

Abstract
We report a bacterial system for the evolution of cyclic peptides that makes use of an expanded set of amino acid building blocks. Orthogonal aminoacyl-tRNA synthetase/tRNACUA pairs, together with a split intein system were used to biosynthesize a library of ribosomal peptides containing amino acids with unique structures and reactivities. This peptide library was subsequently used to evolve an inhibitor of HIV protease using a selection based on cellular viability. Two of three cyclic peptides isolated after two rounds of selection contained the keto amino acid p-benzoylphenylalanine (pBzF). The most potent peptide (G12: GIXVSL; X = pBzF) inhibited HIV protease through the formation of a covalent Schiff base adduct of the pBzF residue with the ϵ-amino group of Lys 14 on the protease. This result suggests that an expanded genetic code can confer an evolutionary advantage in response to selective pressure. Moreover, the combination of natural evolutionary processes with chemically biased building blocks provides another strategy for the generation of biologically active peptides using microbial systems.
Keywords: unnatural amino acid, protein translation, bacterial selection
Bacteria have evolved a wide array of biologically active peptides which consist of the canonical 20 amino acids, as well as nonproteogenic amino acids not typically found in proteins (1–4). These linear and cyclic peptides are biosynthesized either on the ribosome with subsequent enzymatic processing (5, 6) or by the nonribosomal peptide synthetases (1, 7). Considerable effort has focused on reprogramming this natural biosynthetic machinery to generate diverse libraries of structurally complex peptides that can be subsequently selected for a desired biological activity. Although various synthetic and in vitro methods have also been developed to generate large peptide libraries from natural and unnatural amino acids and enrich them using affinity-based methods (e.g., phage and ribosomal display, DNA directed synthesis, and encoded synthetic peptide libraries) (8–13), to date the synthesis and bacterial selection of cyclic peptides containing unnatural amino acid (UAA) building blocks has remained elusive.
The increased proteolytic stability and enhanced binding affinity of conformationally constrained peptides make them attractive biosynthetic targets (14, 15). Toward this end, a method to generate libraries of cyclic peptides that employs circularly permuted inteins which self-catalyze the cyclization of a target peptide has been developed in Escherichia coli (16–22). This method, termed split intein catalyzed ligation of proteins and peptides (SICLOPPS), has been previously used to select inhibitors of hydrolases, methyl transferases, and other proteins (17, 20, 22–24). However, one major disadvantage of this system relative to nonribosomal peptide synthesis is the limited repertoire of functional groups that can be accessed. For example, chemical warheads such as ketoamides, boronates, hydroxamates, and Michael acceptors (which bind covalently to active site residues in proteases, acyl transferases, kinases, etc.), or N-methyl and β-amino acids (which can be used to block proteolysis or restrict conformation) are absent from the canonical 20 amino acids. Toward this end, we report an approach of a template-directed biosynthesis of large libraries of cyclic peptides containing UAAs in bacteria and the use of this library to evolve inhibitors of HIV protease in E. coli.
Results and Discussion
Bacterial Incorporation of Unnatural Amino Acids into Cyclic Peptides.
To test the feasibility of incorporating UAAs into cyclic peptides using the SICLOPPS system, peptides encoding NGXLFC, QRXFVK, and DXSLKR (X represents the nonsense codon, TAG) were inserted between two inteins and cotransformed with the plasmid pEVOL, which encodes p-acetylphenylalanine (pEVOL-pAcF; Fig. 1A). Peptides were expressed under previously described conditions (0.2% arabinose induction, 37 °C, 6 h) in the presence of p-acetylphenylalanine (1 mM). Analysis of the expression cultures by SDS-PAGE and mass spectrometry of butanolic extracts confirmed incorporation of the unnatural amino acid into each peptide (Fig. 1 B and C). Two of the peptides underwent 50–60% intein-mediated cyclization (see SI Appendix), consistent with the efficiencies reported in the literature (21). Yields of the three cyclic peptides and their corresponding inteins were 1.4–10 mg/L (approximately 70% suppression compared to comparable wild-type constructs). In general, we were able to achieve suppression efficiencies of 25–75% (relative to wild-type peptide expression) for many UAAs with a significant fraction of the peptides processing to cyclic structures.
Fig. 1.

Expression of cyclic peptides possessing unnatural amino acids and the bacterial selection of an HIV protease inhibitor. (A) General selection components. Competent cells harboring the pEVOL plasmid (which encodes the tRNA and aminoacyl-tRNA synthetase) and the selection plasmid (encoding both a modified tetracycline antiporter and HIV protease) are transformed with the cyclic peptide library vector. (B) SDS-PAGE demonstrating the expression and processing of the full intein construct (INCycIc) to yield the cyclic peptide and the processed IN fragment. Lanes: 1, D[X]SLKR; 2, NG[X]LFC; and 3, QR[X]FVK. (C) MS analysis of cyclic peptide D[X]SLKR obtained from chitin resin purification (see SI Appendix) with the expected mass at 1,013.0.
Selection for HIV Protease Inhibitors.
We next utilized a bacterial selection to identify cyclic peptides that inhibit a protease of interest. Several such schemes have been described in the literature for protease inhibitors (25–35) in which the viability of E. coli is linked to protease activity (32, 33). For example, Block and Grafstrom (32) introduced an HIV protease recognition sequence into the tetracycline resistance gene (Tn10), a metal-tetracycline/H+ antiporter that contains two functional, homologous integral membrane domains (TetA and TetB) linked by a central cytosolic hinge (36). Insertion of the HIV protease cleavage site (sequence SQNYPIV; ref. 25) between residues 189 and 190 of the flexible hinge region [Tet(HIV)] had little effect on antiporter function; however, when Tet(HIV) is coexpressed with HIV protease, resistance to tetracycline was abolished (31, 32). To adapt this selection to our peptide libraries, we modified a pRSF plasmid to express Tet(HIV) under a constitutive GlnS promoter and HIV protease under an IPTG inducible T7 promoter (Fig. 1A). A control vector was constructed that expresses a cyclic SQNYPIV sequence with inhibitory activity in the SICLOPPS system. This plasmid was cotransformed into E. coli expressing the pRSF-HIV-Tet(HIV) and pEVOL-pAcF plasmids, and cultures were grown with 7 μg/mL tetracycline in the presence and absence of 0.02% arabinose (to induce expression of the HIV protease inhibitor peptide). Only cultures harboring the protease inhibitor grew to saturation. Furthermore, growth was arabinose dependent, indicating that tetracycline resistance resulted from inhibition of the protease by the cyclic peptide (see SI Appendix, Fig. S5).
Next we encoded a library of diverse cyclic hexapeptides containing unnatural amino acids using the SICLOPPS expression system (NNK, where N is A, T, C, or G; and K is G or T, DNA diversity = 1.1 × 109). This design reduces degeneracy, but includes the amber codon, TAG, which is used to encode an unnatural amino acid with a theoretical frequency of approximately 17% per peptide. The enrichment of clones harboring the TAG codon during a selection should indicate that an unnatural amino acid confers a selective advantage. This library was cotransformed into E. coli BL21(DE3) along with the pEVOL 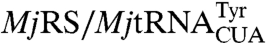 expression system which encodes an evolved orthogonal aminoacyl-tRNA synthetase (aaRS) and a cognate amber suppressor tRNACUA (37) that incorporates a desired unnatural amino acid site-specifically in response to the amber codon (38). A selection was then carried out for cyclic peptides that inhibit HIV protease with p-benzoylphenylalanine (pBzF) as a 21st amino acid. This amino acid has a relatively large hydrophobic surface area and an electrophilic aryl ketone moiety which may form a covalent Schiff base adduct with nucleophilic lysines of the HIV protease target. The pEVOL-pBzF (which encodes pBzF), and pRSF-HIV-Tet were cotransformed into BL21(DE3) E. coli; following transformation of the plasmid encoding the cyclic peptide library, cells were plated on LB agar containing appropriate antibiotics, IPTG (to induce protease expression), arabinose (to induce aaRS and cyclic peptide expression), pBzF (0.5 mM), and tetracycline (7 μg/mL). After 48 h, approximately 500 colonies were present on the plate; of these, 250 were selected, grown in liquid culture, and replica plated with and without arabinose and pBzF. After another round of selection, the surviving colonies were grown and sequenced. Of the 12 clones isolated, eight contained a TAG codon, six encoded GIXVSL (designated hit G12, where X corresponds to TAG), two encoded ILLGYN (hit B7), and one encoded VVIPXI (hit C2) (Fig. 2A and Table 1). Two clones (C2 and G12) required arabinose and pBzF for tetracycline resistance, whereas one clone (B7) only required arabinose (Fig. 2B). These clones were expressed and their sequences were confirmed by MS analysis following butanol extraction (see SI Appendix, Fig. S7).
expression system which encodes an evolved orthogonal aminoacyl-tRNA synthetase (aaRS) and a cognate amber suppressor tRNACUA (37) that incorporates a desired unnatural amino acid site-specifically in response to the amber codon (38). A selection was then carried out for cyclic peptides that inhibit HIV protease with p-benzoylphenylalanine (pBzF) as a 21st amino acid. This amino acid has a relatively large hydrophobic surface area and an electrophilic aryl ketone moiety which may form a covalent Schiff base adduct with nucleophilic lysines of the HIV protease target. The pEVOL-pBzF (which encodes pBzF), and pRSF-HIV-Tet were cotransformed into BL21(DE3) E. coli; following transformation of the plasmid encoding the cyclic peptide library, cells were plated on LB agar containing appropriate antibiotics, IPTG (to induce protease expression), arabinose (to induce aaRS and cyclic peptide expression), pBzF (0.5 mM), and tetracycline (7 μg/mL). After 48 h, approximately 500 colonies were present on the plate; of these, 250 were selected, grown in liquid culture, and replica plated with and without arabinose and pBzF. After another round of selection, the surviving colonies were grown and sequenced. Of the 12 clones isolated, eight contained a TAG codon, six encoded GIXVSL (designated hit G12, where X corresponds to TAG), two encoded ILLGYN (hit B7), and one encoded VVIPXI (hit C2) (Fig. 2A and Table 1). Two clones (C2 and G12) required arabinose and pBzF for tetracycline resistance, whereas one clone (B7) only required arabinose (Fig. 2B). These clones were expressed and their sequences were confirmed by MS analysis following butanol extraction (see SI Appendix, Fig. S7).
Fig. 2.

Cyclic peptides HIV inhibitors isolated from the viability selection. (A) Structures of the three protease inhibitors (G12, C2, and B7). (B) Phenotype of the cyclic peptide HIV protease inhibitors. Plasmids containing the HIV protease inhibitors were isolated and transformed into cells possessing a pEVOL plasmid for the incorporation of an unnatural amino acid (either pEVOL-pBzF or pEVOL-OMeY) and a protease selection plasmid (either HIV or HCV). Liquid cultures were then grown and normalized to an OD600 of 0.1 and replica spotted on selection plates containing IPTG (0.001 mM) and in the presence and absence of arabinose (0.02%) and unnatural amino acid (0.5 mM). After 48 h of growth, the plates were analyzed for colony viability under the described conditions.
Table 1.
Isolated cyclic peptide inhibitors
| Isolate | Nucleotide sequence | Amino acid sequence |
| G12 | GGGATTTAGGTGTCTTTG | G I X V S L |
| D3 | GGGATTTAGGTGTCTTTG | G I X V S L |
| D7 | GGGATTTAGGTGTCTTTG | G I X V S L |
| F2 | GGGATTTAGGTGTCTTTG | G I X V S L |
| B4 | GGGATTTAGGTGTCTTTG | G I X V S L |
| E2 | GGGATTTAGGTGTCTTTG | G I X V S L |
| B7 | ATTTTGCTTGGGTATAAT | I L L G Y N |
| A6 | ATTTTGCTTGGGTATAAT | I L L G Y N |
| C2 | GTGGTTATTCCGTAGATT | V V I P X I |
| D5 | CGGATTGGTATGGCGTGT | R I G M A C |
| F5 | CGATTAAGGTGTCAATGT | R L R C Q C |
| E7 | TATGCTTAGTTTAGTACG | Y A X F S T |
Hit Validation.
To confirm that these clones were selected based on HIV protease inhibition and not an off target effect, HIV protease and its recognition sequence in Tet(HIV) were replaced with hepatitis C (HCV) NS3 protease and an appropriate protease susceptible sequence, respectively [GEAGDDIVPCSMSYTWT; pRSF-HCV/Tet(HCV)]. Hits C2, G12, and B7 were then tested for their ability to confer Tet resistance in this system. None of the peptides conferred resistance to tetracycline when cotransformed with the pEVOL-pBzF plasmid (Fig. 2B). To determine whether pBzF is required in clones C2 and G12, pEVOL-pBzF was replaced with pEVOL-OMeY, which selectively incorporates O-methyltyrosine (OMeY) in response to the TAG codon. Incorporation of OMeY into the cyclic peptide clones C2 and G12 failed to confer tetracycline resistance in the HIV protease selection, whereas tetracycline resistance with the B7 hit was not altered in the presence of the pEVOL-OMeY plasmid.
To determine the IC50 values of the selected cyclic peptides, we chemically synthesized the peptides C2 and G12 in both linear and cyclic forms (CPC Scientific). When analyzed with a standard protease inhibition assay, each peptide inhibited HIV protease activity in the low micromolar range (Fig. 3A); and in both cases, the cyclic form of the peptide inhibited protease activity more potently than its linear analog. The largest difference was observed for peptide G12 with an order of magnitude greater inhibition for the cyclic form (IC50 of 6.67 and 0.96 μM for the linear and cyclic analogs, respectively).
Fig. 3.

Characterization of cyclic peptide HIV protease inhibitors. (A) Inhibition data obtained for the cyclic and linear analogs of GIXVSL (G12), ILLGYN (B7), and VVIPXI (C2). (B) LC/MS deconvoluted mass spectra of either wild-type or K14R mutant HIV protease (0.5 μg) in the presence or absence of cyclic peptide G12 (10 μM) with NaCNBH3 (5 mM) for 16 h at 37 °C. The observed mass (11,776) increases by the mass of G12 only in the case of wild-type protease. The K14R mutant does not appear to form the covalent adduct because only the mass of the protease is detected (10,755 Da). (C) LC/MS trace of tryptic digest for samples containing both HIV protease and cyclic peptide G12, in the presence and absence of NaCNBH3. A new UV absorbance (λmax = 280 nm) is present only when the sample is subjected to NaCNBH3 (5 mM), which corresponds to a tryptic peptide fragment of the HIV protease/cyclic peptide adduct. (D) 8-anilinonapthalene-1-sulfonate analysis of various HIV protease complexes. HIV protease (1.2 μM) was treated either at pH 1.0 (unfolded), or folded in 100 mM NaOAc buffer (pH 5.4) with 1-anilinonaphthalene-8-sulfonic acid (0.8 mM) for 30 min in the presence or absence of cyclic peptide G12 (20 μM). Fluorescence spectra were then taken (λEx = 360 nm).
Cyclic Peptide Inhibitor Mode of Action.
To determine if the electrophilic ketone moiety of pBzF forms a covalent interaction with the protease, cyclic peptides G12 and C2 were incubated for 1 h at 37 °C with recombinantly expressed HIV protease, followed by treatment with NaCNBH3. Analysis of the reactions by mass spectrometry revealed that a covalent adduct is formed between the cyclic peptide and the protease only in the presence of the reductant (see SI Appendix, Fig. S8). In contrast, cyclic peptide B7 did not afford any detectable covalent adduct in the presence or absence of reductant. This result suggests that the electrophilic keto group in the cyclic peptide is a key binding determinant that selectively forms a Schiff base adduct with a nucleophilic residue of HIV protease.
Liquid chromatography (LC) analysis of a tryptic digest of the reduced protease-cyclic peptide revealed a shift in retention time for one of the tryptic fragments compared to an identical digest without reductant (Fig. 3C). Both the UV absorption and the mass of the new tryptic peptide from the HIV protease/G12 adduct correspond with that expected for a reduced Schiff base adduct of the cyclic peptide with the tryptic peptide RPLVTIK*IGGQLK (with the * indicating a modification). The interaction is specific, because none of the other HIV lysines appear to be modified by the cyclic peptide. Interestingly, Lys14 is not in the active site of the protease, the flap region, nor at the dimerization interface. To further investigate the importance of this residue, an HIV mutant was prepared with a K14R mutation. This conservative K14R mutation is found naturally and cannot form a Schiff base adduct. When incubated with G12 in the presence and absence of NaCNBH3, adduct formation was only observed in the HIV protease possessing a lysine at position 14, confirming the importance of this residue (Fig. 3B). Additionally, when analyzed with the protease activity assay, the G12 cyclic peptide failed to inactivate the K14R HIV mutant, whereas the B7 analog (not possessing an UAA) was still capable of inhibition. Thus, formation of a covalent adduct between G12 and HIV protease leads to a loss of protease activity. However, the mechanism of inactivation appears not to involve the active site or dimerization interface. Interestingly, 8-anilinonapthalene-1-sulfonate fluorescence was detected in the presence of 1.2 μM folded HIV protease and 20 μM cyclic peptide, as well as unfolded (pH 1) HIV protease alone (see Fig. 3D). No fluorescence was detected when either the properly folded HIV protease or the cyclic peptide was incubated alone. Thus, it appears that the cyclic peptide may be affecting protease stability by promoting either aggregation or unfolding through covalent association with the protein.
Conclusion
These results demonstrate that the template-directed ribosomal translational machinery can be used to biosynthesize libraries of cyclic peptides containing unnatural amino acids, and that these peptides can be subjected to bacterial selection to identify biologically active cyclic peptides. Interestingly, an unusual mechanism of inhibition resulting from incorporation of the UAA was discovered that involves formation of a covalent Schiff base adduct. We are currently attempting to crystallize the adduct to further characterize the unique interaction between the cyclic peptide and HIV protease, and to better understand the mechanism of inactivation. Nonetheless, this work illustrates that the combination of natural evolutionary processes with synthetic building blocks containing functional groups absent in the common 20 amino acids can provide a selective advantage in an unbiased system.
Methods
Expression of Cyclic Peptides Containing Unnatural Amino Acids.
Selected pBK-In library members containing a TAG mutation were isolated (NG[TAG]LFC, QR[TAG]FVK, and D[TAG]SLKR) and cotransformed with the pEVOL-pAcF plasmid into BL21(DE3) cells. Cultures were grown overnight in 2× YT media (Fisher Scientific) at 37 °C to saturation with chloramphenicol and kanamycin (100 μg/mL). Cultures (500 mL) were diluted to an OD600 of 0.2 and grown to an OD600 of 0.6, induced with 0.2% arabinose and 1 mM pAcF, and grown for 6 h at 37 °C. Cells were lysed and cyclic peptides were purified using Chitin beads according to published protocols (20). Cyclic peptide production was then assessed by both SDS-PAGE and LC/MS analysis (see Fig. 1B and C).
HIV Protease Cyclic Peptide Inhibitor Selection.
Electrocompetent BL21(DE3) cells (competency 3.5 × 108) containing pEVOL-pBzF and pRSF-HIV-Tet(HIV) were transformed with the cyclic peptide library (1.6 mg) and recovered in 2× YT (4 mL) containing pBzF (0.5 mM). The transformations were then plated over 4 LB agar plates (245 × 245 × 25 cm) containing chloramphenicol (20 μg/mL), ampicillin (50 μg/mL), kanamycin (50 μg/mL), IPTG (0.001 mM), arabinose (0.02%), pBzF (0.5 mM), and tetracycline (7 μg/mL) and incubated at 37 °C for 48 h. The colonies were then scraped, diluted, and replated on identical plates for an additional 48 h at 37 °C. Approximately 250 colonies were then individually selected and grown to saturation in 2× YT containing ampicillin, chloramphenicol, and kanamycin. The cultures were diluted to OD600 0.1 and stamped on LB agar plates previously described but in the presence and absence of arabinose (0.02%). After 48 h at 37 °C, 12 colonies that demonstrated differential growth were selected (see SI Appendix, Fig. S6) and grown in liquid culture. The plasmids were then isolated using a Qiagen kit, and the pEVOL-pBzF and pRSF-HIV-Tet(HIV) plasmids were removed by enzymatic digestion (NdeI, SalI, and EcoRI; New England Biolabs; 4 h, 37 °C). Digests were retransformed into DH10B cells and grown on kanamycin (100 μg/mL) plates. Colonies were then grown in liquid culture, and the pBK-In plasmid was isolated using a Qiagen kit and sequenced (see Table 1).
Three sequences were selected (G12, C2, and B7), and retransformed into electrocompetent BL21(DE3) cells harboring either the pEVOL-pBzF or pEVOL-pOMeY and the pRSF-HIV-Tet(HIV) or pRSF-HCV-Tet(HCV) plasmids. Colonies were then grown in liquid culture (2× YT; ampicillin, chlroramphenicol, kanamycin) to saturation, diluted to OD600 0.1, and replica stamped on LB agar plates containing chloramphenicol (20 μg/mL), ampicillin (50 μg/mL), kanamycin (50 μg/mL), IPTG (0.001 mM), tetracycline (7 μg/mL) in the presence or absence of arabinose (0.02%), and with pBzF (0.5 mM), pOMeF (1 mM), or no unnatural amino acid. Incubation at 37 °C for 48 h afforded the assessment of the selectivity of the cyclic peptide inhibitors and requisite of the pBzF unnatural amino acid (see Fig. 2B). To ensure cyclic peptides were produced, the cyclic peptides were expressed in liquid culture as previously described, and analyzed by SDS-PAGE and LC/MS (see SI Text, Fig. S7).
Cyclic Peptide/HIV Protease Interaction.
To ascertain the mechanism of cyclic peptide inhibition, cyclic peptides G12 and C2 (80 μM) were incubated with HIV protease (2.4 μM) for 30 min at 37 °C. The reaction was then divided into aliquots and NaCNBH3 (5 mM) was added to one sample for reduction, while the other sample was left unreduced. The reactions were incubated at 37 °C for 12 h, followed by buffer exchange and analysis by LC/MS on an Agilent 1100 Series LC/MS. The chromatographic peak corresponding to HIV protease (between 6.1 and 6.5 min) was charge deconvoluted using Agilent LC/MS ChemStation software. Deconvolution parameters were set to high Mr = 20,000 and low Mr = 40,000, maximum charge = 50, and minimum peaks in set = 3–8. Error was ± 0.02%, as determined from control samples.
A similar reduction was performed (50 μL) except also in the presence and absence of cyclic peptide; however, this reduction was followed by tryptic digest as urea was added to the reaction [12 M in 25 mM (NH4)HCO3, 50 μL], followed by DTT (200 mM, 5 μL), and incubated at room temperature for 1 h. Additional ammonium bicarbonate buffer was added (900 μL) followed by trypsin (0.2 μg/mL, 5 μL) and the reaction was incubated at 37 °C overnight, then concentrated to near dryness. The excess urea and salts were removed via ZipTip exchange and the tryptic digest was analyzed by LC/MS. Differences in the mass spectrum between the cyclic peptide sample and the control (as well as the different NaCNBH3 treatments) were identified and the masses were correlated to appropriate tryptic fragments.
Supplementary Material
Acknowledgments.
The authors would like to thank Xuemei Han and the Yates Lab for aid in MS characterization, as well as Virginia Seely and Emily Remba for manuscript preparation and submission. This work was funded by Grant 5R01GM062159 from the National Institutes of Health (NIH) (to P.G.S.) and supported in part by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, NIH and the Intramural AIDS-Targeted Program of the Office of the Director, NIH. This is manuscript 21197 of The Scripps Research Institute. D.D.Y. is grateful for an NIH Fellowship F32CA144213 and T.S.Y. is grateful for an Achievement Rewards for College Scientists Scholarship.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1108045108/-/DCSupplemental.
References
- 1.Cane DE, Walsh CT, Khosla C. Biochemistry—harnessing the biosynthetic code: Combinations, permutations, and mutations. Science. 1998;282:63–68. doi: 10.1126/science.282.5386.63. [DOI] [PubMed] [Google Scholar]
- 2.Hancock REW, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 3.Sahl HG, Bierbaum G. Lantibiotics: Biosynthesis and biological activities of uniquely modified peptides from gram-positive bacteria. Annu Rev Microbiol. 1998;52:41–79. doi: 10.1146/annurev.micro.52.1.41. [DOI] [PubMed] [Google Scholar]
- 4.Clardy J, Walsh C. Lessons from natural molecules. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 5.Bayer A, Freund S, Nicholson G, Jung G. Posttranslational backbone modifications in the ribosomal biosynthesis of the glycine-rich antibiotic microcin-B17. Angew Chem Int Ed Engl. 1993;32:1336–1339. [Google Scholar]
- 6.Hansen JN. Antibiotics synthesized by posttranslational modification. Annu Rev Microbiol. 1993;47:535–564. doi: 10.1146/annurev.mi.47.100193.002535. [DOI] [PubMed] [Google Scholar]
- 7.Clardy J, Fischbach MA, Walsh CT. New antibiotics from bacterial natural products. Nat Biotechnol. 2006;24:1541–1550. doi: 10.1038/nbt1266. [DOI] [PubMed] [Google Scholar]
- 8.Koglin A, Walsh CT. Structural insights into nonribosomal peptide enzymatic assembly lines. Nat Prod Rep. 2009;26:987–1000. doi: 10.1039/b904543k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nolan EM, Walsh CT. How nature morphs peptide scaffolds into antibiotics. Chembiochem. 2009;10:34–53. doi: 10.1002/cbic.200800438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sandman KE, Benner JS, Noren CJ. Phage display of selenopeptides. J Am Chem Soc. 2000;122:906–961. [Google Scholar]
- 11.Sattely ES, Fischbach MA, Walsh CT. Total biosynthesis: In vitro reconstitution of polyketide and nonribosomal peptide pathways. Nat Prod Rep. 2008;25:757–793. doi: 10.1039/b801747f. [DOI] [PubMed] [Google Scholar]
- 12.Li SW, Millward S, Roberts R. In vitro selection of mRNA display libraries containing an unnatural amino acid. J Am Chem Soc. 2002;124:9972–9973. doi: 10.1021/ja026789q. [DOI] [PubMed] [Google Scholar]
- 13.Needels MC, et al. Generation and screening of an oligonucleotide-encoded synthetic peptide library. Proc Natl Acad Sci USA. 1993;90:10700–10704. doi: 10.1073/pnas.90.22.10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katsara M, et al. Round and round we go: Cyclic peptides in disease. Curr Med Chem. 2006;13:2221–2232. doi: 10.2174/092986706777935113. [DOI] [PubMed] [Google Scholar]
- 15.Clardy J, Walsh C. Lessons from natural molecules. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 16.Horswill AR, Benkovic SJ. Identifying small-molecule modulators of protein-protein interactions. Curr Protoc Protein Sci. 2006;46:19.15.1–19.15.19. doi: 10.1002/0471140864.ps1915s46. [DOI] [PubMed] [Google Scholar]
- 17.Naumann TA, Tavassoli A, Benkovic SJ. Genetic selection of cyclic peptide Dam methyltransferase inhibitors. Chembiochem. 2008;9:194–197. doi: 10.1002/cbic.200700561. [DOI] [PubMed] [Google Scholar]
- 18.Naumann TA, Savinov SN, Benkovic SJ. Engineering an affinity tag for genetically encoded cyclic peptides. Biotechnol Bioeng. 2005;92:820–830. doi: 10.1002/bit.20644. [DOI] [PubMed] [Google Scholar]
- 19.Horswill AR, Benkovic SJ. Cyclic peptides, a chemical genetics tool for biologists. Cell Cycle. 2005;4:552–555. doi: 10.4161/cc.4.4.1585. [DOI] [PubMed] [Google Scholar]
- 20.Abel-Santos E, Scott CP, Benkovic SJ. Use of inteins for the in vivo production of stable cyclic peptide libraries in E. coli. Methods Mol Biol. 2003;205:281–294. doi: 10.1385/1-59259-301-1:281. [DOI] [PubMed] [Google Scholar]
- 21.Scott CP, Abel-Santos E, Wall M, Wahnon DC, Benkovic SJ. Production of cyclic peptides and proteins in vivo. Proc Natl Acad Sci USA. 1999;96:13638–13643. doi: 10.1073/pnas.96.24.13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tavassoli A, et al. Inhibition of HIV budding by a genetically selected cyclic peptide targeting the Gag-TSG101 interaction. ACS Chem Biol. 2008;3:757–764. doi: 10.1021/cb800193n. [DOI] [PubMed] [Google Scholar]
- 23.Cheng L, et al. Discovery of antibacterial cyclic peptides that inhibit the ClpXP protease. Protein Sci. 2007;16:1535–1542. doi: 10.1110/ps.072933007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tavassoli A, Benkovic SJ. Genetically selected cyclic-peptide inhibitors of AICAR transformylase homodimerization. Angew Chem Int Ed Engl. 2005;44:2760–2763. doi: 10.1002/anie.200500417. [DOI] [PubMed] [Google Scholar]
- 25.Hellen CU. Assay methods for retroviral proteases. Methods Enzymol. 1994;241:46–58. doi: 10.1016/0076-6879(94)41058-5. [DOI] [PubMed] [Google Scholar]
- 26.Zhu W, Williams RS, Kodadek T. A CDC6 protein-binding peptide selected using a bacterial two-hybrid-like system is a cell cycle inhibitor. J Biol Chem. 2000;275:32098–32105. doi: 10.1074/jbc.M001560200. [DOI] [PubMed] [Google Scholar]
- 27.Cloutier SM, et al. Development of recombinant inhibitors specific to human kallikrein 2 using phage-display selected substrates. Eur J Biochem. 2004;271:607–613. doi: 10.1111/j.1432-1033.2003.03963.x. [DOI] [PubMed] [Google Scholar]
- 28.Felber LM, et al. Evaluation of the CFP-substrate-YFP system for protease studies: advantages and limitations. Biotechniques. 2004;36:878–885. doi: 10.2144/04365PT04. [DOI] [PubMed] [Google Scholar]
- 29.Neefjes J, Dantuma NP. Fluorescent probes for proteolysis: Tools for drug discovery. Nat Rev Drug Discov. 2004;3:58–69. doi: 10.1038/nrd1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olsen MJ, et al. Function-based isolation of novel enzymes from a large library. Nat Biotechnol. 2000;18:1071–1074. doi: 10.1038/80267. [DOI] [PubMed] [Google Scholar]
- 31.McCall JO, Kadam S, Katz L. A high capacity microbial screen for inhibitors of human rhinovirus protease 3C. Nat Biotechnol. 1994;12:1012–1016. doi: 10.1038/nbt1094-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Block TM, Grafstrom RH. Novel bacteriological assay for detection of potential antiviral agents. Antimicrob Agents Chemother. 1990;34:2337–2341. doi: 10.1128/aac.34.12.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balint RF, Plooy I. Protease-dependent streptomycin sensitivity in Escherichia-coli—a system for protease inhibitor selection. Nat Biotechnol. 1995;13:507–510. doi: 10.1038/nbt0595-507. [DOI] [PubMed] [Google Scholar]
- 34.Dautin N, Karimova G, Ullmann A, Ladant D. Sensitive genetic screen for protease activity based on a cyclic AMP signaling cascade in Escherichia coli. J Bacteriol. 2000;182:7060–7066. doi: 10.1128/jb.182.24.7060-7066.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hwang YC, Chen W, Yates MV. Use of fluorescence resonance energy transfer for rapid detection of enteroviral infection in vivo. Appl Environ Microbiol. 2006;72:3710–3715. doi: 10.1128/AEM.72.5.3710-3715.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimura T, Yamaguchi A. Asp-285 of the metal-tetracycline/H+ antiporter of Escherichia coli is essential for substrate binding. FEBS Lett. 1996;388:50–52. doi: 10.1016/0014-5793(96)00514-5. [DOI] [PubMed] [Google Scholar]
- 37.Young TS, Ahmad I, Yin JA, Schultz PG. An enhanced system for unnatural amino acid mutagenesis in E. coli. J Mol Biol. 2009;395:361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 38.Liu CC, Schultz PG. Adding new chemistries to the genetic code. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.