

Abstract
Purpose
The goal of this study was to comprehensively identify CpG island methylation alterations between pancreatic cancers and normal pancreata and their associated gene expression alterations.
Experimental Design
We employed Methylated CpG island Amplification followed by CpG island Microarray, a method previously validated for its accuracy and reproducibility, to analyze the methylation profile of 27800 CpG islands covering 21MB of the human genome in nine pairs of pancreatic cancer versus normal pancreatic epithelial tissues as well as in three matched pairs of pancreatic cancer versus lymphoid tissues from the same individual.
Results
This analysis identified 1658 known loci that were commonly differentially methylated in pancreatic cancer compared to normal pancreas. By integrating the pancreatic DNA methylation status with the gene expression profiles of the same samples before and after treatment with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine, and the Histone Deacetylase inhibitor, Trichostatin A, we identified dozens of aberrantly methylated and differentially expressed genes in pancreatic cancers including a more comprehensive list of hypermethylated and silenced genes that have not been previously described as targets for aberrant methylation in cancer.
Conclusion
We expect that the identification of aberrantly hypermethylated and silenced genes will have diagnostic, prognostic and therapeutic applications.
INTRODUCTION
Pancreatic cancer is the fourth most common cause of cancer death in the United States and has the lowest survival rate for any solid cancer. This particularly poor outcome is due in large part to the late presentation of the disease in most patients. Identifying those at risk of developing pancreatic cancer (1, 2) and developing better diagnostic markers of pancreatic neoplasia (3, 4) could improve the early diagnosis of pancreatic cancer and its precursors (5) and allow more patients to undergo curative surgical resection. Previous studies have demonstrated that aberrant gene hyper- and hypo- methylation contributes to pancreatic cancer development and progression (6–11). Furthermore, aberrant methylation increases during neoplastic development among the precursor lesions known as PanINs and IPMNs (11, 12). For example, aberrantly hypermethylated genes have been identified in pancreatic cancer by comparing gene expression profiles of pancreatic cancer cells before and after DNA methylation inhibitor treatment (8) and by using promoter (13) and SNP arrays (13, 14). The detection of aberrantly methylated loci relative to normal tissues could improve the diagnosis of pancreatic cancer (3) and may also identify key regulatory genes and pathways that merit therapeutic targeting (15).
We evaluated the accuracy and reproducibility of the Methylated CpG island Amplification coupled with a genome-wide promoter microarray platform (MCAM) in a pilot study using the pancreatic cancer cell lines Panc-1 and MiaPaca2 (13, 16). In this study, we used MCAM to more comprehensively define the differential methylated genes between pancreatic cancer cells and normal pancreatic tissues. We then compared the methylation profile of our candidate genes with their global gene expression profile in pancreatic cancer cell lines and normal pancreatic epithelial ductal samples, including global gene expression profiles of cell lines before and after treatment with the DNA methyltransferase inhibitor, 5-Aza-dC and the histone deacetylase inhibitor, Trichostatin A. Our aim was to identify genes commonly differentially methylated in pancreatic cancer and to identify the subset of aberrantly methylated genes with aberrant gene expression that represent candidate genes undergoing functional disruption in pancreatic cancer.
MATERIALS AND METHODS
Cell lines and tissue samples
Pancreatic adenocarcinoma cell lines AsPC1, Capan2, MiaPaca2, BxPC3, Capan1, CFPAC1, HS766, Panc1 and Su8686 were cultured under recommended conditions. A32-1, A38-5, Panc215, Panc2.5, Panc2.8, Panc3.014, A2-1, A6L, Panc198, Panc486, and Panc8.13 pancreatic ductal adenocarcinoma cell lines were described previously (17). Immortalized HPDE cells derived from normal human pancreatic ductal epithelium were generously provided by Dr. Ming-Sound Tsao (University of Toronto, Canada). Stored frozen tissues (−80C) of normal lymphoid tissues (lymphocytes or spleen) were obtained from 3 of the patients from whom we had developed a pancreatic cancer cell line (A32-1, A38-5, Panc215).
The frozen primary pancreatic cancer tissues, normal pancreatic and spleen tissues were obtained from patients the time of their pancreatic resection at Johns Hopkins Hospital. Normal pancreatic duct epithelial cells were isolated using laser capture microdissection from the resected pancreata of three patients (mean age, 64 years; range, 59–72 years)who underwent pancreatic resection for benign conditions. Additional normal pancreatic tissues were obtained from patients with pancreatic adenocarcinomas or neuroendocrine neoplasms. DNA was isolated from xenografts of primary pancreatic cancers as previously described (8, 9). Specimens were collected and analyzed with the approval of the Johns Hopkins Committee for Clinical Investigation.
MCA procedure and Agilent CpG island microarrays
MCA was performed as described previously (13). The Human CpG Island ChIP-on-Chip Microarray 244K chip interrogates 27,800 CpG islands covering 21MB with an average of 8 probes per island. Array hybridization was performed by the Johns Hopkins SKCCC Microarray Core Facility. Briefly, 2μg of MCA amplicon were labeled with either Cy3-dUTP or Cy5-dUTP (Perkin Elmer) using the BioPrime DNA Labeling System (Invitrogen). These dye-labeled amplicons were then mixed and co-hybridized to 244K CpG island microarrays, washed, dried and scanned using the Agilent G2505B scanner.
Methylation Specific PCR
Bisulfite-treatment and DNA amplification were performed as described previously (13). Primer sequences are available in Supplemental Table 5.
Affymetrix Exon Arrays
Cells were treated with 1μmol/L of 5-aza-2′-deoxycytidine (5-Aza-dC; Sigma, St. Louis, MO) for 4 days and 1μmol/L of the HDAC inhibitor Trichostatin A (TSA; Sigma) either alone or in combination for 24 hours as previously described (8, 9). Total RNA from cell lines was extracted using mirVana miRNA Isolation Kit (Ambion, Austin, TX) per manufacturer’s protocol before DNA-ase treatment (Ambion).
The Affymetrix Exon Array ST1.0 (Affymetrix, Santa Clara, CA) was used to analyze gene expression profiles in untreated and 5-Aza-dC or TSA-treated cell lines as previously described (18, 19).
Data analysis
For MCAM, data were extracted with Agilent Feature Extraction 9.1 software. Methylation-specific sites were called with Agilent Genomic Workbench Standard Edition 5.0.14 software for methylation (CH3) analysis, which calculates the normalized log2-signal ratios (NLR) and combined Z-scores for each probe. For Affymetrix Exon Array ST1.0, statistical analysis of gene expression array data was completed with Partek Genomic Suite 6.4 software. Raw Affymetrix intensity measurements of all probe sets were background corrected and normalized by the Robust Multichip Average method. Sample relationships were examined using principal component analysis to reveal any technical effects that would encumber the subsequent analysis. Gene expression intensities were summarized by the one-step Tukey’s biweight method. Two-way ANOVA analysis was performed to identify significant expression changes between pancreatic cancer and non cancer samples and between 5-Aza-dC– and/or TSA-treated and untreated cells. Gene network and pathway analysis were done using Ingenuity Pathways Analysis (Ingenuity systems; http://www.ingenuity.com) software. We are in compliance with the Minimum Information about a Microarray Experiment (MIAME) guidelines and have submitted our microarray data set to the NCBI’s Gene Expression Omnibus. They are accessible through GEO Series accession number GSE21163 and GSExxx.
Immunohistochemistry
The expression of ZBTB16 protein was examined by immunohistochemical labeling of tissue microarrays with anti-ZBTB16 (Invitrogen 34–3700; 1:100) polyclonal antibody, counterstained with hematoxylin. Immunohistochemical labeling was scored based on intensity; 0 (negative), 1 (weak) or 2 (strong), and on the percentage of positive epithelial cells as 0 (<5%), 1 (6–25%), 2 (26–50%), 3 (51–75%) or 4 (>76%), respectively. A histo-score was generated as the product of intensity multiplied by area and mean histo-scores of pancreatic ductal adenocarcinoma and normal ductal epithelial cells were compared using Paired Student’s T-Test.
RESULTS AND DISCUSSION
Identification of loci differentially methylated between pancreatic cancer and normal pancreatic tissues
We used Agilent 244K CpG island microarrays to perform 9 MCAM experiments comparing pancreatic cancer cell lines to normal pancreas. The pancreatic cancer cell lines were compared to pooled laser-captured microdissected normal pancreatic ductal tissues (A32-1, A38-5, Panc215, Panc2.5, Panc2.8 and Panc3.014), to normal pancreatic tissue (MiaPaca2 and Capan2), or to the non-neoplastic pancreatic ductal line HPDE (AsPC1).
To identify differentially methylated probes we used the previously validated cutoff (13) of ~4-fold (normalized log2 signal ratios (NLR) ≥2 or ≤−2) differential methylation between pancreatic cancers and normal pancreata (Figure 1). This cut-off was established using the same MCAM strategy comparing the pancreatic cancer cell line Panc-1 to the epithelial line, HPDE and validating the methylation status of 31 genes using bisulfite sequencing and MSP. By this criterion, there were 41,606 probes, representing 10,012 loci with differential methylation in at least one of the nine MCAM experiments, a considerably more extensive list than we identified in our pilot MCA experiment using the 44K array (13). To identify from this list genes commonly methylated in pancreatic cancers, we then calculated the mean of differential methylation ratios of the nine cancer/normal pairs and the combined Z-scores for each of these 41,606 probes and filtered out probes for which the mean of the nine NLRs was ≥ −2 but ≤2 (Note Z scores reflects how far a probe log-ratio value is in relation to the Gaussian distributions of probes with similar melting temperatures on the Agilent array). This criterion allowed us to identify 4672 probes for which the mean combined Z-score was either ≥ −4.67 or ≤ 5.1. To refine our gene list, we then filtered out outlier probes for which the mean combined Z-scores was ≥ −5 but ≤ 5 which left 4,662 probes, corresponding to 1,658 known loci (Supplemental Table 1). Thus, the gene lists identified using the mean NLR of ≥ −2 but ≤ 2 and the mean combined Z-scores was ≥ −5 but ≤ 5 were almost identical. Among these 1,658 loci, 1,226 were hypermethylated (1,206 genes, Supplemental Table 2) and 423 were hypomethylated (379 genes, Supplemental Table 3) in the pancreatic cancer cell lines compared to normal pancreatic tissue.
Figure 1.

An overview of experimental strategy to evaluate differential DNA methylation in pancreatic cancer cells compared to normal pancreas. NP: Normal Pancreas.
We found that 60% of hypermethylated probes were located inside known gene bodies, while 31.3% were located in their promoters, and 8.42% were located downstream of known genes (Figure 2B, left panel). These proportions were very similar among the hypermethylated genes whose expression was downregulated (Figure 2B, right panel) as well as to the overall distribution of probes on the Agilent chip (Figure 2A). We also found a very similar distribution of hypomethylated probes (Figure 2C, left panel). However, when we included only hypomethylated probes that were associated with upregulated genes, we found that 48.5% of probes were located in the promoter region (Figure 2C, right panel). This analysis is consistent with the observation that hypermethylation within the gene body as well as the promoter can result in downregulation, whereas gene upregulation is associated promoter hypomethylation but not gene body hypomethylation.
Figure 2.

Distribution of Agilent Human CpG island 244K probes covering known genes A, global distribution. B, Distribution of hypermethylated probes. C, Distribution of hypomethylated probes.
Since cancer-associated CpG island hypermethylation tends to extensively involve the affected CpG island we used MSP to confirm differential CpG island methylation identified by MCAM. Specifically, to validate our MCAM results we used TMSP to examine the methylation status of 24 aberrantly hypermethylated genes in 69 pancreatic cancers (43 xenografts of primary pancreatic cancers, 15 primary pancreatic cancer tissues, and 11 cell lines (i.e. BxPC3, Capan1, CFPAC1, HS766, Panc1, Su8686, A2-1, A6L, Panc198, Panc486, and Panc8.13), and 16 normal pancreatic tissue. We confirmed the differential methylation of 21 of the 24 genes tested (Supplemental Table 4), demonstrating the robustness of our MCAM strategy.
Several genes were methylated in >70% of pancreatic cancers and 10% or less of normal pancreata by MSP including BMI1, ID4, CACNA1H, IRF5 and SMOC2.
Aberrantly methylated and differentially expressed genes
We have previously examined gene expression in a set of six pancreatic cancer cell lines (A32-1, A38-5, Panc215, Panc2.5, Panc2.8 and Panc3.014) and the pancreatic non-neoplastic line, HPDE, before and after epigenetic treatment (5-Aza-dC, TSA, and the combination) using Affymetrix ST1.0 Exon Arrays (18). Here, we merged this analysis with the results of our MCAM experiments and compared the baseline and post-treatment gene expression profiles of these six pancreatic cancer cell lines focusing on genes shown to be hypermethylated or hypomethylated in our MCAM experiments. We used the two-way ANOVA analysis to identify expression changes between cancer cells and the non-neoplastic line HPDE. We defined genes as differentially expressed based on a fold change criteria of ±2-fold. Among the hypermethylated genes, 96 genes were underexpressed in pancreatic cancer cells compared with HPDE cells and the expression of 675 were induced by 5-Aza-dC and/or TSA treatment either alone or in combination (p-value <0.05) (Supplemental Table 2). 77 genes were both underexpressed and responsive to epigenetic treatment (Table 1). If we employed a more stringent cut-off for underexpressed genes (>3-fold reduction in expression), 37 genes were both underexpressed and responsive to epigenetic treatment (Table 1, bold genes). Among the hypomethylated genes, 24 genes were overexpressed in the pancreatic cancer cell lines compared with non-neoplastic pancreatic duct HPDE cells (Fold Change ≥2) (Table 2) and 243 genes were significantly induced by epigenetic treatment but not necessarily differentially expressed (p-value <0.05 with a Fold-Change >2; Supplemental Table 3).
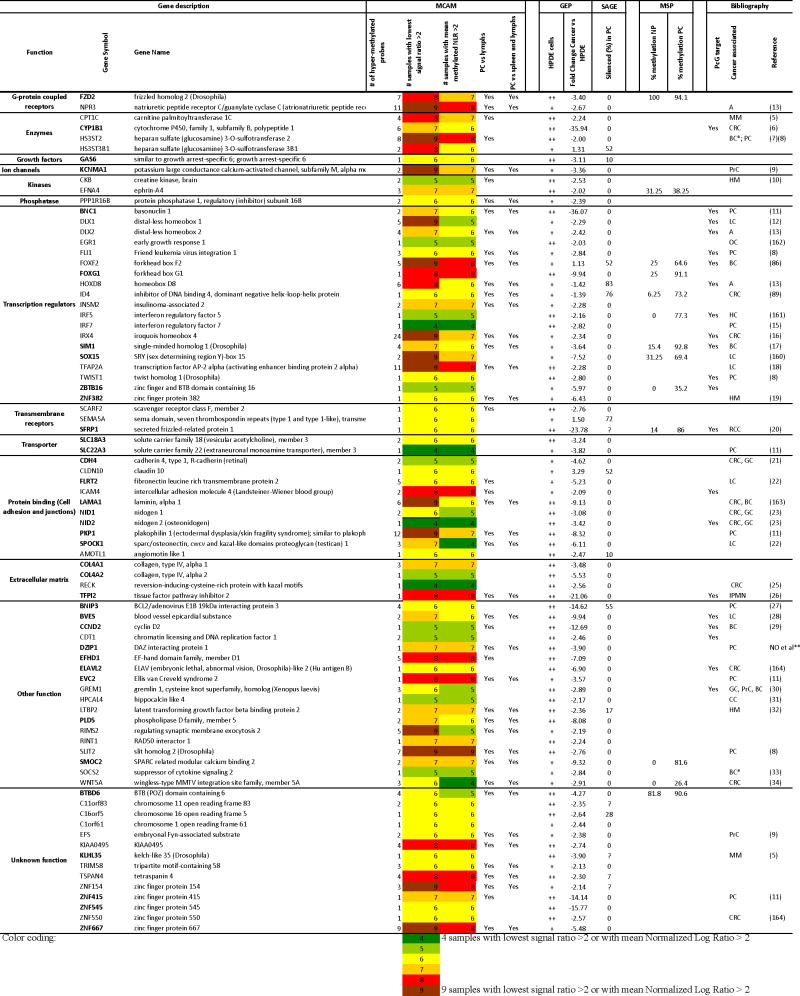
Table 1.
List of hypermethylated and downregulated candidate genes, responsive to epigenetic drug treatment.
|
MCAM: Methylated CpG island Amplification strategy coupled with Microarray platform. PC: Pancreatic Cancer; NP: Normal pancreas; Lymphs: lymphocytes.
GEP: Gene Expression Profiling with Affymetrix Exon Arrays. The HPDE column indicates the level of expression of the gene in normal cells: 0 virtually undetectable signal; +: weak expression;++ strong expression All genes from this list were significantly responsive to 5-aza-dC/TSA treatment based on Two-way ANOVA analysis between 5-aza-dC– +/or TSA-treated and untreated cancer or non cancer cells and P <0.05.
SAGE (Serial Analysis of Gene Expression): The numbers indicate the % of pancreatic cancer samples in which the normalized number of tags was <1 while the number of tags in normal samples was >10 (1).
The numbers indicate the % of pancreatic cancer samples in which the normalized number of tags was <1 while the number of tags in normal samples was >10 (1).
MSP (Methylation-Specific PCR): % of samples in which methylation was detected by MSP
Bibliography: Reference numbers refer to the supplemental list of references in the supplemental materials. PcG: Polycomb group protein targets (2–4).
IPMN: Intraductal Papillary Mucinous Neoplasm; CRC: Colorectal Carcinomas; BC: Breast Cancer; PrC: Prostate Cancer; GC: Gastric Cancer; LC: Lung Cancer; OC: Ovarian Cancer;
HC: hepatocellular carcinoma; RCC: Renal Cell Carcinoma; CC: Cervical Carcinoma; MM: Malignant Melanomas; HM: Hematologic malignancies; A: Astrocytomas.
Biomarkers.
Omura et al, unpublished
Bold genes indicate a fold change lower than -3-fold between cancer cells and HPDE cells.
unpublished data
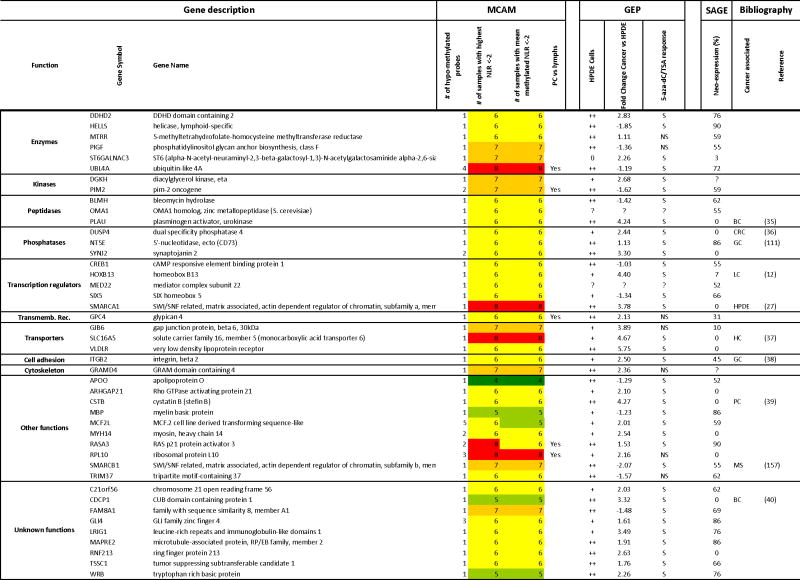
Table 2.
List of hypomethylated and upregulated candidate genes.
|
MCAM: Methylated CpG island Amplification strategy coupled with Microarray platform. PC: Pancreatic Cancer; Lymphs: lymphocytes.
GEP: Gene Expression Profiling with Affymetrix Exon Arrays. The HPDE column indicates the level of expression of the gene in normal cells (0: virtually undetectable signal; +: weak expression; ++ strong expression).
In the 5-aza-dC/TSA column, S indicates a significant response to treatment and NS indicates a not significant response based on a Two-way ANOVA analysis between 5-aza-dC– and/or TSA-treated and untreated cancer or non cancer cells and a P value of <0.05.
SAGE (Serial Analysis of Gene Expression): The numbers indicate the % of pancreatic cancer samples in which the normalized number of tags was >10 while the number of tags in normal samples was <1 (1).
Bibliography: Reference numbers refer to the supplemental list of references in the supplemental materials.
PC: Pancreatic Cancer; HPDE: human pancreatic ductal epithelial cells; CRC: Colorectal Carcinomas; BC: Breast Cancer; GC: Gastric Cancer; LC: Lung Cancer;
HC: hepatocellular carcinoma; MS: Myelodysplastic Syndrome
Color coding:
 4 samples with lowest signal ratio >2 or with mean Normalized Log Ratio > 2
4 samples with lowest signal ratio >2 or with mean Normalized Log Ratio > 2
Even by a modest criteria of ±2-fold for differential expression, only a minority of genes displayed methylation-associated alterations in expression compared to those that underwent aberrant methylation (Table 1 and 2). We previously made this same observation in an MCAM analysis of the Panc-1 vs. HPDE cell lines (13). However, when we next examined the relationship between global methylation and gene expression by comparing mean expression levels in the 6 pancreatic cancer cell lines, we found that hypermethylated genes (Mean of NLR >1) were significantly less expressed in the pancreatic cancer cells than non-hypermethylated genes (Mean of NLR <1, Figure 3A and 3B). And conversely, we found that the expression of hypomethylated genes (Mean of NLR <-1) was significantly higher than non-hypomethylated genes (Mean of NLR >-1, Figure 3A and 3B). These results support the general role of hypermethylation as a mechanism of aberrant silencing in cancer as well as reinforcing the association between localized promoter hypomethylation and gene overexpression.
Figure 3.

Correlation between gene methylation and gene expression levels in pancreatic cancer samples (A32-1, A38-5, Panc215, Panc2.5, Panc2.8 and Panc3.014). A, Boxplots stratifying mean gene expression (intensities values from Affymetrix Exon Array ST1.0) by mean DNA methylation values (NLR from 244K CpG Island Microarrays). For genes with multiple promoter probes the NLRs for each probe were averaged within sample before calculating the promoter region mean at each across the six sample pairs. B, Scatterplot representing for each gene the mean expression fold-change compared to their methylation profile. The line represents the statistically significant association between mean differential DNA methylation and gene expression.
Additionally, we checked the expression profile of our newly identified hypermethylated and hypomethylated genes previously analyzed by the SAGE (serial analysis of gene expression) method (17). The advantage of this SAGE analysis is that it quantified 1–2 million SAGE tags in each of the 24 pancreatic cancer samples 2 laser capture microdissected normal pancreatic duct samples and the non-neoplastic pancreatic duct cell line, HPDE. We defined silenced genes, neo-expressed genes, underexpressed, and overexpressed genes. Silenced genes were defined as genes for which the number of tags was simultaneously <1 in more than 50% of the 24 pancreatic cancer samples and >10 in both normal samples and were added to Table 1. Underexpressed genes were those with a 2-fold decrease in the number of tags in pancreatic cancer compared to either one of the two normal samples (the percentage of pancreatic cancers with underexpression is noted in the SAGE columns of Supplemental Table 2). Neo-expressed genes were defined as genes for which the number of tags was simultaneously >10 in more than 50% of the 24 pancreatic cancer samples and <1 in both normal samples and were added to Table 2. Overexpressed genes were defined as those with a 2-fold increase in the number of tags in pancreatic cancer compared to either one of the two normal samples (the percentage of pancreatic cancer samples with overexpression is noted in Supplemental Table 3).
Genes known to be hypermethylated in pancreatic cancer
Among the list of 1206 candidate hypermethylated genes, were virtually all of the genes we previously identified using MCAM on the Agilent 44K microarray and validated by bisulfite sequencing and MSP in Panc-1 cancer cells, i.e. BAI1, KCNV1, EYA4, BNC1, HOXA5, PAX7, SOX14, TLX3, NRXN1, CNTNAP2, PKP1, ACTA1, MDFI, EVC2, LIN28, NRN1, PENK, m FAM84A, and ZNF415 (13).
We also confirmed the differential methylation profiles of genes whose epigenetic silencing was revealed by our group and others. These genes include NPTX2, CLDN5, LHX1, WNT7A, FOXE1, PAX6, BNIP3, GADD45B (8), HIC1, HS3ST2, TWIST1 (14), IRF7, CCNA1, ALPP, CEBPA (20), CACNA1G (21), CCND2 (22), and TFPI-2 (23).
Our list included the tumor suppressor genes STK11, and WT1, the transcription factor RUNX3, the transcriptional repressor CBFA2T3, the secreted glycoprotein and chemorepulsive factor SLIT2 and the aryl-hydrocarbon receptor repressor, AHRR.
Note that our selection criterion was designed to identify genes that are commonly differentially methylated in pancreatic cancer. Thus, several important genes that are infrequently methylated in pancreatic cancer did not meet our commonly methylated filter including p16/CDKN2A, E-cadherin/CDH1 and hMLH1 since these genes are uncommon targets in pancreatic cancer (24, 25). Among our nine pancreatic cancer/normal pancreas methylomes examined, three had one hypermethylated probe in the CDKN2A promoter. Other genes hypermethylated in pancreatic cancer (RELN (7), MMP1 and MMP14 (26)) did not meet our filter for commonly methylated genes, although several MCAM probes covering these genes were significant for hypermethylation. Finally, some genes hypermethylated in pancreatic cancer were not identified because their promoters were not interrogated by the MCAM method (e.g. SPARC, MMP3, MMP2, MMP9 and MMP7).
Genes involved in stem cell pluripotency are hypermethylated in pancreatic cancer
We found DNA hypermethylation of genes involved in stem cell pluripotency, such as the intestinal stem cell marker and chromatin structure regulator BMI1, the genes encoding bone morphogenetic proteins BMP3, BMP6, the transcription factors FOXD3, CDX2, UTF1, and T, as well as NR5A1, NR5A2, NR2F1, NTRK1, NTRK2, NTRK3, NODAL, SALL4, and SPHK1 genes. Additionally, hypermethylation of FOXA1/A2, BMP7, IGF2BP1, HOXB1 and NR2F2 was associated with gene relative underexpression in pancreatic cancer by SAGE analysis (Supplemental Table 2 and (17)).
The WNT signaling pathway is a target of hypermethylation in pancreatic cancer
Interestingly, numerous actors and mediators of the WNT signaling pathway were found to be hypermethylated in the majority of our pancreatic cancer samples and account for an enrichment ratio of 0.167 in our Ingenuity pathway analysis. As expected, we found hypermethylation involving genes previously shown to be epigenetically silenced both in pancreatic and other types of cancer (14, 27) and encoding either the WNT ligands WNT5A, WNT7A, and WNT9A, or the cell-surface receptor FZD9, as well as the cytoplasmic transducer APC2, the nuclear factors SOX1, SOX7, SOX14, and SOX17, and the pathway inhibitors FRZB, SFRP1, SFRP2, KREMEN2, NKD2 and WIF1. Finally, we also found hypermethylation of the tumor suppressor candidate HIC1, which encodes a transcriptional repressor responsible for abnormal survival circuits through nuclear release of WNT signaling pathway transcription factors (28).
We also found DNA hypermethylation surrounding genes of the WNT pathway and overexpressed in different types of cancer, either in the promoter region (WNT3 and SOX3), within the gene body (FZD1, FZD2, FZD7, SOX15 and SOX18), or both (FZD10, SOX11, and MDFI).
Several hypermethylated WNT pathway genes demonstrated differential gene expression in multiple pancreatic cancer cell lines versus HPDE either by Affymetrix Exon Array (WNT5A, FZD2, SFRP1 and SOX15) or by SAGE analysis (WNT10A, SOX17, SOX18, NKD2, WNT18, FZD1 and FZD7) (Supplemental Table 2 and (17, 18)).
Eleven other WNT pathway genes showed no differential expression by Affymetrix Microarray or SAGE analysis (Supplemental Table 2 and (17, 18)), although 5-Aza-dC treatment increased expression of 7 of these 11 genes (FZD9, APC2, SOX1, SOX3, SOX11, FRZB, and KREMEN2, Supplemental Table 2), suggesting that DNA hypermethylation influences their expression.
Among the WNT Pathway hypermethylated gene members WNT5A, SFRP1 and SOX15 are of particular interest because they had evidence of epigenetic silencing (loss of expression in pancreatic cancers relative to normal pancreas and induced expression with 5-Aza-dC treatment (Supplemental figure 1)). SFRP1 is epigenetically inactivated in colorectal and other cancers (29) and suspected to facilitate tumorigenesis by abrogating WNT signaling.
WNT5A is a member of the non-transforming WNT protein family frequently inactivated by tumor-specific methylation in colorectal cancer (30), although opposing roles of WNT5A in cancer have been described (31). For instance, one study found WNT5A overexpression in pancreatic cancers (32) whereas our gene expression profiling found underexpression of WNT5A compared to HPDE cells (2.9-Fold by Affymetrix Exon Array) and compared to normal pancreatic ductal epithelium by SAGE. Another identified hypermethylated and downregulated gene implicated in pancreatic differentiation was SOX15 (33). To our knowledge, hypermethylation and silencing of SOX15 have not been previously reported in pancreatic cancer.
The finding that the WNT signaling pathway is a common target of DNA methylation in colon and other cancers led Jones and Baylin (34) to propose that loss of epigenetic gatekeepers such as WNT signaling pathway inhibitors locks cells into stem-cell like states that foster abnormal clonal expansion. The pancreatic cancer genome project provided evidence for aberrant WNT pathway signaling during pancreatic tumorigenesis as somatic mutations in the pathway were occasionally identified in pancreatic cancers (17). In this study, the enrichment of aberrant methylation of the WNT pathway gene members supports a significant role for alterations of this pathway during pancreatic tumorigenesis.
Hypermethylation of cell adhesion molecules during pancreatic carcinogenesis
Numerous genes involved in cell adhesion and junctions were also hypermethylated in pancreatic cancer samples, including members of the cadherin superfamily CDH2, CDH4, CDH11, PCDH1, PCDH10, PCDH11Y, PCDH17, PCDH8, PCDHB7, PCDHGA6, PCDHGB1, PCDHGB6, PCDHGC4, and RET. Interestingly, the pancreatic cancer genome project identified occasional somatic mutations in cadherins and protocadherins in pancreatic cancers (17). Of these genes, hypermethylation of CDH4 and PCDHGB6 was associated with gene underexpression in pancreatic cancers by Affymetrix Array or SAGE data, respectively (Table 1 and Supplemental table 2).
Hypermethylation of gene clusters and families as a common feature of human cancer
We found that numerous families of genes were simultaneously hypermethylated in our pancreatic cancer samples compared to normal pancreas (Supplemental Table 2, bold genes). These include families of transcription factors and, in particular, homeobox genes clustered on chromosome 7p15.2 (EVX1, HOXA10, HOXA11, HOXA4, HOXA5, HOXA7, HOXA9), on 17q21.32 (HOXB1, HOXB3, HOXB5, HOXB6, HOXB9), on 12q13.13 (HOXC11, HOXC12, HOXC13, HOXC4, HOXC5), on 2q31.1 (EVX2, HOXD1, HOXD10, HOXD11, HOXD12, HOXD4, HOXD8, HOXD9) (Supplemental Figure 2). These also include chromobox genes on 17q25.3 (CBX2, CBX4, CBX8), Iroquois homeobox genes on 16q12.2 (IRX3, IRX5, IRX6) and on 5p15.33 (IRX1, IRX2, IRX4), and SIX homeobox genes on 2p21 (SIX2 and SIX3).
Hypermethylation of clusters of homeobox genes has been reported in other types of cancer, such as lung cancer and astrocytomas (35, 36), but not in pancreatic cancers. Hypermethylation of a gene region such as the homeobox genes is most likely related to deregulation of the well orchestrated epigenetic machinery involved in the spatiotemporal regulation of these developmental genes. This finding supports other evidence that gene neighborhood is required for an efficient coordination of chromatin modifications during carcinogenesis as well as developmental processes (37, 38). An important contributor to these epigenetic marks is the Polycomb group of proteins, which has been shown to be responsible for the concerted silencing of homeobox gene clusters. From our hypermethylated candidate gene list, we found 288 known targets of Polycomb proteins (Supplemental Table 2), including families of highly homologous genes that do not belong to clusters, such as LIM homeobox, NK2/3/6 homeobox, Paired box, POU class homeobox, sex determining region Y-box and T-box transcription factors, which confirms the very tight link between specific hypermethylation and Polycomb protein complexes occupancy in cancer. However, we found aberrant hypermethylation of several other gene family members including members of the G-protein-coupled receptor superfamily, adenylate cyclases, and voltage-dependant calcium and potassium channels. Sequence similarities between these family members are likely responsible for their similar cancer epigenetic patterns. For example, SINE and LINE repetitive elements are enriched at sites of aberrant hypermethylation (38). These sequence-based contributions to aberrant methylation indicate that selection driven by gene silencing need is not responsible for causing methylation marks to arise during tumorigenesis. The fact that only a relatively small proportion of hypermethylated genes showed evidence of downregulation or silencing (HOXD9, HOXD8, CBX2, IRX2, IRX4, TBX18, SOX15, SOX17, SOX18, PAX8, Supplemental Table 2) supports this hypothesis.
Newly identified hypermethylated and underexpressed candidate genes
Of the 81 hypermethylated and underexpressed genes identified by our analysis, 33 have, to our knowledge, not been previously identified as hypermethylated either in pancreatic or other cancers. The differential methylation of several of these genes including ID4, FOXF2, FOXG1, IRF5, SIM1, SMOC2, and ZBTB16 was confirmed by MSP (Supplemental table 4). Twelve genes demonstrated gene silencing in pancreatic cancer compared to normal pancreatic duct by SAGE (including HOXD8, BNIP3, FOXF2 and ID4), many of which have not been identified as differentially methylated in cancer (Table 1).
Many hypermethylated and underexpressed genes identified have been implicated in tumorigenesis including tumor suppression (RINT1 (39)), proliferation (GAS6, CDT1 (40)), apoptosis (IRF5 (41), C16ORF5/CDIP (42)), cell adhesion (CLDN10, ICAM4, LAMA1, AMOTL1), or transcriptional regulation (FOXG1, INSM2). Interestingly, although SEMA5A was recently proposed as a marker for aggressive pancreatic cancers we find most pancreatic cancers underexpressed SEMA5A. (43). Other genes of note include the Type IV collagen genes COL4A1 and COL4A2 which have not been described as aberrantly methylated in cancer.
Finally, to further evaluate genes identified as both hypermethylated and underexpressed by array analysis, we searched for commercially available antibodies to proteins corresponding to genes on our list (Table 1) and selected an antibody to ZBTB16 for further analysis. The transcription factor ZBTB16 (a.k.a PLZF), has been shown to regulate apoptosis and proliferation, as well as the Hox family of genes during development, and has been proposed as a candidate tumor suppressor gene in acute promyelocytic leukemias. By MSP, we confirmed ZBTB16 promoter methylation in 35.2% of the pancreatic cancer samples tested, while none of the DNAs from 16 normal pancreas samples showed methylation. By immunohistochemistry, ZBTB16 protein expression was not detectable in 38 (15.4%) of 247 pancreatic cancers evaluated. Reduced expression relative to normal pancreas was found in the majority of pancreatic cancers (193/211, 94%) (Mean histo-score 2.7 vs 6.9, p<0.0001; Supplemental Figure 3). Interestingly, aberrant methylation and silencing of ZBTB16 in pancreatic cancers may be functionally significant as it functions to repress miR-221 and miR-222 which is overexpressed in pancreatic cancer cells (45). MiR-221/222 expression is associated with enhanced proliferation and dedifferentiation in melanoma cells (46).
Newly identified hypomethylated and overexpressed candidate genes
Since hypomethylation is associated with gene overexpression in pancreatic and other cancers (9), we also identified candidate hypomethylated genes using the MCAM assay. Of the 379 unique hypomethylated candidate genes identified, 31 have been described as aberrantly methylated in different cancers (Supplemental Table 3). This list confirmed the aberrant hypomethylation of NDN and SMARCA1 genes, previously described in pancreatic cancer (14). Similarly to hypermethylated genes, some known hypomethylated genes did not meet our criteria for frequent hypomethylation. However, when we examined the NLR for probes covering MUC4 and MUC2, genes previously identified as hypomethylated in pancreatic cancer (47), there was evidence for hypomethylation (NLR ≤ −2) in at least one of the nine differential methylomes of pancreatic cancer/normal pancreas studied. The promoter regions of other hypomethylated genes in pancreatic cancers (S100A4, LCN2, TFF2, MSLN CLDN4, SFN and PSCA) (9) were not interrogated by our MCAM method.
Interestingly, while approximately half of the hypermethylated candidate genes had multiple probes meeting criteria for hypermethylation, the vast majority (84.7%) of hypomethylated gene candidates harbored only one hypomethylated probe (mean of the nine NLR ≤ −2 and mean combined Z-scores was ≤ −5) (Supplemental Table 3).
Our hypomethylated gene list contained chromatin modifiers, including the histone methyltransferases SETD8 (lysine) and PRMT1 (arginine), the histone ubiquitin ligase CTR9, the histone demethylase KDM6A, the chromatin assembly protein HIRIP3, and the histone acetyltransferase EP400. These genes were overexpressed in pancreatic cancer samples compared to HPDE cells and/or normal ductal epithelium by SAGE, and HIRIP3 was found to be neo-expressed in 41% of pancreatic cancer samples (Supplemental Table 3 and (17)).
Three oncogenes, JUNB, MYB, and FOS were hypomethylated in our analysis. While FOS undergoes hypomethylation during liver tumorigenesis, JUNB and MYB have not been previously reported to be hypomethylated. Interestingly, MYB was found to be neo-expressed in 31% of pancreatic cancer compared to normal samples by SAGE (Supplemental Table 3 and (17)).
Of the 44 hypomethylated genes identified in our analysis and found to be overexpressed by both ExonArray and SAGE methods, 34 have, to our knowledge, not been described as aberrantly methylated in cancer. Among them, DDHD2, CREB1 and PIM2 have been proposed as oncogenes, while ITGB2 (CD18) and MAPRE2 are involved in cell invasion and metastasis. HELLS is a putative stem cell marker, while MTRR restores methionine synthase for folate metabolism and polymorphisms have been associated with pancreatic cancer risk.
Differentially Methylated loci of pancreatic cancer samples compared to normal lymphoid tissues
To further validate our list of hyper- and hypomethylated candidate markers, we performed three additional MCAM experiments, in which we compared the methylation profiles of pancreatic cancer cell samples with matched normal DNA either from peripheral blood lymphocytes (A32-1 and Panc215) or from normal spleen (A38-5). We used the cutoffs described above for the NLR and combined Z-score to select probes that were hyper- or hypomethylated in pancreatic cancer cells compared to matched lymphocytes or spleen, respectively (Figure 4).
Figure 4.

An overview of experimental strategy to evaluate differential DNA methylation in pancreatic cancer cells compared to normal lymphoid tissue. NP: Microdissected normal pancreatic ductal epithelium; ly: peripheral blood lymphocytes.
We found 11,029 and 14,863 differentially methylated probes in Panc215 and A32-1 samples compared to the patient’s matched lymphocytes DNA, respectively. We found 13,680 differentially methylated probes in A38-5 cancer cells compared to their matched normal spleen. We then compared these probes to those differentially methylated in Panc215, A32-1 and A38-5 cancer cells, vs. the same pooled microdissected normal pancreatic ductal tissue. Since some methylation patterns reflect tissue specific differences, we found more differentially methylated probes when comparing pancreatic cancer DNA to a patient’s matched lymphoid tissue than to unmatched normal pancreatic duct. We found 4,434 and 6,200 differentially methylated probes both in cancer compared to lymphocytes and in cancer compared to normal pancreatic tissue, for Panc215 and A32-1 samples, respectively. Moreover, we found differentially methylated 2,956 probes in both Panc215 and A32-1 samples, and both in cancer compared to lymphocytes and cancer compared to normal pancreatic tissue, corresponding to 786 known loci (Figure 4). Among these loci, 669 unique genes were part of our hypermethylated and hypomethylated candidate lists (Supplementary Tables 2 and 3). These data indicate that while more than half of our differentially methylated candidate genes in pancreatic cancers relative to normal pancreata are also differential methylated relative to peripheral blood lymphocytes, many genes that are methylated in pancreatic cancers are normally methylated in other tissues.
We found differentially methylated 6,858 probes that were both in cancer compared to spleen and cancer compared to normal pancreatic tissue (Figure 4). Among these probes, 1,896 were also differentially methylated in pancreatic cancer cells compared to normal pancreatic tissue and in pancreatic cancer cells compared to lymphocytes. These corresponded to 530 known loci, and 489 genes present in our hypermethylated and hypomethylated gene lists (Supplemental tables 2 and 3).
Among these 489 genes, 32 genes show DNA hypermethylation associated with downregulation in cancer cells compared to normal cells (Table 1). These latter genes, with a more specific hypermethylation profiles in pancreatic cancer tissues are likely to be better candidates as pancreatic cancer markers. Indeed, several of these genes have already been proposed as candidate biomarkers of prognosis (BNC1 (48), SLIT2 (49)), or response to treatment (WNT5A (50)) in other types of cancer.
Conclusion
In summary, we provide a comprehensive list of candidate aberrantly methylated in pancreatic cancers relative to normal pancreatic duct samples, including novel aberrantly methylated and epigenetically silenced genes validated in pancreatic cancer tissue samples, as well as commonly methylated signaling pathways and gene families. We anticipate that our integrated analysis of pancreatic cancer gene methylation and transcription will facilitate the further investigation of pancreatic cancer biomarkers for diagnostic, prognostic and therapeutic applications.
Supplementary Material
Statement of Translational Relevance.
Pancreatic cancer is the fourth leading cause of cancer death in the United States and is characterized by advanced disease at the time of diagnosis and resistance to most therapeutic treatments. Improving our understanding of the molecular mechanisms that contribute to the development of pancreatic cancer at the epigenetic as well as transcriptional level is crucial for the early diagnosis and therefore the treatment of this deadly disease. In this study, we used cancer cell lines derived from pancreatic ductal adenocarcinoma and compared their DNA methylation profile to pancreatic normal tissue as a reference. We found numerous target genes that are either hypermethylated and silenced or hypomethylated and overexpressed in pancreatic cancer versus normal pancreas and that could be used as building blocks for the development of new diagnosis and prognosis biomarkers, as well as therapeutic tools for pancreatic cancer.
Acknowledgments
This work was supported by the National Cancer Institute grants (CA62924, CA120432, RC2CA148346), and the Michael Rolfe Foundation.
References
- 1.Hruban RH, Canto MI, Goggins M, Schulick R, Klein AP. Update on familial pancreatic cancer. Adv Surg. 2010;44:293–311. doi: 10.1016/j.yasu.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsubayashi H, Canto M, Sato N, Klein A, Abe T, Yamashita K, et al. DNA methylation alterations in the pancreatic juice of patients with suspected pancreatic disease. Cancer Res. 2006;66:1208–17. doi: 10.1158/0008-5472.CAN-05-2664. [DOI] [PubMed] [Google Scholar]
- 4.Parsi MA, Li A, Li CP, Goggins M. DNA methylation alterations in endoscopic retrograde cholangiopancreatography brush samples of patients with suspected pancreaticobiliary disease. Clin Gastroenterol Hepatol. 2008;6:1270–8. doi: 10.1016/j.cgh.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canto MI, Goggins M, Hruban RH, Petersen GM, Giardiello FM, Yeo C, et al. Screening for early pancreatic neoplasia in high-risk individuals: a prospective controlled study. Clin Gastroenterol Hepatol. 2006;4:766–81. doi: 10.1016/j.cgh.2006.02.005. quiz 665. [DOI] [PubMed] [Google Scholar]
- 6.Fukushima N, Sato N, Ueki T, Rosty C, Walter KM, Wilentz RE, et al. Aberrant methylation of preproenkephalin and p16 genes in pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am J Pathol. 2002;160:1573–81. doi: 10.1016/S0002-9440(10)61104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato N, Fukushima N, Chang R, Matsubayashi H, Goggins M. Differential and epigenetic gene expression profiling identifies frequent disruption of the RELN pathway in pancreatic cancers. Gastroenterology. 2006;130:548–65. doi: 10.1053/j.gastro.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 8.Sato N, Fukushima N, Maitra A, Matsubayashi H, Yeo CJ, Cameron JL, et al. Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-throughput microarrays. Cancer Res. 2003;63:3735–42. [PubMed] [Google Scholar]
- 9.Sato N, Maitra A, Fukushima N, van Heek NT, Matsubayashi H, Iacobuzio-Donahue CA, et al. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res. 2003;63:4158–66. [PubMed] [Google Scholar]
- 10.Sato N, Matsubayashi H, Abe T, Fukushima N, Goggins M. Epigenetic down-regulation of CDKN1C/p57KIP2 in pancreatic ductal neoplasms identified by gene expression profiling. Clin Cancer Res. 2005;11:4681–8. doi: 10.1158/1078-0432.CCR-04-2471. [DOI] [PubMed] [Google Scholar]
- 11.Sato N, Ueki T, Fukushima N, Iacobuzio-Donahue CA, Yeo CJ, Cameron JL, et al. Aberrant methylation of CpG islands in intraductal papillary mucinous neoplasms of the pancreas. Gastroenterology. 2002;123:365–72. doi: 10.1053/gast.2002.34160. [DOI] [PubMed] [Google Scholar]
- 12.Hong SM, Kelly D, Griffith M, Omura N, Li A, Li CP, et al. Multiple genes are hypermethylated in intraductal papillary mucinous neoplasms of the pancreas. Mod Pathol. 2008;21:1499–507. doi: 10.1038/modpathol.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Omura N, Li CP, Li A, Hong SM, Walter K, Jimeno A, et al. Genome-wide profiling of methylated promoters in pancreatic adenocarcinoma. Cancer Biol Ther. 2008;7:1146–56. doi: 10.4161/cbt.7.7.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan AC, Jimeno A, Lin SH, Wheelhouse J, Chan F, Solomon A, et al. Characterizing DNA methylation patterns in pancreatic cancer genome. Mol Oncol. 2009;3:425–38. doi: 10.1016/j.molonc.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ting AH, McGarvey KM, Baylin SB. The cancer epigenome--components and functional correlates. Genes Dev. 2006;20:3215–31. doi: 10.1101/gad.1464906. [DOI] [PubMed] [Google Scholar]
- 16.Estecio MR, Yan PS, Ibrahim AE, Tellez CS, Shen L, Huang TH, et al. High-throughput methylation profiling by MCA coupled to CpG island microarray. Genome Res. 2007;17:1529–36. doi: 10.1101/gr.6417007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Omura N, Griffith M, Vincent A, Li A, Hong SM, Walter K, et al. Cyclooxygenase-deficient pancreatic cancer cells utilize exogenous sources of prostaglandins. Mol Cancer Res. 2010 doi: 10.1158/1541-7786.MCR-09-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walter K, Omura N, Hong SM, Griffith M, Vincent A, Borges M, et al. Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clin Cancer Res. 2010;16:1781–9. doi: 10.1158/1078-0432.CCR-09-1913. Epub 2010 Mar 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumagai T, Akagi T, Desmond JC, Kawamata N, Gery S, Imai Y, et al. Epigenetic regulation and molecular characterization of C/EBPalpha in pancreatic cancer cells. Int J Cancer. 2009;124:827–33. doi: 10.1002/ijc.23994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ueki T, Toyota M, Sohn T, Yeo CJ, Issa JP, Hruban RH, et al. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000;60:1835–9. [PubMed] [Google Scholar]
- 22.Matsubayashi H, Sato N, Fukushima N, Yeo CJ, Walter KM, Brune K, et al. Methylation of cyclin D2 is observed frequently in pancreatic cancer but is also an age-related phenomenon in gastrointestinal tissues. Clin Cancer Res. 2003;9:1446–52. [PubMed] [Google Scholar]
- 23.Sato N, Parker AR, Fukushima N, Miyagi Y, Iacobuzio-Donahue CA, Eshleman JR, et al. Epigenetic inactivation of TFPI-2 as a common mechanism associated with growth and invasion of pancreatic ductal adenocarcinoma. Oncogene. 2005;24:850–8. doi: 10.1038/sj.onc.1208050. [DOI] [PubMed] [Google Scholar]
- 24.Omura N, Goggins M. Epigenetics and epigenetic alterations in pancreatic cancer. Int J Clin Exp Pathol. 2009;2:310–26. Epub 2008 Nov 15. [PMC free article] [PubMed] [Google Scholar]
- 25.Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57:3126–30. [PubMed] [Google Scholar]
- 26.Sato N, Maehara N, Su GH, Goggins M. Effects of 5-aza-2′-deoxycytidine on matrix metalloproteinase expression and pancreatic cancer cell invasiveness. J Natl Cancer Inst. 2003;95:327–30. doi: 10.1093/jnci/95.4.327. [DOI] [PubMed] [Google Scholar]
- 27.Ying Y, Tao Q. Epigenetic disruption of the WNT/beta-catenin signaling pathway in human cancers. Epigenetics. 2009;4:307–12. doi: 10.4161/epi.4.5.9371. [DOI] [PubMed] [Google Scholar]
- 28.Valenta T, Lukas J, Doubravska L, Fafilek B, Korinek V. HIC1 attenuates Wnt signaling by recruitment of TCF-4 and beta-catenin to the nuclear bodies. EMBO J. 2006;25:2326–37. doi: 10.1038/sj.emboj.7601147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caldwell GM, Jones C, Gensberg K, Jan S, Hardy RG, Byrd P, et al. The Wnt antagonist sFRP1 in colorectal tumorigenesis. Cancer Res. 2004;64:883–8. doi: 10.1158/0008-5472.can-03-1346. [DOI] [PubMed] [Google Scholar]
- 30.Ying J, Li H, Yu J, Ng KM, Poon FF, Wong SC, et al. WNT5A exhibits tumor-suppressive activity through antagonizing the Wnt/beta-catenin signaling, and is frequently methylated in colorectal cancer. Clin Cancer Res. 2008;14:55–61. doi: 10.1158/1078-0432.CCR-07-1644. [DOI] [PubMed] [Google Scholar]
- 31.McDonald SL, Silver A. The opposing roles of Wnt-5a in cancer. Br J Cancer. 2009;101:209–14. doi: 10.1038/sj.bjc.6605174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ripka S, Konig A, Buchholz M, Wagner M, Sipos B, Kloppel G, et al. WNT5A--target of CUTL1 and potent modulator of tumor cell migration and invasion in pancreatic cancer. Carcinogenesis. 2007;28:1178–87. doi: 10.1093/carcin/bgl255. [DOI] [PubMed] [Google Scholar]
- 33.White P, May CL, Lamounier RN, Brestelli JE, Kaestner KH. Defining pancreatic endocrine precursors and their descendants. Diabetes. 2008;57:654–68. doi: 10.2337/db07-1362. [DOI] [PubMed] [Google Scholar]
- 34.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rauch T, Li H, Wu X, Pfeifer GP. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;66:7939–47. doi: 10.1158/0008-5472.CAN-06-1888. [DOI] [PubMed] [Google Scholar]
- 36.Wu X, Rauch TA, Zhong X, Bennett WP, Latif F, Krex D, et al. CpG island hypermethylation in human astrocytomas. Cancer Res. 70:2718–27. doi: 10.1158/0008-5472.CAN-09-3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soshnikova N, Duboule D. Epigenetic regulation of vertebrate Hox genes: a dynamic equilibrium. Epigenetics. 2009;4:537–40. doi: 10.4161/epi.4.8.10132. [DOI] [PubMed] [Google Scholar]
- 38.Estecio MR, Gallegos J, Vallot C, Castoro RJ, Chung W, Maegawa S, et al. Genome architecture marked by retrotransposons modulates predisposition to DNA methylation in cancer. Genome Res. doi: 10.1101/gr.107318.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin X, Liu CC, Gao Q, Zhang X, Wu G, Lee WH. RINT-1 serves as a tumor suppressor and maintains Golgi dynamics and centrosome integrity for cell survival. Mol Cell Biol. 2007;27:4905–16. doi: 10.1128/MCB.02396-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liontos M, Koutsami M, Sideridou M, Evangelou K, Kletsas D, Levy B, et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res. 2007;67:10899–909. doi: 10.1158/0008-5472.CAN-07-2837. [DOI] [PubMed] [Google Scholar]
- 41.Hu G, Barnes BJ. IRF-5 is a mediator of the death receptor-induced apoptotic signaling pathway. J Biol Chem. 2009;284:2767–77. doi: 10.1074/jbc.M804744200. [DOI] [PubMed] [Google Scholar]
- 42.Brown L, Ongusaha PP, Kim HG, Nuti S, Mandinova A, Lee JW, et al. CDIP, a novel pro-apoptotic gene, regulates TNFalpha-mediated apoptosis in a p53-dependent manner. EMBO J. 2007;26:3410–22. doi: 10.1038/sj.emboj.7601779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sadanandam A, Varney ML, Singh S, Ashour AE, Moniaux N, Deb S, et al. High gene expression of semaphorin 5A in pancreatic cancer is associated with tumor growth, invasion and metastasis. Int J Cancer. 127:1373–83. doi: 10.1002/ijc.25166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noetzel E, Veeck J, Niederacher D, Galm O, Horn F, Hartmann A, et al. Promoter methylation-associated loss of ID4 expression is a marker of tumour recurrence in human breast cancer. BMC Cancer. 2008;8:154. doi: 10.1186/1471-2407-8-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kent OA, Mullendore M, Wentzel EA, Lopez-Romero P, Tan AC, Alvarez H, et al. A resource for analysis of microRNA expression and function in pancreatic ductal adenocarcinoma cells. Cancer Biol Ther. 2009;8:2013–24. doi: 10.4161/cbt.8.21.9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Felicetti F, Errico MC, Bottero L, Segnalini P, Stoppacciaro A, Biffoni M, et al. The promyelocytic leukemia zinc finger-microRNA-221/-222 pathway controls melanoma progression through multiple oncogenic mechanisms. Cancer Res. 2008;68:2745–54. doi: 10.1158/0008-5472.CAN-07-2538. [DOI] [PubMed] [Google Scholar]
- 47.Vincent A, van Seuningen I. Mucins, epigenetics and cancer. In: van Seuningen I, editor. The epitheliul mucins: structure/function Roles in cancer and inflammatory diseases. Kerala: Research Signpost; 2008. pp. 95–108. [Google Scholar]
- 48.Morris MR, Ricketts C, Gentle D, Abdulrahman M, Clarke N, Brown M, et al. Identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma. Oncogene. 29:2104–17. doi: 10.1038/onc.2009.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tseng RC, Lee SH, Hsu HS, Chen BH, Tsai WC, Tzao C, et al. SLIT2 attenuation during lung cancer progression deregulates beta-catenin and E-cadherin and associates with poor prognosis. Cancer Res. 70:543–51. doi: 10.1158/0008-5472.CAN-09-2084. [DOI] [PubMed] [Google Scholar]
- 50.Ford CE, Ekstrom EJ, Andersson T. Wnt-5a signaling restores tamoxifen sensitivity in estrogen receptor-negative breast cancer cells. Proc Natl Acad Sci U S A. 2009;106:3919–24. doi: 10.1073/pnas.0809516106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.