thinning as suggested by molecular dynamics simulations. Interactions with membrane lipids, including through the L1 loop acting as a ‘flotation device’, serve to stabilize GlpG in a position tilted ~12 degrees from the membrane normal. This results in slightly deeper submersion of the catalytic serine (in yellow) and TM5. The hydrophobic mismatch between GlpG and membrane also causes deformation of the membrane in the immediate vicinity of GlpG, resulting in a ~4A thinner lipid annulus. The most extreme thinning occurs on the cytosolic side under the L1 loop. Enzyme tilt and membrane thinning may play roles in how substrates approach and are handled by rhomboid proteases.
Many membrane proteins are known to alter the membrane environment in their vicinity [61]. The asymmetric shape and relative sturdiness of the transmembrane core, coupled with the relatively thinner belt of hydrophobic residues that encircle GlpG, also forces lipids in the simulations to reorient their positions to the shape of GlpG [54]. This results in membrane thinning around the first 2 or 3 shells of lipid surrounding the enzyme (Figure 6). Although an extreme thinning to 20A was extrapolated from the crystal structure [37], the actual membrane thickness around GlpG observed in the simulations is nearly twice that estimate, approximately 35A for POPE [54]. Not surprisingly, the thinning is most dramatic under the partially-submerged L1 loop, but overall the result is a 4A thinning around GlpG. This represents approximately a 10% thinning in comparison to the measured 39A glycerol-glycerol distance of membrane POPE lipids.
A necessary caveat of these simulations is that they were conducted in membranes composed entirely of a single phospholipid, POPE or POPC, to simulate the complexity of natural membranes. Cellular membranes are known to be composed of hundreds of different lipid species that vary in headgroup, acyl chain length and degree and position of saturation [62]. It remains to be seen whether mixtures of different lipids might allow lipid species not present in the simulations to adopt preferential positions around GlpG in which the longer POPE lipids were forced to distort. Some early experimental support for this possibility comes from the observation of lipids bound to different parts of the enzyme in the crystal structures, which appear to be of sufficient affinity to be maintained throughout the detergent extraction, purification and crystallization procedure [35]. To what degree the tilt in the simulations is affected by compression of longer lipid remains to be tested. Simulations using more physiological populations of membrane lipids, and further experimental analysis of GlpG directly in lipid membranes using biophysical techniques, could further refine the current view of how lipids interact with the enzyme.
Despite these important caveats, tilting of the molecule in the membrane, coupled with a ~4A thinner lipid annulus, has a number of possible mechanistic implications (Figure 6). First, the active serine may actually be immersed even deeper into the plane of the membrane than previously anticipated: in the POPE membrane simulation the nucleophilic oxygen lies only 5A above the middle of the bilayer [54]. Nevertheless, the active site is well hydrated throughout the simulations, owing to predominance of hydrophilic residue side-chains within the cavity, and prevalence of lipid headgroups.
Secondly, membrane thinning is known to change the tilt angle of transmembrane helices. As such, thinning of the membrane as the substrate approaches the rhomboid enzyme may be important for positioning the substrate for cleavage. Another scenerio suggests that the upper few residues of the substrate may exit the membrane altogether [36], which was also proposed by early studies on site-2 protease [63]. The implications of these models on the mechanism of intramembrane proteolysis have yet to be evaluated functionally.
NOT YOUR TYPICAL SERINE PROTEASE: OUTSTANDING QUESTIONS
At least two further glaring discrepancies remain when comparing rhomboid to other serine proteases. The active site chemistry of serine proteases has been exploited for the development of many classes of inhibitor warheads over nearly half a century of research, some with therapeutic applications [64]. Secondly, studying the biological roles of serine proteases has emphasized that protease activity is exquisitely regulated at the protein level; serine proteases are seldom made in active form [2]. On both levels, rhomboid proteases could not be more different from their soluble serine protease cousins: no selective inhibitors of rhomboid proteases exist, and no direct mechanism of regulation is known for any rhomboid protease.
Prospects for rhomboid inhibition
Protease inhibitors have served as exquisite probes for studying protease mechanisms, as well as being invaluable tools for pharmacological approaches for uncovering the roles proteases play in different processes and organisms [65]. These approaches are particularly important when studying such an unusual membrane-immersed active site, and a family of proteases with hundreds of members conserved in all forms of life, yet whose functions are understood in less than a dozen organisms [13]. Unfortunately, rhomboid proteases have proven to be unexpectedly tricky in the area of inhibitor design.
Initial inhibitor profiling in living cells revealed that rhomboid protease activity is sensitive to only a small subset of serine protease inhibitors [9]. Many serine protease inhibitors are either highly cytotoxic, unstable, or membrane impermeable, therefore a reasonable fear was that this initial inhibitor profile was incomplete despite being carefully controlled. Yet development of a defined, pure enzyme reconstitution system conclusively demonstrated that only isocoumarins are consistent and potent inhibitors of all rhomboid proteases tested [28]. Although not all classes of inhibitors have yet been exhausted, classical serine protease warheads including sulphonyl fluorides (e.g. PMSF, AEBSF) and phosphonates (e.g. DFP), frustratingly proved unable to exert any apparent effect on rhomboid activity even when preincubated in millimolar concentrations with pure enzyme. Unfortunately, isocoumarins are masked electrophiles that are unstable and quite cytotoxic, which limits their application in cellular investigations.
The limited reactivity of rhomboid with serine protease inhibitors was suspected to result from a failure of the compounds to gain access to the protease active site. However, the ensuing rhomboid structures showed that the catalytic machinery is open to the outside environment through a large, aqueous cavity [33–35, 38]. In fact, chemical accessibility probes subsequently demonstrated that a cysteine in place of the catalytic serine can be directly labeled with both membrane-permeable and membrane-impermeable thiol-reactive compounds of a similar size as serine protease inhibitors [46].
The failure of serine protease inhibitor prototypes to influence rhomboid activity may result from its unusual active site stereochemistry. One unexpected implication of the gating mechanism is that the substrate polypeptide approaches the catalytic apparatus from the ‘si’ face, which is stereochemically opposite to most serine proteases. Indeed, α/β-hydrolases, which include β-lactamases, lipases and bacterial leader peptidases, also attack their substrates from the ‘si’ face, and are refractory to inhibition by classical serine protease inhibitors [66]. The only active-site directed warheads that were found to inhibit leader peptidase are β-lactams, and these compounds deserve consideration for testing as rhomboid inhibitor prototypes.
Compounds that do not depend on catalytic cooperation from rhomboid may provide an alternate and more likely source of inhibitors. Substrate-mimicking helical peptides are capable of high potency as competitive inhibitors of both γ-secretase and signal peptide peptidase [67, 68]. Preliminary investigation indicates that helical peptides can also be developed as rhomboid protease inhibitors (S. Urban, unpublished observations). These encouraging early-generation compounds warrant further experimental development and scrutiny.
Future advances in understanding the mechanistic peculiarities of rhomboid proteases is likely to provide new and more informed avenues for inhibitor development. Such compounds will undoubtedly be of high utility in future mechanistic and biological investigations, and could even find eventual applications in the therapeutic realm [16].
Regulation
A notable contradiction in our understanding of rhomboid function is the apparent lack of mechanisms for their regulation. Unlike all other intramembrane proteases, rhomboid proteins cleave full-length proteins and therefore do not depend on a prior cleavage by a site-1 protease [29, 69]. It is this site-1 cleavage that usually serves as a point of regulation for intramembrane proteolysis [10, 70].
Currently the only sources of regulation for rhomboid catalysis rely on transcription of the rhomboid gene, or physical separation of substrate from protease in the cell [71]. The best example of transcriptional regulation is the rhomboid-1 protease during Drosophila embryogenesis, where its strong and dynamic mRNA expression pattern has long served as an atlas for EGF signaling [72]. Separation of substrate and protease is exemplified by Toxoplasma and Plasmodium parasites, in which adhesin substrates are segregated in internal organelles while rhomboid is restricted to the surface of the parasite [19, 20, 73]. Only at the end of the invasion program do adhesins encounter rhomboid protease at the parasite surface as a way of dismantling the junction formed between the host cell and invading parasite.
It is unusual for a catalytic activity to be regulated so indirectly, and it remains possible that current examples reflect circumstances in which regulation has become specialized to meet a particular need, rather than being representative of rhomboid regulation in general. This is underscored by attempting to apply current paradigms to GlpG. E. coli GlpG derives its name from being in the glycerol-3-phosphate (Glp) operon [74], but this gene structure is not conserved among bacteria, and GlpG is not thought to function in glycerol metabolism. Therefore, its transcription profile could be inappropriate for regulating its activity. Separation of substrate from protease in different compartments is also unlikely to be a source of rhomboid regulation in prokaryotic organisms.
The challenge is to understand how a membrane-embedded active site can be regulated. One possibility is regulation by the membrane environment itself, and this has been demonstrated experimentally for many rhomboid proteases, but only in vitro [28]. It remains to be seen whether this potential mechanism is indeed used in a cellular context, and if so, whether it is a more common mechanism for these proteases as a family. In reality, one significant but under-appreciated obstacle to understanding rhomboid regulation is the lack of clues due to the limited biological examples where rhomboid function is defined [13].
CONCLUDING PERSPECTIVES
Despite sharing some catalytic properties with their soluble counterparts as a result of convergent evolution, intramembrane proteases are a different class of enzymes that are only beginning to be studied mechanistically. This area is particularly exciting because the enzymatic properties of intramembrane proteases are likely to be fundamentally different as a result of their immersion in the membrane. Perhaps the most conspicuous issue has been how water is delivered to the active site, but past this apparent structural novelty, more interesting and less understood are the unique properties that the unusual milieu of the membrane imparts on these enzymes. Realizing a sophisticated understanding of these properties requires detailed investigation from multiple disciplines, and for the first time the tools for this analysis are becoming available. As these exciting new avenues are undertaken, two fundamental caveats must be taken into account.
There is an inherent danger in studying the detailed mechanism of an enzyme like GlpG whose cellular function, and even natural substrate, remain entirely unknown [29, 51]. For example, all substrates that have been used to interrogate GlpG specificity have been heterologous [28, 55], while our current view of GlpG position in a membrane is based on simulations with POPE, a lipid in which GlpG displays low activity [28] (Moin and Urban, unpublished observations). Indeed, PE is known as a non-bilayer lipid because it cannot form a membrane bilayer by itself due to its shape. In all of these investigations the greater challenge is to be certain that the exact parameters that are being measured are directly relevant to the natural environment and physiological function of GlpG.
It is also important to avoid the temptation of extrapolating lessons learned with GlpG to other rhomboid proteins without deliberate experimental evaluation. Eukaryotic rhomboid proteases could have important structural differences, because they contain a seventh transmembrane segment that can either precede of follow the six transmembrane core, as well as longer extramembraneous domains [11]. Moreover, GlpG reacts unlike any other rhomboid protease to reconstitution into different lipid environments [28]. Mechanistic diversity among rhomboid proteases also has already been encountered: Plasmodium falciparum rhomboid-4 that has been implicated in parasite invasion cannot cleave canonical substrates such as Spitz [18], while radically different substrate cleavage site and requirements have been proposed for the mitochondrial rhomboid [75]. Indeed, given the fact that rhomboid proteins can even be hard to recognize based on basic sequence analysis alone [76], variation in their biochemical properties should be expected. On an optimistic note, these differences could hold opportunities to target pathogenic versus homeostatic rhomboid enzymes differentially for therapeutic intervention. Detailed, but cautious, biochemical interrogation may well be the best strategy for tackling the therapeutic promise that these core enzymes might hold for human health.
Acknowledgments
I am grateful to the members of my laboratory, collaborators, and the many colleagues worldwide for making this field such a vibrant and exciting area of research. Research in my laboratory is funded through grants from the National Institutes of Health (R01AI066025), and career awards from the Burroughs-Wellcome Fund, the Packard Foundation, and the Howard Hughes Medical Institute. Structure images were rendered with MacPymol using PDB coordinates 2NRF (most GlpG images), 2IRV (GlpG with lipid in active site), 2IC8 (closed form of GlpG), 2NR9 (H. influenzae GlpG) and 1MOX (Spitz EGF).
References
- 1.Lopez-Otin C, Overall CM. Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol. 2002;3:509–519. doi: 10.1038/nrm858. [DOI] [PubMed] [Google Scholar]
- 2.Lopez-Otin C, Bond JS. Proteases: multifunctional enzymes in life and disease. J Biol Chem. 2008;283:30433–30437. doi: 10.1074/jbc.R800035200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polgar L. The catalytic triad of serine peptidases. Cell Mol Life Sci. 2005;62:2161–2172. doi: 10.1007/s00018-005-5160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blow DM, Birktoft JJ, Hartley BS. Role of a buried acid group in the mechanism of action of chymotrypsin. Nature. 1969;221:337–340. doi: 10.1038/221337a0. [DOI] [PubMed] [Google Scholar]
- 5.Blow D. So do we understand how enzymes work? Structure. 2000;8:R77–81. doi: 10.1016/s0969-2126(00)00125-8. [DOI] [PubMed] [Google Scholar]
- 6.Blow DM. The tortuous story of Asp … His … Ser: structural analysis of alpha-chymotrypsin. Trends Biochem Sci. 1997;22:405–408. doi: 10.1016/s0968-0004(97)01115-8. [DOI] [PubMed] [Google Scholar]
- 7.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 8.Rawson RB, Zelenski NG, Nijhawan D, Ye J, Sakai J, Hasan MT, Chang TY, Brown MS, Goldstein JL. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol Cell. 1997;1:47–57. doi: 10.1016/s1097-2765(00)80006-4. [DOI] [PubMed] [Google Scholar]
- 9.Urban S, Lee JR, Freeman M. Drosophila rhomboid-1 defines a family of putative intramembrane serine proteases. Cell. 2001;107:173–182. doi: 10.1016/s0092-8674(01)00525-6. [DOI] [PubMed] [Google Scholar]
- 10.Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 11.Koonin EV, Makarova KS, Rogozin IB, Davidovic L, Letellier MC, Pellegrini L. The rhomboids: a nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol. 2003;4:R19. doi: 10.1186/gb-2003-4-3-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kinch LN, Ginalski K, Grishin NV. Site-2 protease regulated intramembrane proteolysis: sequence homologs suggest an ancient signaling cascade. Protein Sci. 2006;15:84–93. doi: 10.1110/ps.051766506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urban S. Rhomboid proteins: conserved membrane proteases with divergent biological functions. Genes Dev. 2006;20:3054–3068. doi: 10.1101/gad.1488606. [DOI] [PubMed] [Google Scholar]
- 14.Urban S, Lee JR, Freeman M. A family of Rhomboid intramembrane proteases activates all membrane-tether EGF ligands in Drosophila. EMBO J. 2002;21:4277–4286. doi: 10.1093/emboj/cdf434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urban S, Brown G, Freeman M. EGF receptor signaling protects smooth-cuticle cells from apoptosis during Drosophila ventral epidermis development. Development. 2004;131:1835–1845. doi: 10.1242/dev.01058. [DOI] [PubMed] [Google Scholar]
- 16.Urban S. Making the cut: central roles of intramembrane proteolysis in pathogenic microorganisms. Nature Reviews Microbiology. 2009;7:411–423. doi: 10.1038/nrmicro2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stevenson LG, Strisovsky K, Clemmer KM, Bhatt S, Freeman M, Rather PN. Rhomboid protease AarA mediates quorum-sensing in Providencia stuartii by activating TatA of the twin-arginine translocase. Proc Natl Acad Sci U S A. 2007;104:1003–1008. doi: 10.1073/pnas.0608140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker RP, Wijetilaka R, Urban S. Two Plasmodium Rhomboid Proteases Preferentially Cleave Different Adhesins Implicated in All Invasive Stages of Malaria. PLoS Pathog. 2006;2:e113, 922–932. doi: 10.1371/journal.ppat.0020113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Donnell RA, Hackett F, Howell SA, Treeck M, Struck N, Krnajski Z, Withers-Martinez C, Gilberger TW, Blackman MJ. Intramembrane proteolysis mediates shedding of a key adhesin during erythrocyte invasion by the malaria parasite. J Cell Biol. 2006;174:1023–1033. doi: 10.1083/jcb.200604136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brossier F, Jewett TJ, Sibley LD, Urban S. A spatially localized rhomboid protease cleaves cell surface adhesins essential for invasion by Toxoplasma. Proc Natl Acad Sci U S A. 2005;102:4146–4151. doi: 10.1073/pnas.0407918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baxt LA, Baker RP, Singh U, Urban S. An Entamoeba histolytica rhomboid protease with atypical specificity cleaves a surface lectin involved in phagocytosis and immune evasion. Genes Dev. 2008;22:1636–1646. doi: 10.1101/gad.1667708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- 23.Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci U S A. 2003;100:6382–6387. doi: 10.1073/pnas.1037392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Urban S, Schlieper D, Freeman M. Conservation of intramembrane proteolytic activity and substrate specificity in eukaryotic and prokaryotic Rhomboids. Curr Biol. 2002;12:1507–1512. doi: 10.1016/s0960-9822(02)01092-8. [DOI] [PubMed] [Google Scholar]
- 25.Kanaoka MM, Urban S, Freeman M, Okada K. An Arabidopsis Rhomboid homolog is an intramembrane protease in plants. FEBS Lett. 2005;579:5723–5728. doi: 10.1016/j.febslet.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 26.Gallio M, Sturgill G, Rather P, Kylsten P. A conserved mechanism for extracellular signaling in eukaryotes and prokaryotes. Proc Natl Acad Sci U S A. 2002;99:12208–12213. doi: 10.1073/pnas.192138799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urban S, Freeman M. Substrate specificity of rhomboid intramembrane proteases is governed by helix-breaking residues in the substrate transmembrane domain. Mol Cell. 2003;11:1425–1434. doi: 10.1016/s1097-2765(03)00181-3. [DOI] [PubMed] [Google Scholar]
- 28.Urban S, Wolfe MS. Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity. Proc Natl Acad Sci U S A. 2005;102:1883–1888. doi: 10.1073/pnas.0408306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maegawa S, Ito K, Akiyama Y. Proteolytic action of GlpG, a rhomboid protease in the Escherichia coli cytoplasmic membrane. Biochemistry (Mosc) 2005;44:13543–13552. doi: 10.1021/bi051363k. [DOI] [PubMed] [Google Scholar]
- 30.Lemberg MK, Menendez J, Misik A, Garcia M, Koth CM, Freeman M. Mechanism of intramembrane proteolysis investigated with purified rhomboid proteases. EMBO J. 2005;24:464–472. doi: 10.1038/sj.emboj.7600537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng L, Yan H, Wu Z, Yan N, Wang Z, Jeffrey PD, Shi Y. Structure of a site-2 protease family intramembrane metalloprotease. Science. 2007;318:1608–1612. doi: 10.1126/science.1150755. [DOI] [PubMed] [Google Scholar]
- 32.Urban S, Shi Y. Core principles of intramembrane proteolysis: comparison of rhomboid and site-2 family proteases. Curr Opin Struct Biol. 2008;18:432–441. doi: 10.1016/j.sbi.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Z, Yan N, Feng L, Oberstein A, Yan H, Baker RP, Gu L, Jeffrey PD, Urban S, Shi Y. Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat Struct Mol Biol. 2006;13:1084–1091. doi: 10.1038/nsmb1179. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- 35.Ben-Shem A, Fass D, Bibi E. Structural basis for intramembrane proteolysis by rhomboid serine proteases. Proc Natl Acad Sci U S A. 2007;104:462–466. doi: 10.1073/pnas.0609773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Ha Y. Open-cap conformation of intramembrane protease GlpG. Proc Natl Acad Sci U S A. 2007;104:2098–2102. doi: 10.1073/pnas.0611080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Maegawa S, Akiyama Y, Ha Y. The role of L1 loop in the mechanism of rhomboid intramembrane protease GlpG. J Mol Biol. 2007;374:1104–1113. doi: 10.1016/j.jmb.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemieux MJ, Fischer SJ, Cherney MM, Bateman KS, James MN. The crystal structure of the rhomboid peptidase from Haemophilus influenzae provides insight into intramembrane proteolysis. Proc Natl Acad Sci U S A. 2007;104:750–754. doi: 10.1073/pnas.0609981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature. 2004;429:834–840. doi: 10.1038/nature02592. [DOI] [PubMed] [Google Scholar]
- 40.Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature. 2004;429:841–847. doi: 10.1038/nature02656. [DOI] [PubMed] [Google Scholar]
- 41.Baker RP, Young K, Feng L, Shi Y, Urban S. Enzymatic analysis of a rhomboid intramembrane protease implicates transmembrane helix 5 as the lateral substrate gate. Proc Natl Acad Sci U S A. 2007;104:8257–8262. doi: 10.1073/pnas.0700814104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Urban S, Baker RP. In vivo analysis reveals substrate-gating mutants of a rhomboid intramembrane protease display increased activity in living cells. Biol Chem. 2008;389:1107–1115. doi: 10.1515/BC.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Senes A, Ubarretxena-Belandia I, Engelman DM. The Calpha ---H…O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc Natl Acad Sci U S A. 2001;98:9056–9061. doi: 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Senes A, Gerstein M, Engelman DM. Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J Mol Biol. 2000;296:921–936. doi: 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- 45.Li J, Ahvazi B, Szittner R, Meighen E. Mutation of the nucleophilic elbow of the Lux-specific thioesterase from Vibrio harveyi. Biochem Biophys Res Commun. 2000;275:704–708. doi: 10.1006/bbrc.2000.3362. [DOI] [PubMed] [Google Scholar]
- 46.Maegawa S, Koide K, Ito K, Akiyama Y. The intramembrane active site of GlpG, an E. coli rhomboid protease, is accessible to water and hydrolyses an extramembrane peptide bond of substrates. Mol Microbiol. 2007;64:435–447. doi: 10.1111/j.1365-2958.2007.05679.x. [DOI] [PubMed] [Google Scholar]
- 47.Lemberg MK, Bland FA, Weihofen A, Braud VM, Martoglio B. Intramembrane proteolysis of signal peptides: an essential step in the generation of HLA-E epitopes. J Immunol. 2001;167:6441–6446. doi: 10.4049/jimmunol.167.11.6441. [DOI] [PubMed] [Google Scholar]
- 48.Carter P, Wells JA. Dissecting the catalytic triad of a serine protease. Nature. 1988;332:564–568. doi: 10.1038/332564a0. [DOI] [PubMed] [Google Scholar]
- 49.Dodson G, Wlodawer A. Catalytic triads and their relatives. Trends Biochem Sci. 1998;23:347–352. doi: 10.1016/s0968-0004(98)01254-7. [DOI] [PubMed] [Google Scholar]
- 50.Ekici OD, Paetzel M, Dalbey RE. Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008;17:2023–2037. doi: 10.1110/ps.035436.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clemmer KM, Sturgill GM, Veenstra A, Rather PN. Functional characterization of Escherichia coli GlpG and additional rhomboid proteins using an aarA mutant of Providencia stuartii. J Bacteriol. 2006;188:3415–3419. doi: 10.1128/JB.188.9.3415-3419.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.White SH. Rhomboid intramembrane protease structures galore! Nat Struct Mol Biol. 2006;13:1049–1051. doi: 10.1038/nsmb1206-1049. [DOI] [PubMed] [Google Scholar]
- 53.Freeman M. Structural biology: enzyme theory holds water. Nature. 2006;444:153–155. doi: 10.1038/nature05305. [DOI] [PubMed] [Google Scholar]
- 54.Bondar AN, del Val C, White SH. Rhomboid protease dynamics and lipid interactions. Structure. 2009;17:395–405. doi: 10.1016/j.str.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akiyama Y, Maegawa S. Sequence features of substrates required for cleavage by GlpG, an Escherichia coli rhomboid protease. Mol Microbiol. 2007;64:1028–1037. doi: 10.1111/j.1365-2958.2007.05715.x. [DOI] [PubMed] [Google Scholar]
- 56.Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301:610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 57.White SH, Wimley WC. Hydrophobic interactions of peptides with membrane interfaces. Biochim Biophys Acta. 1998;1376:339–352. doi: 10.1016/s0304-4157(98)00021-5. [DOI] [PubMed] [Google Scholar]
- 58.Zhou XW, Blackman MJ, Howell SA, Carruthers VB. Proteomic analysis of cleavage events reveals a dynamic two-step mechanism for proteolysis of a key parasite adhesive complex. Mol Cell Proteomics. 2004;3:565–576. doi: 10.1074/mcp.M300123-MCP200. [DOI] [PubMed] [Google Scholar]
- 59.Opitz C, Di Cristina M, Reiss M, Ruppert T, Crisanti A, Soldati D. Intramembrane cleavage of microneme proteins at the surface of the apicomplexan parasite Toxoplasma gondii. EMBO J. 2002;21:1577–1585. doi: 10.1093/emboj/21.7.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Howell SA, Hackett F, Jongco AM, Withers-Martinez C, Kim K, Carruthers VB, Blackman MJ. Distinct mechanisms govern proteolytic shedding of a key invasion protein in apicomplexan pathogens. Mol Microbiol. 2005;57:1342–1356. doi: 10.1111/j.1365-2958.2005.04772.x. [DOI] [PubMed] [Google Scholar]
- 61.Phillips R, Ursell T, Wiggins P, Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature. 2009;459:379–385. doi: 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Munro S. Lipid rafts: elusive or illusive? Cell. 2003;115:377–388. doi: 10.1016/s0092-8674(03)00882-1. [DOI] [PubMed] [Google Scholar]
- 63.Ye J, Dave UP, Grishin NV, Goldstein JL, Brown MS. Asparagine-proline sequence within membrane-spanning segment of SREBP triggers intramembrane cleavage by site-2 protease. Proc Natl Acad Sci U S A. 2000;97:5123–5128. doi: 10.1073/pnas.97.10.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walker B, Lynas JF. Strategies for the inhibition of serine proteases. Cell Mol Life Sci. 2001;58:596–624. doi: 10.1007/PL00000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paulick MG, Bogyo M. Application of activity-based probes to the study of enzymes involved in cancer progression. Curr Opin Genet Dev. 2008;18:97–106. doi: 10.1016/j.gde.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Paetzel M, Dalbey RE, Strynadka NC. Crystal structure of a bacterial signal peptidase in complex with a beta-lactam inhibitor. Nature. 1998;396:186–190. doi: 10.1038/24196. [DOI] [PubMed] [Google Scholar]
- 67.Bihel F, Das C, Bowman MJ, Wolfe MS. Discovery of a Subnanomolar helical D-tridecapeptide inhibitor of gamma-secretase. J Med Chem. 2004;47:3931–3933. doi: 10.1021/jm049788c. [DOI] [PubMed] [Google Scholar]
- 68.Sato T, Nyborg AC, Iwata N, Diehl TS, Saido TC, Golde TE, Wolfe MS. Signal peptide peptidase: biochemical properties and modulation by nonsteroidal antiinflammatory drugs. Biochemistry (Mosc) 2006;45:8649–8656. doi: 10.1021/bi060597g. [DOI] [PubMed] [Google Scholar]
- 69.Lee JR, Urban S, Garvey CF, Freeman M. Regulated intracellular ligand transport and proteolysis control EGF signal activation in Drosophila. Cell. 2001;107:161–171. doi: 10.1016/s0092-8674(01)00526-8. [DOI] [PubMed] [Google Scholar]
- 70.Urban S, Freeman M. Intramembrane proteolysis controls diverse signalling pathways throughout evolution. Curr Opin Genet Dev. 2002;12:512–518. doi: 10.1016/s0959-437x(02)00334-9. [DOI] [PubMed] [Google Scholar]
- 71.Freeman M. Rhomboid proteases and their biological functions. Annu Rev Genet. 2008;42:191–210. doi: 10.1146/annurev.genet.42.110807.091628. [DOI] [PubMed] [Google Scholar]
- 72.Bier E, Jan LY, Jan YN. rhomboid, a gene required for dorsoventral axis establishment and peripheral nervous system development in Drosophila melanogaster. Genes Dev. 1990;4:190–203. doi: 10.1101/gad.4.2.190. [DOI] [PubMed] [Google Scholar]
- 73.Dowse TJ, Pascall JC, Brown KD, Soldati D. Apicomplexan rhomboids have a potential role in microneme protein cleavage during host cell invasion. Int J Parasitol. 2005;35:747–756. doi: 10.1016/j.ijpara.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 74.Yang B, Larson TJ. Multiple promoters are responsible for transcription of the glpEGR operon of Escherichia coli K-12. Biochim Biophys Acta. 1998;1396:114–126. doi: 10.1016/s0167-4781(97)00179-6. [DOI] [PubMed] [Google Scholar]
- 75.Herlan M, Bornhovd C, Hell K, Neupert W, Reichert AS. Alternative topogenesis of Mgm1 and mitochondrial morphology depend on ATP and a functional import motor. J Cell Biol. 2004;165:167–173. doi: 10.1083/jcb.200403022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lemberg MK, Freeman M. Functional and evolutionary implications of enhanced genomic analysis of rhomboid intramembrane proteases. Genome Res. 2007;17:1634–1646. doi: 10.1101/gr.6425307. [DOI] [PMC free article] [PubMed] [Google Scholar]