

Summary
Aging is inevitably accompanied by gradual and irreversible innate endothelial dysfunction. In this study, we tested the hypothesis that accentuation of glucose metabolism via the aldose reductase (AR) pathway contributes to age-related vascular dysfunction. AR protein and activity levels were significantly increased in aged vs. young aortic homogenates from Fischer 344 rats. Immunostaining revealed that the principal site of increased AR protein was the aortic endothelium as well as smooth muscle cells. Studies revealed that endothelial-dependent relaxation (EDR) in response to acetylcholine was impaired in aged rats compared to young rats and that treatment with the AR inhibitor (ARI) zopolrestat significantly improved EDR in aged rats. Methylglyoxal (MG), a key precursor of advanced glycation endproducts (AGEs), was significantly increased in the aortas of aged rats vs. young rats. Consistent with central roles for AR in generation of MG in aging, ARI treatment significantly reduced MG levels in aged rat aorta to those in young rats. Treatment of aged rats with soluble(s) RAGE, a soluble form of the chief signal transduction receptor for AGEs, RAGE, significantly improved EDR in aged rats, thus establishing the contribution of age-related increases in AGEs to endothelial dysfunction. These findings reveal that significant increases in AR expression and activity in aged rat vasculature linked to endothelial dysfunction may be mitigated, at least in part, via ARI and that aging-linked increased flux via AR generates AGEs; species which transduce endothelial injury consequent to their interaction with RAGE. These data demonstrate for the first time that AR mediates aging-related vascular dysfunction, at least in part, via RAGE.
Keywords: aging, aldose reductase, endothelial dysfunction, RAGE, receptors
Introduction
Aging, even in the absence of diseases that generally accompany aging, confers the largest risk factor for cardiovascular disease in human subjects (Al-Shaer et al., 2006). Progressive increases in innate vascular dysfunction with aging have been demonstrated in humans and animals (Blackwell et al., 2004; Brandes et al., 2005; Chinellato et al., 1991; Csiszar et al., 2002; Geary & Buchholz, 2003; Hongo et al., 1988; Kung & Luscher, 1995; Muller-Delp et al., 2002; Murohara et al., 1991). It is likely that fundamental metabolic and biochemical changes occur over time in aging vasculature, resulting in alterations in substrate metabolism and ATP levels, factors that may contribute to vascular dysfunction (Al-Shaer et al., 2006; Headrick, 1998; Kates et al., 2003; McMillin et al., 1993). This study proposes that altered glucose metabolism may be one of these fundamental changes that contribute to age-related vascular dysfunction.
Aldose reductase (AR) is the first enzyme of the polyol pathway and plays a key role in the biochemical and molecular signaling response to glucose in vascular and inflammatory cells linked to the pathogenesis and progression of vascular dysfunction. AR reduces glucose to sorbitol; sorbitol is reduced to fructose by the enzyme sorbitol dehydrogenase. While the polyol pathway has been investigated for its role in diabetic complications, recent studies have demonstrated its role in mediating myocardial ischemia-reperfusion injury even in the absence of diabetes (Hwang et al., 2004, 2002, 2005). Recent studies also support the critical role of AR in accelerating atherosclerosis in diabetes (Vikramadithyan et al., 2005). Additionally, earlier studies have demonstrated the effectiveness of AR inhibitors in improving endothelium-dependent relaxation (EDR) in hyperglycemia (Cameron & Cotter, 1992; Keegan et al., 2000). We hypothesize that this alteration in AR pathway enzymes lead to increased substrate flux and thereby creates a heightened susceptibility to vascular disease.
A critical consequence of flux via the AR pathway is the generation of precursors of advanced glycation endproducts (AGEs), specifically methylglyoxal (MG) and 3-deoxyglucose (3-DG) (Hamada et al., 1996; Kato et al., 1989; Lal et al., 1995; Thornalley, 1996, 1998). Additionally, studies have demonstrated that inhibition of the AR pathway results in reduction in AGEs (Hamada et al., 2000; Lal et al., 1995; Nakamura et al., 2003). AGEs may contribute to the impairment of EDR by increasing oxidative stress and reducing the bioavailability of nitric oxide (Bucala et al., 1991). An accumulation of AGEs in the vasculature has been shown to accompany increasing age (Brett et al., 1993; Shapiro et al., 2008).
The purpose of this investigation was to examine whether the AR pathway and its influence in modulating AGE precursors play a role in age-related vascular dysfunction. Specifically, we examined whether inhibition of AR activity and suppression of the effects of the chief AGE signal transduction receptor, RAGE, improved EDR in aged Fischer 344 rats.
Results
Upregulation of the AR pathway with aging
In our first studies, we sought to determine whether aging affected levels and activity of AR. Western blotting revealed a 2.9 ± 0.4 fold increase in AR protein expression in the aorta of aged rats when compared with young rats (P < 0.05) (Fig. 1a). AR inhibitor (ARI) treatment for 10 days prior to sacrifice resulted in a decrease in AR protein levels (Fig. 1a).
Fig. 1.

Activity and expression of polyol pathway enzyme aldose reductase (AR) is increased in aged aortae (A) Western blotting revealed 1.9 ± 0.4 fold increase in aortic AR protein levels in aged rats compared with young rats (n = 5 and 6/group respectively; P < 0.05). AR inhibitor (ARI)-treated rats showed a significant decrease in AR protein levels. (B) Aortic tissue levels of sorbitol were significantly higher in aorta from aged animals (n = 3) than in young (n = 5). Treatment with ARI reduced the sorbitol levels in aged aorta. (C) Similar changes were observed in fructose levels as well (n = 5 young rats; n = 3 aged rats). (D) AR activity is significantly greater in aortas from aged animals when compared with young animals.
Consistent with increased AR protein expression, increased AR activity was observed, as tissue levels of sorbitol were significantly higher in aorta from aged animals compared to young animals (Fig. 1b), and treatment with ARI reduced the sorbitol levels in aged aorta. We also examined aorta levels of fructose, as an additional measure of AR activity. Fructose is produced by the action of sorbitol dehydrogenase on sorbitol. Similar changes were observed in fructose levels as well (Fig. 1c). Furthermore, AR enzyme activity was directly measured. These studies revealed an increase in AR enzyme activity in aged rat aorta homogenates compared with young rat (P < 0.05; Fig. 1d).
To localize the principal cell types expressing AR in the aged aorta, immunostaining was performed using anti-AR immunoglobulin G. Immunostaining for AR in aortic sections localized AR protein expression to the aortic endothelium and smooth muscle cells of aged rats when compared with young rats (Fig. 2a,b respectively).
Fig. 2.
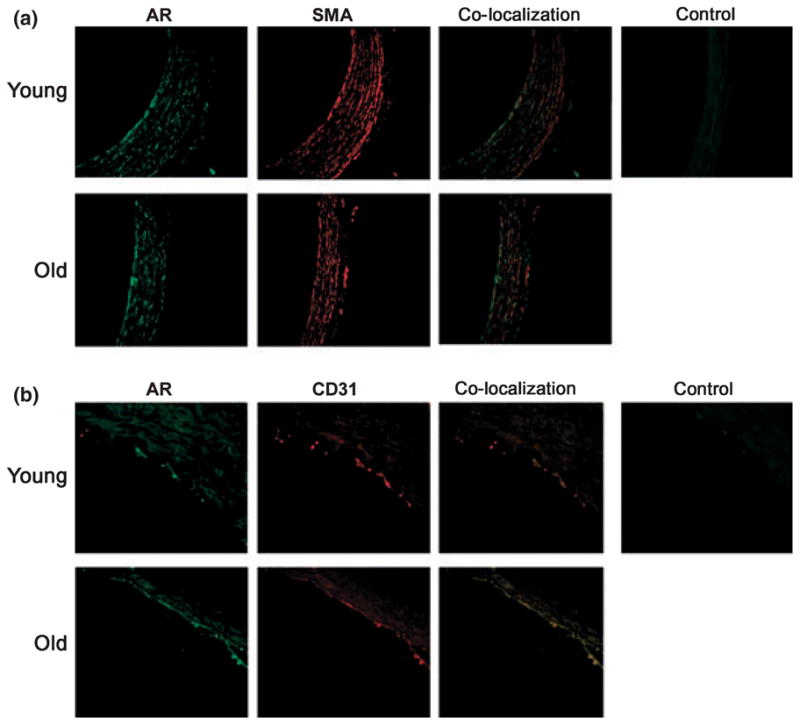
Immunostaining reveals aldose reductase (AR) expression in aortic endothelial cells and smooth muscle cells of aged animals. Formalin-embedded sections of aortas from young and aged rats were probed with anti-AR IgG, anti-alpha smooth muscle actin (SMA) (a) and anti CD31 (b) IgG, respectively, and visualized after using appropriate second antibody and fluorescent conjugated IgG. Images were visualized under 40× magnification. Immunostaining localized AR to the aortic endothelial and smooth muscle cells in aged animals. Aorta sections stained with appropriate nonimmune serum are presented as negative control.
Methylglyoxal
A central consequence of increased AR activity is increased production of major AGE precursors, such as MG. Levels of MG were significantly higher in aortas of aged rats compared with young animals (P < 0.05). Treatment of the rats with ARI dramatically reduced MG levels in aged rat aorta to levels seen in young rats (P < 0.05) (Fig. 3). In parallel with increased MG levels, aortas of aged rats displayed significantly higher levels of the chief AGE signaling receptor RAGE antigen vs. young rats by western blotting; P < 0.05 (Fig. 4a). In line with roles for ARI in reducing levels of MG in the aortas of aged rats, RAGE expression was also reduced in aged rat aortas after treatment with ARI; P < 0.05 (Fig. 4a). Circulating levels of carboxymethyl lysine (CML)-AGEs were determined by western blots containing standard CML-AGEs on plasma samples from young and old rats. CML-AGEs were significantly increased in aged vs. young rat plasma (P < 0.05; Fig. 4b). Treatment with ARI reduced circulating levels of plasma CML-AGEs as well (P < 0.05; Fig. 4b).
Fig. 3.

AGE precursor MG is increased in aged rat aortas. MG levels were significantly higher in aged (n = 6) rats when compared with young (n = 4) rats (P = 0.0016). MG levels were significantly lower in the abdominal aortas of aged rats that received AR inhibitor treatment (n = 6, P < 0.001).
Fig. 4.

Western blot studies reveal increases in RAGE expression in aged rat aortas. (a) RAGE protein expression was significantly higher in aged rat aortas (n = 6) when compared with young rat aortas (n = 6) (P < 0.01). RAGE protein expression was significantly lower in the abdominal aortas of aged rats that received ARI treatment (n = 6, P < 0.05). (b) AGE concentrations in mouse sera were determined by western blot using monoclonal antibody reacting with N-carboxy methyl lysine. Appropriate CML-bovine serum albumin containing modified lysine was used as a standard for comparing the band intensity. (n = 6 per group, respectively; P < 0.05).
Endothelium-dependent relaxation: effect of aging and roles for AR/RAGE
To begin to determine the potential roles of AR and AGE–RAGE pathways in endothelial dysfunction of aging, we assessed EDR in aortic rings retrieved from aged vs. young rats. Baseline EDR in response to acetylcholine (Ach) was significantly impaired in aged rats compared with young rats (P < 0.05; Fig. 5a). The EDR response to sodium nitroprusside was similar in both young and aged rats (data not shown). Vasoconstriction to phenylephrine was similar in both young and aged rats and revealed no significant differences between the age groups (Fig. 5b).
Fig. 5.

Endothelial-dependent relaxation (EDR) changes in aged and young rat aortas. (a) EDR in response to acetylcholine was significantly impaired in aged rats (n = 7) compared with young rats (n = 6; P < 0.05). (b) Vasoconstriction to phenylephrine was similar in both young and aged rats.
Consistent with pathogenic roles for AR in EDR dysfunction in aging, administration of ARI to aged rats for 10 days resulted in improved EDR in response to Ach compared with aged rats receiving no treatment (P < 0.05, in young, aged, aged + ARI groups, Fig. 6). Furthermore, consistent with pathogenic roles for RAGE ligands in mediating EDR dysfunction in aged rat aortic rings, EDR was significantly improved in aged rats treated with the ligand binding decoy of RAGE, sRAGE, for 10 days (Fig. 7) compared with control aged rats (P < 0.05, in young, aged, aged + 10 day sRAGE groups). It should be noted that sRAGE treatment did not alter AR activity in young and old aortas (data not shown). The maximal response elicited by ACh was different between young and old aortas, EC50 being the concentration of ACh that elicited 50% of the maximal response significantly correlated with improved endothelial function after ARI/sRAGE treatment. ARI and sRAGE improved the relaxation elicited by 1.65 × 10−7 ± 0.36 × 10−7 M ACh or higher in old aortas and all treated groups, and by 1.65 × 10−7 ± 0.61 × 10−7 M ACh or higher in sRAGE-treated group. These data suggest that decreased ACh-induced vasorelaxation in aortas from aged rats may result from impairment of endothelial nitric oxide synthase and endothelium-derived signaling pathways. This effect is prevented by ARI and sRAGE.
Fig. 6.

Oral administration of ARI improves endothelial-dependent relaxation (EDR) to acetylcholine in aged rats. EDR was significantly improved in aged rats given ARI zopolrestat compared with control aged rats (P < 0.05, in young, aged, aged + ARI groups, n = 6,7,6, respectively).
Fig. 7.

Administration of sRAGE improves endothelial-dependent relaxation (EDR) to acetylcholine in aged rats. EDR was significantly improved in aged rats given sRAGE compared with control aged rats (P < 0.05, in young, aged, aged + sRAGE groups, n = 6, 7, 6, respectively).
Oxidative stress: Effect of aging and role of ARI
Fresh aortic lysates from young and old rats were analyzed for total GSH and GSSG as an indicator of oxidative stress. Aortas from old rats had significantly lower Reduced glutathione/Oxidized glutathione ratio compared to aortas from young rats (P < 0.05; Fig. 8). ARI was very effective in improving the GSH/GSSG ratio over a 10-day treatment period. (P < 0.05; Fig. 8). We measured an additional index of oxidative stress in these tissues. As Akt phosphorylates eNOS and therefore is able to promote NO bioavailability and impact EDR in large vessels, we determined the ratio of phosphorylated to total Akt in aortas from young and old rats. As shown in Fig. 9, the p-Akt/T-Akt ratio was significantly higher in aortic samples from young rats compared to aged rat aortas (P < 0.05), and treatment with ARI significantly improved the p-Akt/tT-Akt ratio in aged rat aortas (P < 0.05).
Fig. 8.
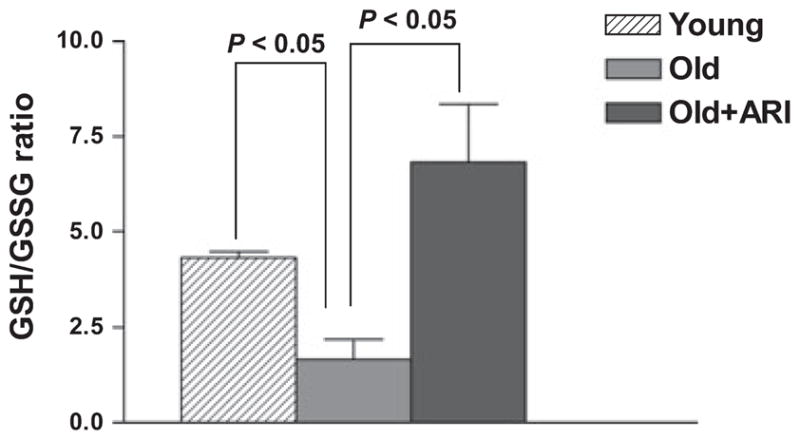
Oxidative stress changes in aged and young rat aortas. Reduced glutathione and oxidized glutathione were analyzed in lysate from young, old and ARI treated aortas. (n = 6,6 and 7, respectively; P < 0.05). Aged aortas had significantly lower GSH/GSSG; ARI improved the GSH/GSSG ratio.
Fig. 9.
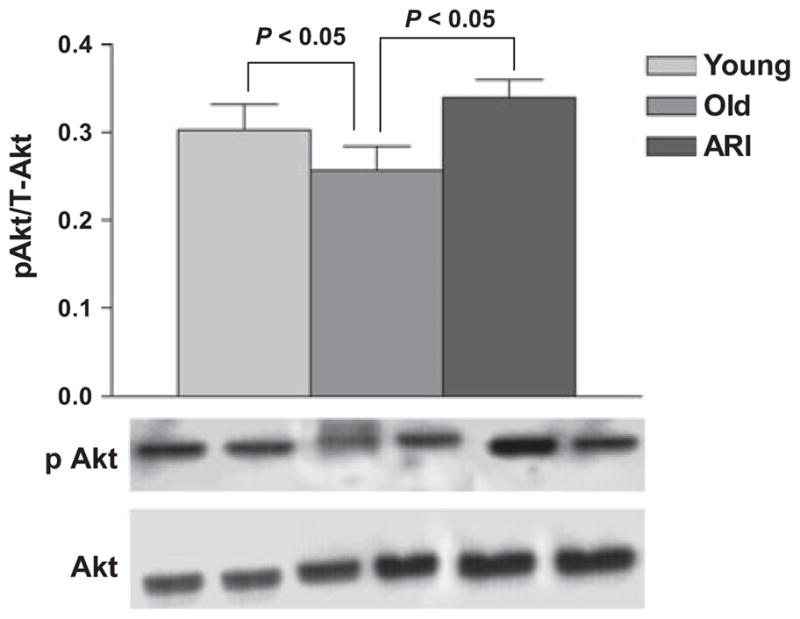
Effect of aging and ARI on aorta levels of phosphorylated Akt. Western blotting was performed on lysate proteins from young, aged and ARI treated aortas using antibodies to phosphorylated followed by total Akt. Although aging was associated with reduced phosho/total Akt ratio, ARI significantly improved phosphorylation of Akt. (n = 5 and 6 per group, respectively; P < 0.05).
Discussion
Although multiple studies have suggested that endothelial function declines with aging, the present study is the first to link changes in glucose metabolism to the pathogenesis of endothelial-dependent relaxation impairment in natural aging. While it is known that chronic elevation of cytosolic glucose drives metabolic flux via the AR pathway (Hwang et al., 2003), the present findings revealed that natural aging, even in the absence of the common aging-associated disorder hyperglycemia, also results in enhanced flux via AR, and, thereby, increased accumulation of AR pathway products in the vasculature. Our experiments demonstrate the novel observation that the primary enzyme in the polyol pathway, AR, exhibits increased protein expression and enzyme activity in aged Fischer 344 rat aortas compared with young rat aortas. Further evidence of increased flux via the AR pathway was that the products of this pathway, fructose and sorbitol, were significantly higher in aged animals aorta. Immunostaining of aortas localized AR protein expression to the endothelium and smooth muscle layer of aged rats. This expression of AR in the endothelium of aged vasculature led us to probe the hypothesis that increased flux via AR mediated, at least in part, age-related endothelial dysfunction.
Levels of MG, a precursor to AGEs, were higher in aged vs. young Fischer 344 rat aortas. Administration of ARI significantly lowered MG levels in aged animals, indicating that age-related increases in this AGE precursor and subsequent activation of RAGE, is mediated, at least in part, via the AR pathway. These results are in contrast to studies that have shown that AR can catalyze reduction in AGE precursors (Vander Jagt et al., 1992; Baba et al., 2009). Others also showed that genetic deletion of AR increased AGE accumulation and atherosclerotic lesion formation in young mice (Baba et al., 2009). It is important to note that numerous differences exist between our models, the most important of which is our study of aging rats; this may explain the link between AR and AGE generation in our studies. Given that AGEs transduce their effects largely via the signal transduction receptor RAGE, we postulated that age-related vascular dysfunction mediated via the AR pathway exerts its effects in part via RAGE. Consistent with this concept, administration of sRAGE attenuated age-related endothelial dysfunction in the aged rat aortic rings. Indeed, our findings revealed increased circulating CML-AGEs in aged vs. young rats, thereby providing evidence for circulating ligands likely bound by exogenously added sRAGE in these studies.
These data provide strong evidence that glucose metabolism is altered in natural, disease-free aging in the vasculature. It has been shown that the AR pathway and RAGE play vital roles in the pathogenesis of cardiovascular disease. Previous work supports the critical role of AR in accelerating atherosclerosis in diabetes (Vikramadithyan et al., 2005). Additionally, studies by other groups have demonstrated the effectiveness of AR inhibitors on improving EDR in hyperglycemic models (Cameron & Cotter, 1992; Keegan et al., 2000). Critically, Fischer 344 rats do not display typical aging-associated diseases, such as insulin resistance or frank hyperglycemia. Thus, our findings reveal for the first time that increased metabolic flux via the AR pathway may account, at least in part, for the increased vulnerability of aged vasculature to dysfunction in the absence of aging-associated diseases.
Our findings confirm previous investigations revealing that EDR in response to acetylcholine was impaired in aged rats compared with young rats (Brown et al., 2006; Celermajer et al., 1994; Ibarra et al., 1995; Shirasaki et al., 1986; Tschudi et al., 1996; van der Loo et al., 2000). In our studies, we did not observe an increased contractile response to phenylephrine between young vs. aged rats. Similar investigations have observed either an increase or no change in vasoconstrictor response in aged animals compared with young. It is possible that differences in these findings may be related to the strain of animals tested or, perhaps, unrecognized factors in the animals’ diet that may have accounted for altered vasoconstrictor responses.
In the present study, we examined the impact of inhibiting metabolic flux via the AR pathway by utilizing an AR inhibitor and also by inhibiting the consequences of AGE interaction with RAGE. ARI treatment significantly improved EDR in aged animals. Additionally, antagonism of the receptor for AGEs also significantly improved EDR in aged animals. EDR in response to sodium nitroprusside remained intact in all young, aged and treated animals, suggesting that the differences in EDR between the groups were attributed to endothelium-dependent mechanisms.
The focus of the current investigation was to probe potential links between the AR pathway and age-related vascular dysfunction. The bioavailability of nitric oxide, the major effector of EDR, is scavenged by oxidative stress. AR activation may generate oxidative stress by several means: (i) AR activation depletes NADPH, which is a necessary cofactor for glutathione reductase, thereby leading to diminished cellular antioxidant capacity; (ii) the conversion of sorbitol to fructose requires NAD+ which is converted to NADH, causes increased cytosolic Reduced Nicotanimadie Adenine Dinucleotide/Nicotanimadie Adenine Dinucleotide ratio, a mechanism to generate reactive oxygen species (ROS); and (iii) AR-driven activation of phospholipase, together with increased generation of oxidant species, leads to the production of oxidized lipids. Studies have shown that oxidized lipids can impair EDR in vascular tissues (Shaul, 2003). Here, we show decreases in Reduced glutathione GSH/GSSG ratio in aged aortas (vs. young) and attenuation of these changes with ARI treatment. Furthermore, we show that ARI improves the ratio of phosphorylated to total Akt in the aged aortas; phosphorylated Akt is a key kinase in regulating eNOS phosphorylation and the bioavailability of NO. Overall, the data are consistent with AR impacting EDR, in part, via changes in oxidant stress.
AR-driven changes in glucose metabolism generate MG, a central precursor in formation of a range of AGEs. The primary mechanism of AGE impact on vascular and cellular properties occurs through interaction with the receptor for AGEs; one consequence of AGE–RAGE interaction is the generation of oxidative stress. AGEs have been shown to generate ROS through activation of NADPH oxidase as well as possibly through mitochondrial sources (Basta et al., 2005; Coughlan et al., 2009; Wautier et al., 2001). We propose that AGE-triggered generation of ROS results in the quenching of NO, which is the primary vasorelaxing substance linked to EDR (Bucala et al., 1991).
It is essential to note that even in apparently healthy human subjects, endothelial dysfunction accompanies the aging process. In addition to endothelial dysfunction, the changes that occur in human vasculature during aging include large artery thickening, and stiffness as well as the resulting increase in systolic and pulse pressure (Lakatta, 2003). Aging vascular tissues including vessels and myocardium display increased AGE formation even in the absence of diabetes in humans and rats (Black-well et al., 2004; Brandes et al., 2005). The ability of AGEs to cross-link with proteins may account for increases in arterial stiffness seen in aging. Indeed, the potent impact of ALT-711, an AGE cross-link breaker, in both aged non-human primates and human subjects, provides strong support for the contribution of AGEs to aging-linked vascular dysfunction. Here, we showed for the first time that increased AR flux may critically link AGE formation to innate vascular aging.
In conclusion, the primary findings of this study include (i) AR expression and activities are increased in aged vasculature, (ii) AR inhibition improves EDR in aged rats and (iii) inhibition of RAGE improves EDR in aged rats. We posit that the alteration in AR pathway enzymes that occurs in aging leads to increased substrate flux via this pathway and creates a heightened susceptibility to vascular disease, probably at least in part via RAGE. AR-driven generation of CML-AGEs and MG, a key precursor of AGEs, may impact EDR via RAGE. Although multiple pathways in age-related endothelial dysfunction are implicated by these findings, this study specifically elucidates for the first time the potential role of AR in age-related vascular disease. Importantly, our findings underscore the potential of ARI and RAGE antagonism as novel strategies to ameliorate age-related vascular dysfunction.
Experimental procedures
All studies were performed with the approval of the Institutional Animal Care and Use of Laboratory Animals Committee at Columbia University, New York. This investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85–23, 1996). The study was conducted in young (4 months) and aged (24–26 months) Fischer 344 rats purchased from the National Institutes of Aging colony. Rats were divided into four groups: young (n = 9), aged (n = 13), aged + ARI treatment (n = 6) and aged + sRAGE treatment (n = 4). ARI treatment was administered to aged rats via oral gavage at a dose of 40 mg zopolrestat/animal/twice daily for a period of 10 days (Mylari et al., 1992, 1991). Soluble RAGE (sRAGE) blocks the interaction of AGEs with RAGE and hence acts as an antagonist for RAGE. sRAGE was administered by intraperitoneal route at a dose of 500 μg/animal per day for 10 days (Bucciarelli et al., 2002; Park et al., 1998).
Biochemical analysis of the AR pathway
Aortic homogenates were analyzed for AR protein expression by western blotting. Protein extracts were separated on a 5%SDS-PAGE gel and immobilized on polyvinylidene difluoride membrane. Nonspecific binding of the membrane was blocked with 5% nonfat dry milk (Bio-Rad, Richmond, CA, USA). The membrane was incubated in Phosphate Buffered Saline containing 1% BSA and rabbit anti-AR IgG at 4 °C overnight and washed three times in Phosphate Buffered Saline with 0.1% Tween-20. The blot was then further incubated in PBS containing anti-goat IgG coupled with horseradish peroxidase. The membranes were visualized using the ECL method. The blot signals were digitized and subjected to densitometric scanning using a standard NIH image program (v 1.62, Bethesda, MD, USA). Other primary antibodies used include anti-mouse/rat RAGE (R &D systems inc), anti phospho Akt, total Akt IgG (Cell signaling, Danvers, MA, USA), anti CML monoclonal antibody (COSMO Bio Co., Tokyo, Japan) and anti β-actin IgG (BD Biosciences, San Jose, CA, USA).
Levels of sorbitol and fructose were measured as described previously by our group (Hwang et al., 2003, 2002). AR activities were measured in aortic tissue homogenates using spectrophotometric techniques as described previously (Hwang et al., 2003).
GSH/GSSG ratios were measured in cleared aortic extracts using a commercially available BiotechR GSH-412 TM kit (Oxis Research, Portland, OR, USA) as per manufacturer’s instruction.
Immunohistochemistry
Immunohistochemistry to detect AR antigen in rat aorta was performed as previously described (Hwang et al., 2004) in three young and three aged Fischer 344 rats. Immunostaining was performed on 10% formalin-embedded sections (6 μm) The deparaffinized sections were stained with a primary AR antibody or platelet endothelial cell adhesion molecule-1 (CD31) antibody or anti-alpha smooth muscle actin and then incubated with a biotinylated secondary immunoglobulin (IgG; 1:250; Vector Laboratories Inc., Burlingame, CA, USA), followed by incubation with fluorescein-avidin D/Texas red-avidin D. The signals of individual and merged images for antigen detection were obtained using a fluorescent microscope with epifluorescent illumination (excitation wavelength 488 nm for fluorescein-avidin D, 568 nm for Texas Red-avidin D; Bio-Rad Laboratories Inc., Richmond, CA, USA). Rabbit IgG (Zymed, Invitrogen) or omission of the primary antibody was used as negative control.
Biochemical analysis of the AGE–RAGE interaction
3-Methylglyoxal (3-MG), a precursor in the formation of AGEs, was measured in the neutralized perchloric acid extracts of hearts by high-performance liquid chromatography (HPLC) methods according to previously published procedures (Hwang et al., 2004, 2002; Ohmori et al., 1987).
Assessment of endothelial function
Fischer 344 rats were anesthetized using sodium pentabarbitol (50 mg kg−1; i.p.) (Abbott Laboratory, Cranbury, NJ, USA). The descending thoracic aorta was carefully excised. The aorta was flushed twice with fresh cold, oxygenated Krebs Henseleit buffer containing NaCl 118, KCl 5.4, MgCl2 1.2, CaCl2 2.5, NaHCO3 22, NaH2PO4 1.2 and glucose 10.1 mM and placed in a dissecting tray filled with the same buffer. The aorta was carefully dissected from periadventitial tissue and fat and then segmented into rings (4–5 mm length). The ring proximal to that was snap frozen in liquid nitrogen and stored at −80 °C for further analysis by western blotting. The remaining aortic rings were then mounted using two tungsten wire triangles in conventional 20-mL organ bath chambers (Experimentria, Budapest, Hungary) containing Krebs-Henseleit solution maintained at 37 °C and bubbled with a mixture of 95% oxygen and 5% carbon dioxide at pH 7.4. A preload tension of 2 g was applied to the rings. The tissues were allowed to equilibrate for 90 min during which time the organ bath chamber was filled with fresh buffer three times. Changes in isometric tension were recorded with a force transducer (Experimentria, Hungary). At the end of the equilibration period, the aortic rings were precontracted with phenylephrine (PE) (10−5 M). Baseline EDR dose–response curves to acetylcholine (10−8 to 10−4 M) were obtained by adding the agonist into the organ bath in an incremental manner. Rings were washed three times and allowed to recover for one hour. Dose–response curves to phenylephrine (10−8–10−5 M) were then obtained. The % relaxations for individual rings were calculated based on ratio of differences in tension owing to maximum contraction in the presence of phenylephrine and tension in the presence of acetylcholine (10−8–10−4 M) to that of maximum contraction (PE) and baseline tension. Finally, dose–response curves to sodium nitroprusside (10−9–10−4 M) were obtained to confirm that the vascular smooth muscle tissue of the rings was intact.
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). Repeated measures of analysis of variance were used to determine differences in dose response between groups (Stat-View). A P value <0.05 was regarded as significant.
Acknowledgments
This work was supported by grants from the U.S.P.H.S. PO1 AG 026467. We are grateful to Ms. Latoya Woods for her expert assistance in manuscript preparation.
References
- Al-Shaer MH, Choueiri NE, Correia ML, Sinkey CA, Barenz TA, Haynes WG. Effects of aging and atherosclerosis on endothelial and vascular smooth muscle function in humans. Int J Cardiol. 2006;109:201–206. doi: 10.1016/j.ijcard.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Baba SP, Barski OA, Ahmed Y, O’Toole TE, Conklin DJ, Bhatnagar A, Srivastava S. Reductive metabolism of AGE precursors: a metabolic route for preventing AGE accumulation in cardiovascular tissue. Diabetes. 2009;58:2486–97. doi: 10.2337/db09-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basta G, Lazzerini G, Del Turco S, Ratto GM, Schmidt AM, De Caterina R. At least 2 distinct pathways generating reactive oxygen species mediate vascular cell adhesion molecule-1 induction by advanced glycation end products. Arterioscler Thromb Vasc Biol. 2005;25:1401–1407. doi: 10.1161/01.ATV.0000167522.48370.5e. [DOI] [PubMed] [Google Scholar]
- Blackwell KA, Sorenson JP, Richardson DM, Smith LA, Suda O, Nath K, Katusic ZS. Mechanisms of aging-induced impairment of endothelium-dependent relaxation: role of tetrahydrobiopterin. Am J Physiol Heart Circ Physiol. 2004;287:H2448–H2453. doi: 10.1152/ajpheart.00248.2004. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Fleming I, Busse R. Endothelial aging. Cardiovasc Res. 2005;66:286–294. doi: 10.1016/j.cardiores.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, Nowygrod R, Neeper M, Przysiecki C, Shaw A, Migheli A, Stern D. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- Brown KA, Chu Y, Lund DD, Heistad DD, Faraci FM. Gene transfer of extracellular superoxide dismutase protects against vascular dysfunction with aging. Am J Physiol Heart Circ Physiol. 2006;290:H2600–H2605. doi: 10.1152/ajpheart.00676.2005. [DOI] [PubMed] [Google Scholar]
- Bucala R, Tracey KJ, Cerami A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J Clin Invest. 1991;87:432–438. doi: 10.1172/JCI115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, Goova MT, Moser B, Kislinger T, Lee DC, Kashyap Y, Stern DM, Schmidt AM. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 2002;106:2827–2835. doi: 10.1161/01.cir.0000039325.03698.36. [DOI] [PubMed] [Google Scholar]
- Cameron NE, Cotter MA. Impaired contraction and relaxation in aorta from streptozotocin-diabetic rats: role of polyol pathway. Diabetologia. 1992;35:1011–1019. doi: 10.1007/BF02221675. [DOI] [PubMed] [Google Scholar]
- Celermajer DS, Sorensen KE, Spiegelhalter DJ, Georgakopoulos D, Robinson J, Deanfield JE. Aging is associated with endothelial dysfunction in healthy men years before the age-related decline in women. J Am Coll Cardiol. 1994;24:471–476. doi: 10.1016/0735-1097(94)90305-0. [DOI] [PubMed] [Google Scholar]
- Chinellato A, Pandolfo L, Ragazzi E, Zambonin MR, Froldi G, De Biasi M, Caparrotta L, Fassina G. Effect of age on rabbit aortic responses to relaxant endothelium-dependent and endothelium-independent agents. Blood Vessels. 1991;28:358–365. doi: 10.1159/000158882. [DOI] [PubMed] [Google Scholar]
- Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, Sourris KC, Tan AL, Fukami K, Thallas-Bonke V, Nawroth PP, Brownlee M, Bierhaus A, Cooper ME, Forbes JM. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol. 2009;20:742–752. doi: 10.1681/ASN.2008050514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, Kaley G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. 2002;90:1159–1166. doi: 10.1161/01.res.0000020401.61826.ea. [DOI] [PubMed] [Google Scholar]
- Geary GG, Buchholz JN. Selected contribution: effects of aging on cerebrovascular tone and [Ca2+]i. J Appl Physiol. 2003;95:1746–1754. doi: 10.1152/japplphysiol.00275.2003. [DOI] [PubMed] [Google Scholar]
- Hamada Y, Araki N, Horiuchi S, Hotta N. Role of polyol pathway in nonenzymatic glycation. Nephrol Dial Transplant. 1996;11(Suppl 5):95–98. doi: 10.1093/ndt/11.supp5.95. [DOI] [PubMed] [Google Scholar]
- Hamada Y, Nakamura J, Naruse K, Komori T, Kato K, Kasuya Y, Nagai R, Horiuchi S, Hotta N. Epalrestat, an aldose reductase inhibitor, reduces the levels of Nepsilon-(carboxymethyl)lysine protein adducts and their precursors in erythrocytes from diabetic patients. Diabetes Care. 2000;23:1539–1544. doi: 10.2337/diacare.23.10.1539. [DOI] [PubMed] [Google Scholar]
- Headrick JP. Aging impairs functional, metabolic and ionic recovery from ischemia-reperfusion and hypoxia-reoxygenation. J Mol Cell Cardiol. 1998;30:1415–1430. doi: 10.1006/jmcc.1998.0710. [DOI] [PubMed] [Google Scholar]
- Hongo K, Nakagomi T, Kassell NF, Sasaki T, Lehman M, Vollmer DG, Tsukahara T, Ogawa H, Torner J. Effects of aging and hypertension on endothelium-dependent vascular relaxation in rat carotid artery. Stroke. 1988;19:892–897. doi: 10.1161/01.str.19.7.892. [DOI] [PubMed] [Google Scholar]
- Hwang YC, Sato S, Tsai JY, Yan S, Bakr S, Zhang H, Oates PJ, Ramasamy R. Aldose reductase activation is a key component of myocardial response to ischemia. FASEB J. 2002;16:243–245. doi: 10.1096/fj.01-0368fje. [DOI] [PubMed] [Google Scholar]
- Hwang YC, Bakr S, Ellery CA, Oates PJ, Ramasamy R. Sorbitol dehydrogenase: a novel target for adjunctive protection of ischemic myocardium. FASEB J. 2003;17:2331–2333. doi: 10.1096/fj.03-0128fje. [DOI] [PubMed] [Google Scholar]
- Hwang YC, Kaneko M, Bakr S, Liao H, Lu Y, Lewis ER, Yan S, Ii S, Itakura M, Rui L, Skopicki H, Homma S, Schmidt AM, Oates PJ, Szabolcs M, Ramasamy R. Central role for aldose reductase pathway in myocardial ischemic injury. FASEB J. 2004;18:1192–1199. doi: 10.1096/fj.03-1400com. [DOI] [PubMed] [Google Scholar]
- Hwang YC, Shaw S, Kaneko M, Redd H, Marrero MB, Ramasamy R. Aldose reductase pathway mediates JAK-STAT signaling: a novel axis in myocardial ischemic injury. FASEB J. 2005;19:795–797. doi: 10.1096/fj.04-2780fje. [DOI] [PubMed] [Google Scholar]
- Ibarra M, Meneses A, Ransanz V, Castillo C, Hong E. Changes in endothelium-dependent vascular responses associated with spontaneous hypertension and age in rats. Arch Med Res. 1995;26:S177–S183. [PubMed] [Google Scholar]
- Kates AM, Herrero P, Dence C, Soto P, Srinivasan M, Delano DG, Ehsani A, Gropler RJ. Impact of aging on substrate metabolism by the human heart. J Am Coll Cardiol. 2003;41:293–299. doi: 10.1016/s0735-1097(02)02714-6. [DOI] [PubMed] [Google Scholar]
- Kato H, Hayase F, Shin DB, Oimomi M, Baba S. 3-Deoxyglucosone, an intermediate product of the Maillard reaction. Prog Clin Biol Res. 1989;304:69–84. [PubMed] [Google Scholar]
- Keegan A, Jack AM, Cotter MA, Cameron NE. Effects of aldose reductase inhibition on responses of the corpus cavernosum and mesenteric vascular bed of diabetic rats. J Cardiovasc Pharmacol. 2000;35:606–613. doi: 10.1097/00005344-200004000-00014. [DOI] [PubMed] [Google Scholar]
- Kung CF, Luscher TF. Different mechanisms of endothelial dysfunction with aging and hypertension in rat aorta. Hypertension. 1995;25:194–200. doi: 10.1161/01.hyp.25.2.194. [DOI] [PubMed] [Google Scholar]
- Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107:490–497. doi: 10.1161/01.cir.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- Lal S, Szwergold BS, Taylor AH, Randall WC, Kappler F, Wells-Knecht K, Baynes JW, Brown TR. Metabolism of fructose-3-phosphate in the diabetic rat lens. Arch Biochem Biophys. 1995;318:191–199. doi: 10.1006/abbi.1995.1220. [DOI] [PubMed] [Google Scholar]
- van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Luscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–1744. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillin JB, Taffet GE, Taegtmeyer H, Hudson EK, Tate CA. Mitochondrial metabolism and substrate competition in the aging Fischer rat heart. Cardiovasc Res. 1993;27:2222–2228. doi: 10.1093/cvr/27.12.2222. [DOI] [PubMed] [Google Scholar]
- Muller-Delp J, Spier SA, Ramsey MW, Lesniewski LA, Papadopoulos A, Humphrey JD, Delp MD. Effects of aging on vasoconstrictor and mechanical properties of rat skeletal muscle arterioles. Am J Physiol Heart Circ Physiol. 2002;282:H1843–H1854. doi: 10.1152/ajpheart.00666.2001. [DOI] [PubMed] [Google Scholar]
- Murohara T, Yasue H, Ohgushi M, Sakaino N, Jougasaki M. Age related attenuation of the endothelium dependent relaxation to noradrenaline in isolated pig coronary arteries. Cardiovasc Res. 1991;25:1002–1009. doi: 10.1093/cvr/25.12.1002. [DOI] [PubMed] [Google Scholar]
- Mylari BL, Larson ER, Beyer TA, Zembrowski WJ, Aldinger CE, Dee MF, Siegel TW, Singleton DH. Novel, potent aldose reductase inhibitors: 3,4-dihydro-4-oxo-3-[[5-(trifluoromethyl)-2-benzothiazolyl] methyl]-1-phthalazineacetic acid (zopolrestat) and congeners. J Med Chem. 1991;34:108–122. doi: 10.1021/jm00105a018. [DOI] [PubMed] [Google Scholar]
- Mylari BL, Beyer TA, Scott PJ, Aldinger CE, Dee MF, Siegel TW, Zembrowski WJ. Potent, orally active aldose reductase inhibitors related to zopolrestat: surrogates for benzothiazole side chain. J Med Chem. 1992;35:457–465. doi: 10.1021/jm00081a006. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Yamazaki K, Satoh A, Urakaze M, Kobayashi M, Yamabe H, Osawa H, Shirato K, Sugawara T, Nakamura M, Tamura M, Okumura K. Effects of eparlestat on plasma levels of advanced glycation end products in patients with type 2 diabetes. In Vivo. 2003;17:177–180. [PubMed] [Google Scholar]
- Ohmori S, Mori M, Kawase M, Tsuboi S. Determination of methylglyoxal as 2-methylquinoxaline by high-performance liquid chromatography and its application to biological samples. J Chromatogr. 1987;414:149–155. doi: 10.1016/0378-4347(87)80033-6. [DOI] [PubMed] [Google Scholar]
- Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr, Chow WS, Stern D, Schmidt AM. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- Shapiro BP, Owan TE, Mohammed SF, Meyer DM, Mills LD, Schalkwijk CG, Redfield MM. Advanced glycation end products accumulate in vascular smooth muscle and modify vascular but not ventricular properties in elderly hypertensive canines. Circulation. 2008;118:1002–1010. doi: 10.1161/CIRCULATIONAHA.108.777326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul PW. Endothelial nitric oxide synthase, caveolae and the development of atherosclerosis. J Physiol. 2003;547:21–33. doi: 10.1113/jphysiol.2002.031534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaki Y, Su C, Lee TJ, Kolm P, Cline WH, Jr, Nickols GA. Endothelial modulation of vascular relaxation to nitrovasodilators in aging and hypertension. J Pharmacol Exp Ther. 1986;239:861–866. [PubMed] [Google Scholar]
- Thornalley PJ. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification – a role in pathogenesis and antiproliferative chemotherapy. Gen Pharmacol. 1996;27:565–753. doi: 10.1016/0306-3623(95)02054-3. [DOI] [PubMed] [Google Scholar]
- Thornalley PJ. Glutathione-dependent detoxification of alphaoxoaldehydes by the glyoxalase system: involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chem Biol Interact. 1998:111–112. 137–151. doi: 10.1016/s0009-2797(97)00157-9. [DOI] [PubMed] [Google Scholar]
- Tschudi MR, Barton M, Bersinger NA, Moreau P, Cosentino F, Noll G, Malinski T, Luscher TF. Effect of age on kinetics of nitric oxide release in rat aorta and pulmonary artery. J Clin Invest. 1996;98:899–905. doi: 10.1172/JCI118872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Jagt DL, Robinson B, Taylor KK, Hunsaker LA. Reduction of trioses by NADPH-dependent aldo-keto reductases. Aldose reductase, methylglyoxal, and diabetic complications. J Biol Chem. 1992;267:4364–4369. [PubMed] [Google Scholar]
- Vikramadithyan RK, Hu Y, Noh HL, Liang CP, Hallam K, Tall AR, Ramasamy R, Goldberg IJ. Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. J Clin Invest. 2005;115:2434–2443. doi: 10.1172/JCI24819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab. 2001;280:E685–E694. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]