

Abstract
Current views of multidrug (MD) recognition focus on large drug-binding cavities with flexible elements. However, MD recognition in BmrR is supported by a small, rigid drug-binding pocket. Here, a detailed description of MD binding by the noncanonical BmrR protein is offered through the combined use of X-ray and solution studies. Low shape complementarity, suboptimal packing, and efficient burial of a diverse set of ligands is facilitated by an aromatic docking platform formed by a set of conformationally fixed aromatic residues, hydrophobic pincer pair that locks the different drug structures on the adaptable platform surface, and a trio of acidic residues that enables cation selectivity without much regard to ligand structure. Within the binding pocket is a set of BmrR-derived H-bonding donor and acceptors that solvate a wide range of ligand polar substituent arrangements in a manner analogous to aqueous solvent. Energetic analyses of MD binding by BmrR are consistent with structural data. A common binding orientation for the different BmrR ligands is in line with promiscuous allosteric regulation.
Keywords: ligand binding, ligand responsive transcription factor, molecular recognition, multidrug resistance, multispecificity
Multidrug (MD) efflux protects nearly all living cells against a barrage of cytotoxic chemicals (1, 2). High levels of MD efflux can extend broad chemoprotection to drug-targeted cells, which in turn are rendered resistant to lethal doses of multiple, unrelated drug therapies. The transport of antimicrobial and antifungal agents has been established as a primary cause of multidrug resistance (MDR) in pathogenic strains of Escherichia coli, Staphylococcus aureus, Pseudomonas aeruginosa, and Candida albicans (3). In human cells, increased export of chemicals by P-glycoprotein (Pgp) and numerous MDR-associated proteins can also confer simultaneous resistance to a multitude of antitumor agents and has been linked to chemotherapy failure (4). MDR-related transport is controlled largely by two cellular functions, namely those enacted by drug-responsive gene regulators and efflux pumps (3). Quorum sensing regulators are also known to influence MD efflux pump expression (5). For the former, promiscuous chemical recognition facilitates broad cellular responses that target drug-like compounds for export while leaving native biological ligands untouched. Elucidating the basis of MDR functions requires detailed descriptions of MD recognition. To date, an incomplete understanding of how MDR proteins interact with structurally and chemically diverse drugs continue to hinder efforts to evade, inhibit and control MDR activities.
Crystallographic studies of drug binding by MDR proteins have enabled improved descriptions of MD recognition (6, 7). Indeed, details made available through structural data connect MD recognition to well-known physical and chemical features of binding. Moreover, contributions to multispecificity are now more recognizable due to the increasing availability of X-ray structures of MDR systems with different architectures and binding-site designs. However, both multispecificity and partial selectivity remain poorly defined due to insufficient analyses of MD interactions with individual MDR proteins. Current views of MD recognition are dominated by the canonical “multisite” model, which highlights structural and biochemical aspects of drug binding by regulators and transporters. Binding plasticity and adaptability are central to this model and, in many cases, are believed to derive largely from large drug-binding cavities composed of distinct, overlapping “minipockets” and flexible protein elements (SI Appendix, Fig. S1). For QacR (7), TtgR (8), AcrB (9), Pgp (10), and other characterized MDR systems, these features are present and appear to be critical for multifaceted binding, which includes the accommodation of dissimilar drug structures and bound configurations. Descriptions of MD recognition are currently limited to the canonical model. However, recent data suggest that it offers only a partial picture of multispecific drug binding.
BmrR is a gene regulator with a verified MDR role in Bacillus subtilis. It controls the expression of the Bmr efflux pump in response to a diverse array of cationic antibiotics, dyes, and disinfectants, which are also transported by Bmr and other efflux pumps (11). Although BmrR has been widely viewed as a prototype for investigating MD recognition, only a limited number of binding studies have been reported. Importantly, the data available include crystal structures of three BmrR-drug complexes that suggest possible departures from the canonical MD-binding model wherein MD recognition occurs in a small, rigid drug pocket (12, 13). Irreconcilable differences between BmrR and the canonical view suggest alternative binding models; one has been proposed based on the limited amount of data available (14). The model considers two binding components (Fig. 1A). One, coined the “hydrophobic (Hb) slot,” provides a common anchoring point for different drugs through contacts with rigid, nonpolar ligand moieties. The second, called the “hydrophilic (Hp) cavity,” offers extended binding versatility due to its concave structure and solvent exposure. The Hb slot–Hp cavity combination appears to provide another biological solution to MD recognition. The present study replaces this model with one that provides a molecular depiction of MD recognition in BmrR. Interestingly, the Hb slot–Hp cavity elements are retained.
Fig. 1.

Structural and solution-based analyses of drug recognition by BmrR. (A) Schematic of noncanonical MD-binding model. The drug pocket is divided into two functional units: a hydrophobic slot located at the top front of the pocket and an adaptable, solvent-accessible, polar vestibule. (B) The chemical structures of the probes used in this study. The color and abbreviated naming scheme shown is used throughout the paper. All (crystallographic) figures were generated using Pymol (40).
Because of its noncanonical features, BmrR offers an alternative framework to investigate MD recognition. To better understand MD binding by the small, rigid BmrR pocket, solution and crystallographic approaches have been employed to investigate BmrR interactions with medically important ligands, including puromycin (PUR), ethidium (ET), tetracycline (TET), 4-aminoqualdine (4AQ), kanamycin (KAN), and acetylcholine (Ach) (Fig. 1B). Due to the broad range of structural, chemical, and binding properties exhibited, the selected probe set facilitates the most extensive analyses of MD recognition for BmrR to date. Furthermore, by combining structural with solution-binding data for the diverse ligands, we are able to elucidate key features of MD recognition and dissect important contributions to MD binding.
Results
The drug-bound BmrR structures were solved by molecular replacement and refined using data collected to 2.8–3.2 Å. The structure of BmrR bound to tetraphenylphosphonium (TPP) and DNA (residue side chains, ligands, and solvent excluded) was used as the starting model for the refinements. Initial rigid-body, group B factor, and positional refinements, combined with simulated annealing, returned well-defined, continuous protein electron density for the six complexes studied (15–17). All ligand-soaking experiments produced large positive electron density peaks in the putative BmrR drug pocket (Fig. 2 A–C); mock ligand soaks produced only weak patches of density. Although the resolution limit of X-ray data prevents unambiguous assignment of the ligand-bound orientations, those observed in the refined structural models offer the best fits to the ligand-derived density based on real space correlation coefficients (SI Appendix, Table S1). The docking modes depicted also result in reasonable burial of apolar and polar surface areas, H-bonding geometries, and long-range electrostatic interaction distances. Alternative ligand-binding modes were explored. However, they did not offer improved fits to the electron density, nor did they better reflect chemical principles or the BmrR drug recognition properties (SI Appendix, Fig. S2). Table 1 presents the data collection, refinement, and model geometry statistics for the series of ligand-bound structures.
Fig. 2.

Crystallographic analysis of MD recognition by BmrR. (Left) Simulated annealing omit maps (Fobs - Fcalc) of wild-type BmrR bound to (A) 4AQ, (B) puromycin, and (C) acetylcholine. Each is contoured at 1.0σ. (D) Ligands are shown in the space-filling representation with the oxygen and nitrogen atoms colored red and blue, respectively. Drug carbon atoms are colored according the scheme used in Fig. 1B. An “all-ligand-bound” representation of binding is shown at the far right. The binding cavity surface was generated using CASTp (41). (E) Relative bound positions of the BmrR ligands. (F) Superposition of the drug-bound BmrR structures reveals pocket rigidity. Coloring scheme is described in Fig. 1B legend. (G) H-bonding contacts in the BmrR pocket. Tyr residues are colored magenta; the H bonds and distances are depicted in green.
Table 1.
Data collection and refinement
| BmrR•PUR•DNA |
BmrR•ET•DNA |
BmrR•TET•DNA |
BmrR•4AQ•DNA |
BmrR•KAN•DNA |
BmrR•ACh•DNA |
|
| Data collection | ||||||
| Space group | P4322 | P4322 | P4322 | P4322 | P4322 | P4322 |
| Cell dimensions | ||||||
| a, b, c, Å | 106.5, 106.5, 145.4 | 106.6, 106.6, 145.7 | 106.7, 106.7, 146.2 | 106.5, 106.5, 146.5 | 106.9, 106.9, 145.8 | 106.4, 106.4, 145.6 |
| α, β, γ, ° | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Rsym* | 0.12 (0.68) | 0.11 (0.73) | 0.13 (0.79) | 0.11 (0.74) | 0.13 (0.73) | 0.13 (0.69) |
| I/σI* | 10.9 (1.1) | 12.4 (1.3) | 9.4 (1.0) | 12.6 (1.5) | 13.9 (1.5) | 10.0 (1.1) |
| Completeness,* % | 92.2 (60.8) | 89.5 (80.0) | 95.8 (68.2) | 91.0 (52.0) | 95.15 (69.3) | 91.7 (50.2) |
| Redundancy | 3.8 (2.4) | 7.1 (3.3) | 5.1 (3.0) | 6.6 (5.1) | 3.6 (2.4) | 6.2(3.0) |
| Refinement | ||||||
| I/σI = 1.0, Å | 37.7-2.8 | 37.7-2.9 | 44.3-2.9 | 44.4-3.2 | 44.2-3.0 | 45.2-3.1 |
| (I/σI = 2.0), Å | (37.7-3.0) | (37.7-3.1) | (44.3-3.1) | (44.4-3.2) | (44.2-3.2) | (45.2-3.3) |
| No. reflections | 17,051 | 14,575 | 15,217 | 11,986 | 13,958 | 13,749 |
| Rwork/Rfree, % | 23.2/26.7 | 20.2/24.7 | 23.9/26.9 | 24.8/29.4 | 21.6/25.9 | 23.8/28.3 |
| rmsd | ||||||
| Bond lengths, Å | 0.017 | 0.014 | 0.022 | 0.006 | 0.015 | 0.013 |
| Bond angles, ° | 1.846 | 1.394 | 1.595 | 1.12 | 1.79 | 1.68 |
| Ramachadran analysis: Protein geometry | ||||||
| Preferred regions, % | 97.8 | 98.5 | 98.2 | 98.2 | 99.6 | 99.3 |
| Allowed regions, % | 2.2 | 1.5 | 1.8 | 1.8 | 0.4 | 0.7 |
| Outlier, % | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
Statistics of BmrR-drug complex data collection and refinement. Data integration and scaling was carried out using the program HKL and refinement was initially carried using the program CNS (15) and completed in Refmac5 of the CCP4 package (16). Model building and refinement was performed using Coot (42). Structure quality was analyzed using the MolProbity (43) server and Procheck-CCP4 (16). 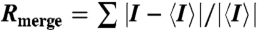 ;
; 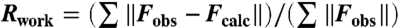 ;
; 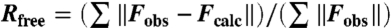 , where all reflections (10% of the data) belong to a random test set.
, where all reflections (10% of the data) belong to a random test set.
*Values in parentheses refer to the highest resolution shell.
Structural details of MD recognition by BmrR are summarized in the SI Appendix, Fig. S3 and Tables S2–S4. They include similar ligand accessible surface area (ASA) burial (ca. 79 ± 2%) and shape complementarity (Sc) values (0.58 ± 0.02) for the different BmrR–ligand complexes (18). The values calculated for BmrR are somewhat reduced relative to those found for ligand-specific ligand prototypes, which bury cognate ligands ca. 90% and exhibit Sc values > 0.7 (19, 20) (SI Appendix, Table S2). In agreement with X-ray structures of QacR and other MDR proteins, drug recognition by BmrR is dominated by apolar contacts involving aromatic and hydrophobic residues (SI Appendix, Fig. S3 and Table S3). Unlike canonical MDR systems, the recognition of diverse ligands is restricted to a common set of residues lining the BmrR drug pocket (SI Appendix, Table S3). Polar moieties also contribute to MD recognition. The PUR, TET, 4AQ, KAN, and ACh probes reveal unique H-bonding contacts and defined long-range electrostatic interactions with Glu253 that range from 4.5 to 6 Å (see Fig. 3).
Fig. 3.

MD recognition by BmrR. Binding of the (A) PUR, (B) ET, (C) AQ, (D) TET, (E) KAN, and (F) ACh ligands. Only the key residues are shown. Protein carbons are colored light blue. The distances between E253 and PURN, 4.8 Å; ETN5, 5.5 Å; 4AQN4, 2.9 Å; TETN4, 4.1 Å; KANN1, 3.5 Å.; and AchN1, 4.5 Å.
A solution-binding approach based on the intrinsic BmrR fluorescence was also used to examine MD recognition by BmrR (SI Appendix, Fig. S4). For all probes, ligand-dependent quenching effects afforded binding isotherms that reflect identical, independent binding-site behavior (SI Appendix, Fig. S4). Nonidentical Kd values are obtained for the structurally distinct probes (SI Appendix, Table S2). The tightest binders of the selected probes include the strong cations, ET (1.9 μM) and ACh (6.6 μM). The weak cations, PUR and KAN, bound less tightly with Kd values of 17 and 28 μM, respectively. In its zwitterionic form, the TET probe bound with a Kd of 51 μM. The small, weakly cationic 4AQ is the weakest binder with a Kd of 210 μM. The binding constants obtained for TPP and RHO6G (SI Appendix, Fig. S5) reproduce those reported previously (12, 13).
Discussion
An extended set of diverse ligands has been used to probe MD recognition in BmrR. Compared to the previous studies, the ligand set employed here is larger, more diverse, and better resembles those used routinely to investigate MDR functions (21). Solution-binding results, including the range of Kd values observed, are in line with promiscuous drug recognition, the matching series of BmrR crystal structures, and a MDR role for BmrR (11). Importantly, the ligand-bound structures reveal similar docking locations for all six probes. The confined nature of drug binding is emphasized by the superimposed set of structures (Fig. 2E). By localizing the key determinants of MD recognition to a common interaction surface, the newly solved BmrR structures confirm departures from the canonical MD-binding model implicated previously by the BmrR complexes with RHO6G, TPP, and berberine (BER) (12, 13) (SI Appendix, Fig. S5). Whereas canonical MDR prototypes interact with different drugs using large, flexible cavities (7) (1,100–1,600 Å3, SI Appendix, Fig. S1), BmrR supports MD recognition in a rigid pocket about half the volume (ca.650 Å3, Fig. 2D). Despite the differences in drug pocket size, X-ray and solution-binding results suggest similar degrees of ligand promiscuity for BmrR and canonical MDR proteins (11, 22). This result is significant considering current descriptions of MD recognition and the large emphasis placed on drug pocket size and flexibility.
The superimposed BmrR structures also highlight the rigidity of the drug pocket. Residues involved in binding respond negligibly to the different ligand structures and show an average rmsd of 0.45 Å over all side-chain atoms (Fig. 2F). This apparent rigidity is unlikely an artifact of the ligand-soaking procedure because cocrystallization experiments with ligands, BmrR, and DNA gave identical results. Instead, BmrR pocket rigidity appears to be linked in part to H-bonding interactions between the Tyr residues lining the drug pocket and nearby backbone and side-chain atoms (Fig. 2G). Each Tyr hydroxyl group engages in one or two H bonds with heavy atom separations ranging from 2.4 to 3.4 Å; estimated angles range from approximately 100° to 180°. These contacts are observed in all BmrR structures determined to date (6, 13, 23).
Key Features of MD Recognition in the Noncanonical BmrR.
Disconnects between the canonical MD-binding model and BmrR expose gaps in our current knowledge of MD recognition by MDR proteins. However, these gaps are somewhat closed by the BmrR structures that highlight the dominance of familiar MD-binding elements. Those most prevalent include contributions from aromatic and aliphatic residues (Fig. 3 A–F and SI Appendix, Table S3). Both are well suited for MD recognition and play well-established roles in multispecific interactions with antigens (24, 25), odorants (26), and xenobiotics (7). The aromatic contribution to structure and binding in the BmrR drug pocket is striking. Although typically regarded as individual components, the spatial arrangement of the aromatic side chains is more consistent with all six residues functioning as a cooperative binding unit. Together, they offer a rigid, ring-like scaffold for the different drugs to dock (Fig. 4). Although the surface formed by the aromatic side chains is not entirely smooth, the broad range of interactions with PUR, ET, TET, 4AQ, KAN, and ACh underscores the versatility of the docking platform. The conformational rigidity of the BmrR drug-pocket aromatics contrasts with QacR and other MDR proteins that use flexible Tyr and Phe side chains for multispecific binding. To the contrary, pocket rigidity appears to be a necessary feature of MD recognition in BmrR. Recently, Tyr residues were shown to offer binding specificity in immunoglobulins with a restricted set of antigen combining site residues (27, 28). This specificity role likely requires the participation of the phenolic hydroxyl groups. Drug docking to the aromatic platform in BmrR exhibits no such requirements.
Fig. 4.

Structural basis of drug binding and multispecificity. Aromatic side chains lining the BmrR drug pocket form a rigid, aromatic platform for diverse drugs to dock. (A) PUR and (B) ET, as well as the other drugs (not shown), are clamped onto the platform through interactions with the hydrophobic pincer formed by V147 and I255. Aromatic residues are shown as magenta sticks; residues of the hydrophobic pincer are shown as orange sticks; the small dots are used to illustrate the pincer residue space-filling volumes. (C and D) Top and side views of both hydrophobic elements interacting with PUR, ET, and KAN.
Although less represented, aliphatic side chains interact extensively with the BmrR probes (Fig. 4). Like the drug-binding aromatics, two aliphatic residues are presented as a single binding unit. Indeed, V147 and I255 appear to function as a hydrophobic pincer that anchors a variety ligands on to the aromatic drug-docking platform. I255 caps the narrow side of the scaffold; V147 sits near the edge of the opposite face, such that drugs are granted full access to the wider side of the docking surface (Fig. 4 A, B, and D). Only two polar side chains are located within 5 Å of bound ligands, including N149 and E253 (Fig. 3). A low participation of polar residues in BmrR is consonant with the idea that polar contacts enforce specificity and undermine promiscuous ligand recognition.
Ligand Burial and Suboptimal Ligand Packing in MD Recognition.
Together, the residue contributions to MD recognition in BmrR bury approximately 70–80% of the ligand ASAs (SI Appendix, Table S2). Although such values have been used to indicate efficient ligand packing, the low Sc values (18) obtained for the BmrR complexes suggest otherwise (SI Appendix, Table S2). Comparisons of the protein and ligand ASA burial support the Sc calculations. Larger ligand burial values are observed for all complexes, which suggest that the ligand contacts ensued upon binding (or burial within the BmrR pocket) involve only a fraction of their ASAs (SI Appendix, Table S4). Importantly, low complementarity of BmrR to its ligands is an essential feature of MD recognition and is likely the basis for the ca. 80% ligand burial threshold. In aggregate, the data indicate that whereas ligand burial does not maximize binding contacts, poor packing can be supplemented largely by excluding water from noncontacting drug surfaces. Although several BmrR–ligand complexes suggest the accommodation of water molecules (Fig. 2D and SI Appendix, Fig. S8), the described burial strategy apparently mediates favorable binding for a broad range of compounds.
Ligand burial appears to be a key component of MD recognition in BmrR. For TPP, low ligand burial (59%) is likely the basis of weak binding despite its hydrophobic and cationic properties (SI Appendix, Table S1). Moreover, analyses of binding and ligand burial reveal linear correlations between ln Kd and both the total and nonpolar ASA buried for the BmrR probes employed here and in previous structural and solution-binding studies (SI Appendix, Fig. S6). Slope values of -6.10 ± 3.0 cal/(mol·Å2) and -6.23 ± 2.3 cal/(mol·Å2) are obtained, respectively, indicating favorable effects on binding due to ligand burial and the dominance of apolar contributions. The nonpolar contributions revealed for BmrR are suboptimal based on the range reported [30–50 cal/(mol·Å2)] for solute transfer, protein folding, and binding studies (29, 30). The low energetics [ca. 10 cal/(mol·Å2)] agree with the inefficient ligand packing and incomplete ligand burial in BmrR. An analysis of ligand efficiency in BmrR reveals a slope of 0.22 with a modest correlation coefficient of 0.54 (SI Appendix, Fig. S7). This value is similar to those observed for inhibitors of protein–protein interactions, protein kinases, and many proteases. As such, the energetics of MD recognition are comparable to those achieved by drug discovery and inhibitor design efforts (31, 32). Despite notable differences, QacR displays a similar relationship between ligand binding and burial (SI Appendix, Fig. S6C). Other MDR proteins will likely exhibit similar mechanisms and energetics for MD binding.
Polar and Electrostatic Contributions to MD Recognition.
Although the burial of polar atoms generally opposes binding and folding, the dissection of ligand ASA burial into its nonpolar and polar components reveals little to no adverse effects on binding from the burial of the latter (SI Appendix, Fig. S8). In addition, the data may suggest an ASA burial threshold of ca. 150 Å2, below which polar effects appear favorable; above this arbitrary value, polar interactions display their anticipated properties. The capacity of BmrR to bury polar atoms without energetic costs is somewhat unexpected considering the nonpolar character of the drug pocket. Interestingly, a similar value of approximately 160 Å2 has been suggested as the optimal polar ASA for potentially successful drug candidates (molecular mass of approximately 500 Da) (33). The role of water is currently unknown for BmrR; insights await higher resolution structures (see SI Appendix, Fig. S9).
The X-ray structures presented here highlight three features, all of which extend binding versatility to polar and electrostatic contacts. These include E253, which is a proposed determinant of cation selectivity; N149, the lone drug-contacting polar residue identified to date; and the Tyr residues lining the drug pocket.
Issues related to cation neutralization by E253 have been only partially addressed using ligands with delocalized charges (13). However, those employed in this study show spatially defined charges, enabling the influences of E253 to be more clearly revealed. BmrR structures with PUR, ET, TET, KAN, and ACh place the ligand cationic nitrogen atoms within 3.5–6 Å of E253 (Fig. 3 A, B, and D–F). Although not close enough to be called salt bridges, these distances fall within the well-established window of long-range electrostatic interactions (34, 35). The exception is 4AQ, which instead appears to maximize packing, which results in an 4AQN1-E253Oε distance of 6.3 Å (Fig. 3C). In addition to optimal packing, favorable electrostatics are facilitated by a 2.9 Å E253Oε-4AQN4 H bond and the 4AQ π-system. Unlike 4AQ, TET binding produces an acceptable E253Oε-TETN4 distance of 4.7 Å. In this case, a proton gain at TETN4 and a proton loss at TETO3 result in attractive (4.7 Å) and repulsive (7.4 Å) interactions with E253 (Fig. 3D). Clearly, the former is favorable enough to offset the repulsive effects, which appear to be dissipated only partially by an H bond with N149.
The X-ray data suggest that D47 and E266 may also play roles in the promiscuous binding of cationic ligands. The acidic trio is symmetrically disposed around the drug pocket, with each being equidistant from the drug-pocket center (Fig. 5). The residues also define a plane that slices through the pocket in an arrangement that enables electrostatic stabilization for a broad array of bound ligands with cationic centers that show moderate approaches to one or more of the E253, D47, and E266 side chains (Fig. 5B). Based on the X-ray data, the buried E253 residue appears to dominate cation recognition for BmrR (Fig. 5 C and D). However, the prevalence of shorter cation-E253 distances may reflect a bias in the probe sets chosen for X-ray and solution studies. Indeed, recent mutagenesis data support electrostatic roles for the less buried D47 and E266 residues (13).
Fig. 5.

Electrostatic contributions to MD recognition include a trio of acidic residues. (A and B) Side views of the E253-D47-E266 trio and the PUR (yellow) and TPP (silver) ligands. The protein residues are shown in the space-filling representation. Both ligands are depicted as sticks with their cationic centers highlighted as spheres. (C and D) Front and side views of the electrostatic feature along with the relative positions of cationic centers (shown as spheres) of the ligands used to probe MD recognition in BmrR. The cationic centers (spheres) are colored according the color scheme described in the Fig. 1 legend.
H-Bonding Contributions to MD Recognition.
Previously solved BmrR structures reveal a single H bond with the BER ligand (13). Using a probe set with expanded H-bonding properties, we show that BmrR is highly adaptive to polar interactions (Fig. 3). H-bonding elements in the drug pocket interact with the majority of the buried polar atoms on PUR, ET, 4AQ, and KAN. PUR and KAN engage in distinct sets of three H bonds (Fig. 3 A and G). TET donates an H bond to N149 (Fig. 3D). Alternative N149 conformations enable the donation of an H bond to the BER, KAN, and ACh probes.
MD recognition is rarely devoid of polar contributions, owing to their importance in drug action and drug-receptor specificity (32, 33). For MDR functions to be effective, complementary contacts must be presented to drugs with H-bonding atoms. BmrR presents a redundant set of H-bond donor and acceptor atoms that appears to be capable of satisfying different ligand H-bonding arrangements in a rigid drug pocket without compromising preexisting interactions (SI Appendix, Fig. S10). As a result, polar interactions are “supplied” on a need basis without large energetic penalties. In contrast, the canonical QacR drug pocket contains at least three Glu and two Asn that use drug-dependent rotamer geometries (7). For BmrR, the availability of such elements is consistent with the analysis of polar ASA burial effects on binding and the favorable accommodation of polar ligand groups.
Several mechanisms for multispecificity have been described (36). One involves a single, rigid pocket that interacts with unrelated ligands through nonidentical contacts. Another requires pocket flexibility and “induced-fit” recognition. A third strategy focuses on a less flexible pocket with distinct minipockets that interacts differentially with diverse ligands. The crystal structures of BmrR bound to our extended probe set appears to be consistent with the first mechanism and includes contributions from promiscuous elements composed of multiple symmetrically disposed residues that function as single drug-binding units. These include a rigid platform, a hydrophobic pincer pair, and a trio of acidic residues. In aggregate, these elements interact extensively with a diverse set of drugs, burying each approximately 80%. Their features are also consistent with multispecific binding, dominant apolar contributions, and cation selectivity. The ligand-binding orientations appear to be dictated by auxiliary contacts that specify the Hb slot and Hp cavity regions of the BmrR drug pocket. P144, Y229, and F224 compose the outer edge of the former, whereas residues below the V147-I255 pincer specify the concave shape, solvent exposure, and binding properties of the latter. The ligand-docking modes revealed here and in previous studies implicate major roles for the Hb slot–Hp cavity combination (Fig. 6). As predicted, planar and rigid ligand moieties presented by our extended probe set interact with the Hb slot. In contrast, small hydrophilic ligands with no rigid or planar moieties (Fig. 6F) show interactions only with the Hp cavity, which binds vastly different moieties of PUR, ET, TET, and 4AQ. This mode of binding may be important for allosteric regulation of the promiscuous BmrR switch.
Fig. 6.

Auxiliary contributions to drug docking in BmrR. (A–E) Space-filling representation of (A) PUR, (B) ET, (C) 4AQ, (D) TET, (E) KAN, and (F) ACh show elements that define the hydrophobic slot and hydrophilic cavity in the BmrR drug pocket dictate the orientation of drug binding. Elements defining the opening of the hydrophobic slot are shown as light pink spheres. The Val-Ile pincer pair are depicted as orange sphere. Characteristics of the hydrophilic cavity are governed by its solvent exposure and the presence of H-bonding elements.
The combination of solution binding and X-ray studies has elucidated key features of MD recognition in a system that presents a binding scheme very different to views of the currently dominant, canonical MD-binding model. Although BmrR does not employ a large drug-binding cavity or flexible protein elements, its drug recognition features highlight key issues regarding the recognition of structurally and chemically diverse compounds. Our current knowledge of MD recognition remains rudimentary. The discovery of additional structural solutions to MD recognition will be important toward better understanding the functions of systems that influence drug action.
Methods
Protein Preparation.
The His-tagged version of the bmrR gene was amplified by PCR from B. subtilis genome using gene specific primers, subcloned into the pBad vector and transformed into E. coli BL21(DE3) cells. Cells were harvested by centrifugation and the pellet was resuspended in lysis buffer (30 mM phosphate, 1.5 mM EDTA, 5% glycerol, 200 mM imidazole, 100 μM protease inhibitor cocktail, 50 μg/mL lysozyme, and 100 uM PMSF). BmrR was then purified by HisTrap HP, Heparin HP, and HiTrap Q HP (GE Healthcare) chromatography. BmrR was subsequently concentrated and subjected to gel filtration on a Superdex 200 (GE Healthcare) in a buffer containing 10 mM Hepes, 300 mM NaCl, 500 μM EDTA, and 10% glycerol.
Crystallization and Data Collection.
Crystals of BmrR bound to a 23 bp DNA duplex containing the bmr promoter sequence were produced as described previously (15). Ligand complexes were produced by soaking preexisting crystals (24 h) in solutions containing 1 M sodium malonate pH 7.0, 0.05% jeffamine, and 2 mM of each ligand: 4-aminoquinaldine, ethidium bromide, puromycin, tetracycline, and kanamycin A (Sigma Aldrich). The ligand-soaked crystals were stabilized and cryoprotected using a solution containing 1 M sodium malonate, 0.05% jeffamine, and 30% glycerol. These crystals were flash frozen in liquid nitrogen. All diffraction data were collected at the Stanford Synchotron Radiation Light Source, beamline 9-2.
Data Reduction and Refinement.
The diffraction data were indexed, reduced, and scaled using the program HKL 2000. After scaling, phases were obtained by molecular replacement by rigid-body refinement using CNS (17) and the structure of BmrR bound to TPP and DNA (accession code IR8E, 2.4 Å) (16) as a starting model. At this point, 5.0% of the data were removed to be used for cross-validation (RFREE). Several rounds of simulated annealing, B-factor refinement, positional refinement, and model building were carried out using CNS (17) and Coot (37). After the refinements converged in CNS, the structure was further refined using the program Refmac5 (CCP4 program suite) (18). The ligands were then modeled into the excess density observed in the BmrR drug pocket, resulting in a 1–2% decrease in RFREE. Fixed translation libration screw (TLS) parameters were determined using the TLS motion detection server (38) and then used in the subsequent rounds of model building and structure refinement. In the last stages of the refinements, waters were added to models. The criteria used to judge their validity included H-bonding geometries and B factors (less than 80). The quality of the final structures were validated using the Molprobity server (39), which shows 97% of all residues in the most favored region of the Ramachadran plot; no residues were found in an unfavorable conformation.
Supplementary Material
Acknowledgments.
S.B. is a National Science Foundation (NSF) graduate research fellowship awardee (DGE-0707427). H.W. is a recipient of The Arnold and Mabel Beckman Young Investigator Award. This work is also supported by a NSF CAREER Award (MCB-0953430). Intensity data was collected at the Sanford Synchrotron Laboratory, beamline 9-2.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3Q5P, 3Q5R, 3Q5S, 3Q3D, 3Q2Y, 3Q1M).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1104850108/-/DCSupplemental.
References
- 1.Nikaido H. Multidrug resistance in bacteria. Annu Rev Biochem. 2009;78:119–146. doi: 10.1146/annurev.biochem.78.082907.145923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007;446:749–757. doi: 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- 3.Grkovic S, Brown MH, Skurray RA. Regulation of bacterial drug export systems. Microbiol Mol Biol Rev. 2002;66:671–701. doi: 10.1128/MMBR.66.4.671-701.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gutmann DA, Ward A, Urbatsch IL, Chang G, van Veen HW. Understanding polyspecificity of multidrug ABC transporters: Closing in on the gaps in ABCB1. Trends Biochem Sci. 2010;35:36–42. doi: 10.1016/j.tibs.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang S, Lopez CR, Zechiedrich EL. Quorum sensing and multidrug transporters in Escherichia coli. Proc Natl Acad Sci USA. 2006;103:2386–2391. doi: 10.1073/pnas.0502890102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heldwein EE, Brennan RG. Crystal structure of the transcription activator BmrR bound to DNA and a drug. Nature. 2001;409:378–382. doi: 10.1038/35053138. [DOI] [PubMed] [Google Scholar]
- 7.Schumacher MA, et al. Structural mechanisms of QacR induction and multidrug recognition. Science. 2001;294:2158–2163. doi: 10.1126/science.1066020. [DOI] [PubMed] [Google Scholar]
- 8.Alguel Y, et al. Crystal structures of multidrug binding protein TtgR in complex with antibiotics and plant antimicrobials. J Mol Biol. 2007;369:829–840. doi: 10.1016/j.jmb.2007.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE. Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science. 2003;300:976–980. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
- 10.Aller SG, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmed M, Borsch CM, Taylor SS, Vazquez-Laslop N, Neyfakh AA. A protein that activates expression of a multidrug efflux transporter upon binding the transporter substrates. J Biol Chem. 1994;269:28506–28513. [PubMed] [Google Scholar]
- 12.Newberry KJ, Brennan RG. The structural mechanism for transcription activation by MerR family member multidrug transporter activation, N terminus. J Biol Chem. 2004;279:20356–20362. doi: 10.1074/jbc.M400960200. [DOI] [PubMed] [Google Scholar]
- 13.Newberry KJ, et al. Structures of BmrR-drug complexes reveal a rigid multidrug binding pocket and transcription activation through Tyrosine expulsion. J Biol Chem. 2008;283:26795–26804. doi: 10.1074/jbc.M804191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wade H. MD recognition by MDR gene regulators. Curr Opin Struct Biol. 2010;20:489–496. doi: 10.1016/j.sbi.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 15.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 16.Dodson EJ, Winn M, Ralph A. Collaborative Computational Project, number 4: Providing programs for protein crystallography. Methods Enzymol. 1997;277:620–633. doi: 10.1016/s0076-6879(97)77034-4. [DOI] [PubMed] [Google Scholar]
- 17.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 19.Hugot M, et al. A structural basis for the activity of retro-Diels-Alder catalytic antibodies: Evidence for a catalytic aromatic residue. Proc Natl Acad Sci USA. 2002;99:9674–9678. doi: 10.1073/pnas.142286599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Li H, Yang F, Smith-Gill SJ, Mariuzza RA. X-ray snapshots of the maturation of an antibody response to a protein antigen. Nat Struct Biol. 2003;10:482–488. doi: 10.1038/nsb930. [DOI] [PubMed] [Google Scholar]
- 21.Lewinson O, Adler J, Sigal N, Bibi E. Promiscuity in multidrug recognition and transport: The bacterial MFS Mdr transporters. Mol Microbiol. 2006;61:277–284. doi: 10.1111/j.1365-2958.2006.05254.x. [DOI] [PubMed] [Google Scholar]
- 22.Peters KM, Sharbeen G, Theis T, Skurray RA, Brown MH. Biochemical characterization of the multidrug regulator QacR distinguishes residues that are crucial to multidrug binding and induction of qacA transcription. Biochemistry. 2009;48:9794–9800. doi: 10.1021/bi901102h. [DOI] [PubMed] [Google Scholar]
- 23.Schumacher MA, Miller MC, Brennan RG. Structural mechanism of the simultaneous binding of two drugs to a multidrug-binding protein. EMBO J. 2004;23:2923–2930. doi: 10.1038/sj.emboj.7600288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Padlan EA. On the nature of antibody combining sites: Unusual structural features that may confer on these sites an enhanced capacity for binding ligands. Proteins. 1990;7:112–124. doi: 10.1002/prot.340070203. [DOI] [PubMed] [Google Scholar]
- 25.Mariuzza RA, Phillips SE, Poljak RJ. The structural basis of antigen-antibody recognition. Annu Rev Biophys Biophys Chem. 1987;16:139–159. doi: 10.1146/annurev.bb.16.060187.001035. [DOI] [PubMed] [Google Scholar]
- 26.Bianchet MA, et al. The three-dimensional structure of bovine odorant binding protein and its mechanism of odor recognition. Nat Struct Biol. 1996;3:934–939. doi: 10.1038/nsb1196-934. [DOI] [PubMed] [Google Scholar]
- 27.Fellouse FA, et al. Molecular recognition by a binary code. J Mol Biol. 2005;348:1153–1162. doi: 10.1016/j.jmb.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 28.Birtalan S, Fisher RD, Sidhu SS. The functional capacity of the natural amino acids for molecular recognition. Mol Biosyst. 2010;6:1186–1194. doi: 10.1039/b927393j. [DOI] [PubMed] [Google Scholar]
- 29.Chothia C. Nature of accessible and buried surfaces in proteins. J Mol Biol. 1976;105:1–14. doi: 10.1016/0022-2836(76)90191-1. [DOI] [PubMed] [Google Scholar]
- 30.Sharp KA, Nicholls A, Fine RF, Honig B. Reconciling the magnitude of the microscopic and macroscopic hydrophobic effects. Science. 1991;252:106–109. doi: 10.1126/science.2011744. [DOI] [PubMed] [Google Scholar]
- 31.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 32.Kuntz ID, Chen K, Sharp KA, Kollman PA. The maximal affinity of ligands. Proc Natl Acad Sci USA. 1999;96:9997–10002. doi: 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abad-Zapatero C. Ligand efficiency indices for effective drug discovery. Expert Opin Drug Discov. 2007;2:469–488. doi: 10.1517/17460441.2.4.469. [DOI] [PubMed] [Google Scholar]
- 34.Barlow DJ, Thornton JM. Ion-pairs in proteins. J Mol Biol. 1983;168:867–885. doi: 10.1016/s0022-2836(83)80079-5. [DOI] [PubMed] [Google Scholar]
- 35.Sharp KA, Honig B. Electrostatic interactions in macromolecules: Theory and applications. Annu Rev Biophys Biophys Chem. 1990;19:301–332. doi: 10.1146/annurev.bb.19.060190.001505. [DOI] [PubMed] [Google Scholar]
- 36.Mariuzza RA. Multiple paths to multispecificity. Immunity. 2006;24:359–361. doi: 10.1016/j.immuni.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 37.Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15:327–332. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- 38.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 39.Bevington P, Robinson DK. Data Reduction and Error Analysis for the Physical Sciences. New York: McGraw-Hill; 1992. p. 328. [Google Scholar]
- 40.DeLano WL. The Pymol Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 41.Binkowski TA, Naghibzadeh S, Liang J. CASTp: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2003;31:3352–3355. doi: 10.1093/nar/gkg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 43.Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: Structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.