

Abstract
Septic bacterial pneumonias are a major cause of death worldwide. Several of the highest priority bioterror concerns, including anthrax, tularemia and plague, are caused by bacteria that acutely infect the lung. Bacterial resistance to multiple antibiotics is increasingly common. Although vaccines may be our best defense against antibiotic-resistant bacteria, there has been little progress in the development of safe and effective vaccines for pulmonary bacterial pathogens. The gram-negative bacterium Yersinia pestis causes pneumonic plague, an acutely lethal septic pneumonia. Historic pandemics of plague caused millions of deaths, and the plague bacilli’s potential for weaponization sustains an ongoing quest for effective countermeasures. Subunit vaccines have failed, thus far, to fully protect non-human primates. In mice, they induce the production of antibodies that act in concert with type 1 cytokines to deliver high-level protection, however, the Y. pestis antigens recognized by cytokine-producing T cells have yet to be defined. Here, we report that Y. pestis YopE is a dominant antigen recognized by CD8 T cells in C57BL/6 mice. After vaccinating with live attenuated Y. pestis and challenging intranasally with virulent plague, nearly 20% of pulmonary CD8 T cells recognize this single, highly conserved antigen. Moreover, immunizing mice with a single peptide, YopE69-77, suffices to confer significant protection from lethal pulmonary challenge. These findings suggest YopE could be a valuable addition to subunit plague vaccines and provide a new animal model in which sensitive, pathogen-specific assays can be used to study CD8 T cell-mediated defense against acutely lethal bacterial infections of the lung.
Introduction
Bacterial infections of the lung are a major cause of morbidity and mortality worldwide (1, 2). Many species of opportunistic bacteria, including Streptococcus, Staphylococcus, Klebsiella, Acinetobacter and Pseudomonas, cause considerable morbidity in both community and hospital settings. Bacterial strains resistant to multiple antibiotics are increasingly common. Moreover, several of the United States Centers for Disease Control and Prevention’s highest priority bioterror concerns, including anthrax, tularemia and plague, are caused by acutely lethal bacterial infections of the lung.
Vaccines may be our best defense against disease outbreaks caused by antibiotic-resistant bacteria. Yet, there has been little progress in the quest for safe and effective vaccines for bacterial pathogens of the lung. One notable exception is Streptococcus pneumoniae, for which antibody-based vaccines have been approved. However, growing evidence suggests that optimal defense against pulmonary bacteria often requires the concerted action of both antibody and cell-mediated immunity (3). Unfortunately, for most acutely lethal bacterial pathogens that infect the human lung, we know remarkably little about the antigens recognized by protective T cells. This deficit hampers efforts to study cellular defense mechanisms and develop subunit vaccines that prime both humoral and cellular immunity.
Plague is an exceptionally virulent disease caused by the gram-negative facultative intracellular bacterium Yersinia pestis (4, 5). Plague has taken the lives of hundreds of millions of humans over the course of recorded history, most famously during the 14th century’s “Black Death” pandemic that killed one-third of Europe’s population. Humans usually acquire the infection via fleabites, after which the plague bacilli gain access to regional lymph nodes, overwhelm innate defense mechanisms, replicate explosively, and cause the painfully swollen buboes that characterize bubonic plague. Unless treated with antibiotics, bubonic plague typically progresses to bacteremia and sepsis. Once the bacilli reach the human lung, they may be transmitted person-to-person via infectious respiratory droplets (6). Untreated pneumonic plague is thought to be 100% fatal. Death from primary pneumonic plague occurs within 2–6 days of infection, and antibiotics are ineffective unless administered within 24 hours of symptom onset. Plague outbreaks are uncommon today but the need for countermeasures remains high because antibiotic-resistant strains have been described (7), the plague bacilli’s genetic makeup is mutable and evolving (8), and Cold War era scientists developed the means to intentionally aerosolize infectious Y. pestis (6).
Vaccination with live attenuated Y. pestis protects against pneumonic plague in animal models (9, 10). Live attenuated plague vaccines are available in some countries, including Russia (11), however, they have never been licensed in the United States and Europe, presumably due to the significant safety concerns (11–14). Subunit vaccines containing the F1 protein, a component of the bacilli’s capsule-like surface, and LcrV, a component of its plasmid-encoded type III secretion system, demonstrate considerable efficacy in several animal models of pneumonic plague (11, 14), but they confer little protection in others, most notably in African green monkey models (15). Mechanistic studies in the mouse have established that subunit vaccines protect by inducing production of F1/LcrV-specific antibodies, however, optimal protection also requires the type 1 cytokines IFNγ and TNFα (16–18). These findings suggest that subunit vaccines might demonstrate improved efficacy if they prime Y. pestis-specific memory T cells capable of producing type 1 cytokines, in addition to inducing production of F1/LcrV-specific antibody.
To facilitate the development of such vaccines, we sought to identify protective T cell antigens. Here, we demonstrate that Y. pestis YopE is a dominant antigen recognized by CD8 T cells in mice immunized with live attenuated Y. pestis. Moreover, we found that immunization with a single peptide, YopE69-77, suffices to confer remarkable CD8-mediated protection against lethal pulmonary challenge. We report the first model in which sensitive, quantitative, pathogen-specific assays can be applied to the study of CD8 T cell-mediated defense against acutely lethal bacterial infections of the lung.
Materials and Methods
Mice
C57BL/6 wild-type mice and B cell-deficient μMT mice (6 to 8 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME) and then bred at the Trudeau Institute Animal Breeding Facility after embryo rederivation. Experimental mice were matched for age and sex and cared for according to Trudeau Institute Animal Care and Use Committee guidelines.
Bacteria
Strains D27 (pCD1+, pPCP+, pMT+) and D28 (pCD1-, pPCP+, pMT+), two pigmentation locus (pgm)-deficient variants of Y. pestis strain KIM, were provided by Dr. Robert Brubaker (Michigan State University, East Lansing, MI). A pgm-deficient variant of Y. pestis strain CO92 (pCD1+, pPCP+, pMT+) was provided by Dr. James B. Bliska (State University of New York at Stony Brook, Stony Brook, NY). Attenuated strain D27-pLpxL was prepared by transforming strain D27 with plasmid pLpxL (10), which was provided by Drs. Egil Lien and Jon Goguen (University of Massachusetts Medical School, Worcester, MA). For challenge infections, strain D27 was grown overnight at 26°C in Bacto heart infusion broth supplemented with 2.5 mM CaCl2, diluted to an optical density of 0.1 at 620nm, re-grown for 3–4 hours at 26°C, quantified by measuring the optical density, and resuspended in saline at the desired concentration. The number of bacteria in the inoculating dose was confirmed by plating. For immunizations, strain D27-pLpxL was prepared as described for strain D27, except the broth was supplemented with 100 μg/ml ampicillin (10). To prepare heat-killed bacteria, Y. pestis strains were grown overnight at 26°C, diluted to an optical density of 0.1 at 620nm, re-grown for 3–4 hours at 26°C or for 4–5 hours at 37°C, quantified by measuring the optical density, resuspended in saline and then inactivated by heating to 60°C for 1 hour. E. coli strain 018:K1 was grown at 37°C and then inactivated by heating to 60°C for 1 hour.
Generation of Y. pestis-specific CD8 T cell clones
Antigen-presenting cells (APC) pulsed with Y. pestis were prepared by harvesting splenocytes from naïve C57BL/6 mice, treating with 50 μg/ml mitomycin C (Sigma) for 30 minutes at 37°C in complete medium (DMEM supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM non-essential amino acid, 1 mM penicillin-streptomycin and 55 μM b-mercaptoethanol), washing with complete medium, and then incubating at 37°C for 2 hours with heat-killed Y. pestis strain D27 grown at 37°C, using an APC:bacterium ratio of 1:5. The D27-pulsed APC were used to stimulate immune splenocytes harvested from C57BL/6 mice that were vaccinated intranasally with live attenuated Y. pestis strain D27-pLpxL (2×106 CFU) and then boosted 62 and 92 days later by challenging intranasally with Y. pestis strain D27 (2×105 CFU). Thirty-five days after the final boost, the splenocytes were harvested and cultured with the D27-pulsed APC in complete medium at an APC:splenocyte ratio of 1:1. After 48 hours, recombinant human IL-2 (Peprotech, Rocky Hill, NJ) was added to the culture at a final concentration of 20 U/ml. Culture medium was replenished every other day with fresh IL-2-containing medium. After 2 weeks, cells were cloned by limiting dilution in 96-well flat-bottom plates containing D27-pulsed APC prepared as described above. The resultant T cell clones were maintained by re-exposure to D27-pulsed APC every two weeks, followed by expansion in IL-2-containing medium. Clone phenotypes were determined by flow cytometry using fluorochrome-conjugated antibodies specific for mouse CD3, CD4 and CD8.
T cell assays
Cloned T cells (1×104/well) were cultured in 96-well flat-bottom plates containing mitomycin C-treated splenic APC (1×105/well) and the indicated peptides or heat-killed bacteria in a total volume of 200 μl complete medium. Culture supernatants (100 μl) were collected after 48 hours of culture and IFNγ levels were measured by ELISA using an OptEIA kit (BD Bioscience, San Diego, CA). The supernatant removed from the cultures was replaced with fresh complete medium and 0.2 μCi of [3H]-thymidine. Cells were incubated for another 24 hours before harvesting DNA and measuring [3H]-thymidine incorporation using a Luminescence Counter (Perkin Elmer, Waltham, MA). The stimulation index was calculated as: (cpm in culture with antigen) ÷ (cpm in culture without antigen).
E. coli expressing recombinant Y. pestis proteins
A set of 4,021 individual E. coli clones containing the open reading frames from Y. pestis strain KIM was obtained from the Pathogen Functional Genomics Resource Center. Selected clones were converted to expression format in plasmid pDEST17 using Gateway Technology (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. This plasmid enables the expression of recombinant proteins under the transcriptional control of the bacteriophage T7 promoter in frame with an N-terminal 6×His tag. The plasmid was transferred to E. coli strain BL21-AI and the bacteria were grown in Luria broth containing 100 μg/ml ampicillin and 0.2% L-arabinose to induce production of recombinant proteins. Heat-killed bacteria were obtained by heating cultures to 60°C for 1 hour. Expression of recombinant protein was confirmed by Western blotting using anti-His tag antibody.
Synthetic peptides
All peptides were synthesized by New England Peptide (Gardner, MA). A set of 42 overlapping Y. pestis YopE peptides was generated; each peptide contained 15 contiguous amino acids of the YopE protein and overlapped its neighbors by 10 amino acids. In addition, peptides predicted to bind H 2-Kb MHC class I molecules were identified using RANKPEP (http://bio.dfci.harvard.edu/RANKPEP/), SYFPEITHI (http://www.syfpeithi.de/home.htm) and BIMAS (http://www-bimas.cit.nih.gov/molbio/hla_bind/) algorithms. For immunizations, YopE69-77 (H2N-SVIGFIQRM-OH) and control peptide OVA257-264 (H2N-SIINFEKL-OH) were synthesized and purified (>95%) at larger scale.
Intracellular cytokine staining
Lungs were perfused with saline containing heparin, minced, and digested with collagenase and DNase as described previously (10). Cells were stimulated with plate-bound anti-CD3 (clone 145-2C11, 2 μg/ml) or peptides (10 nM) for 5 hours in complete medium containing brefeldin A (Sigma; 12.5 μg/ml) and stained with anti-CD4-allophycocyanin (clone RM4-5) and anti-CD8-peridinin chlorophyll protein (clone 53-6.7), followed by fixation, permeabilization, and intracellular staining with anti-TNFα-fluorescein isothiocyanate (clone MP6-XT22), and anti-IFNγ-phycoerythrin cyanine dye 7 (clone XMG1.2). Data were collected on a Beckton Dickinson FACSCanto II and analyzed using FlowJo version 8.8 software (Tree Star, Inc.). The TNFα- and IFNγ-specific mAb were purchased from eBioscience and all other staining mAb were purchased from BD Bioscience.
MHC class I tetramer reagents and staining
Allophycocyanin-conjugated MHC class I peptide tetramers KbYopE69-77 were generated by the Trudeau Institute Molecular Biology Core Facility. Lung cells were prepared as described above. PBL were obtained from blood collected by cardiac puncture using heparin-treated syringes. Red blood cells were lysed by treatment with 0.85% ammonium chloride solution prior to tetramer staining. To stain with tetramers, cells were treated with Fc Block (clone 2.4G2, 1 μg) for 15 minutes at 4°C, washed, and incubated with tetramers for 1 hour at room temperature. Each tetramer concentration was optimized empirically. After washing again, cells were stained with anti-CD4-fluorescein isothiocyanate (clone RM4-5) and anti-CD8-peridinin chlorophyll protein (clone 53-6.7) for 30 minutes at 4°C. Data were collected on a Beckton Dickinson FACSCanto II and analyzed using FlowJo software. For each sample, at least 10,000 CD8-positiveevents were collected.
Immunizations using bone marrow-derived dendritic cells
Bone marrow was harvested from the femurs of C57BL/6 mice. Red blood cells were lysed as described above and 2×106 cells were cultured in bacteriological petri dishes containing 10 ml complete medium supplemented with 20 ng/ml recombinant murine granulocyte/monocyte colony-stimulating factor (Peprotech). On day 3, an additional 10 ml of the supplemented medium was added to the cultures. On day 6, 10 ml of the medium was removed and replaced with 10 ml of fresh, supplemented medium. On day 8, the non-adherent cells were collected, centrifuged, resuspended at 2×106/ml in complete medium containing 1 μM peptide, and cultured for another 24 hours. Then, the pulsed dendritic cells were washed, resuspended in neat DMEM, and injected intravenously into mice (5×105 cells/mouse in 200 μl). The mice were challenged with Y. pestis strain D27 14 days later.
Immunizations using cholera toxin adjuvant
Mice were lightly anesthetized by isoflurane and immunized intranasally with a 15 μl saline solution containing 1 or 10 μg peptide and 1 μg cholera toxin (List Biological Laboratory, Campbell, CA). Mice were immunized on days 0, 7, and 21, and challenged with Y. pestis strain D27 on day 37 or day 56.
Challenge infections
Mice were lightly anesthetized by isoflurane and infected intranasally with 20 or 200 median lethal doses (MLD) Y. pestis strain D27 in 30 μl saline. The intranasal MLD of Y. pestis strain D27 is approximately 1×104 CFU when the bacteria are grown and administered as described above. For all survival studies, recumbent mice were considered moribund and euthanized. For measurement of bacterial burden, mice were euthanized at the indicated day after infection. Liver and lung tissues were harvested and plated for CFU determination as described previously (19).
CD8 T cell depletion
CD8 T cells were depleted by treating mice with 1 mg rat IgG2b mAb specific for mouse CD8 (clone 2.43). The mAb was diluted in saline and administered as two intraperitoneal doses of 500 μg each on the day before and the day after challenge. Control mice received an equal quantity of isotype-matched rat IgG2b mAb (clone LTF-2). All mAbs were supplied by Bio X Cell (West Lebanon, NH).
Statistics
Statistical analyses were performed using the program Prism 4.0 (GraphPad Software, Inc.). Survival data were analyzed by log rank tests and cell numbers/percentages were analyzed by students t-test. Bacterial burden was analyzed by nonparametric test (Kruskal-Wallis); CFU less than the detection limit of our assay were assigned values 0.2 log below the detection limit.
Results
YopEis a dominant antigen recognized by the Y. pestis-specific CD8 T cells
To facilitate the identification of Y. pestis antigens recognized by protective T cells, we generated T cell clones from C57BL/6 mice that were immunized with live attenuated Y. pestis strain D27-pLpxL and then challenged 2 and 3 months later with virulent Y. pestis strain D27. After another month, the T cells from surviving mice were expanded in vitro using splenocytes as APC and heat-killed Y. pestis strain D27 as a source of antigen. The resulting T cell line was cloned by limiting dilution.
To assess the antigen specificity of the T cell clones, we measured their response to various Y. pestis strains. The strains included D27, a pgm-deficient member of biovar Mediavalis; D28, a derivative of D27 that lacks the pCD1 virulence plasmid; and CO92 pgm-, a pgm-deficient member of biovar Orientalis. All of the CD8 T cell clones produced IFNγ and proliferated in response to strains D27 and CO92 pgm-, but not to strain D28, suggesting that they recognized a pCD1-dependent antigen (Figure 1A). Since temperature regulates the expression of many Y. pestis genes (20), we also grew each strain at either 26°C or 37°C before assaying T cell responses. The CD8 T cell clones only produced IFNγ and proliferated in response to Y. pestis bacilli grown at 37°C (Figure 1A). We also observed that supplementing cultures with antibodies reactive with mouse MHC class I H2-Kb, but not H2-Db, suppressed the response to strain D27 (data not shown). Together, these findings indicated that the CD8 T cell clones recognize a temperature-regulated, H2-Kb-restricted antigen encoded by, or dependent upon, the pCD1 plasmid.
Figure 1. Y. pestis-specific CD8 T cell clones recognizes YopE69-77.

(A) CD8 T cell clone YP-39 was assayed for responsiveness to heat-killed E. coli and Y. pestis strains grown at either 26°C or 37°C. The tested strains included D27 (biovar Mediavalis); D28, a D27 derivative lacking the pCD1 plasmid; and CO92 pgm- (biovar Orientalis). The clone specifically produced IFNγ (as measured by ELISA) and proliferated (as measured by incorporation of [3H]-thymidine) in response to pCD1+ Y. pestis strains grown at 37°C. SI; stimulation index. (B) CD8 T cell clone YP-39 was assayed for responsiveness to Y. pestis strain D27 and individual E. coli strains induced to express the indicated pCD1-encoded proteins. The clone responded specifically to an E. coli strain expressing Y. pestis YopE. (C) CD8 T cell clone YP-60 was assayed for responsiveness to 42 individual Y. pestis YopE peptides at a concentration of 100 nM. Each 15 amino acid peptide overlaps its neighbors sequence by 10 amino acids. The T cell clone responded specifically to YopE peptide #14, which contains amino acids 66–80 (i.e. YopE66-80; H2N-VAHSVIGFIQRMFSE-OH). (D) CD8 T cell clone YP-60 was assayed for responsiveness to Y. pestis YopE66-80, YopE69-77 (H2N-SVIGFIQRM-OH), YopE69-76 (H2N-SVIGFIQR-OH) and a homologous peptide from Y. enterocolitica (Yent YopE66-80; H2N-MARSAIEFIKRMFSE-OH). Each peptide was assayed at concentrations of 100 and 10 nM. The clone specifically recognized Y. pestis YopE66-80 and YopE69-77.
To identify the protein antigen, we measured the response of the CD8 T cell clones to individual pCD1-encoded proteins expressed in E. coli. We focused initially on proteins whose expression is known to be up-regulated when Y. pestis bacilli are grown at 37°C (20). As shown in Figure 1B, the CD8 T cell clones responded specifically to E. coli expressing Y. pestis YopE. To decisively demonstrate that the clones recognized YopE and to identify the specific epitope(s) recognized by the clones, we synthesized overlapping peptides encompassing the entire sequence of the Y. pestis YopE protein. Each peptide was 15 amino acids in length and overlapped its neighboring peptides by 10 amino acids. The CD8 T cell clones all responded specifically to YopE66-80 (VAHSVIGFIQRMFSE) (Figure 1C). We used computer algorithms to predict peptides within YopE66-80 that were likely to bind H2-Kb. As shown in Figure 1D, the CD8 T cell clones responded to YopE69-77 (SVIGFIQRM) but not YopE69-76 (SVIGFIQR), and the response to YopE69-77 remained strong at very low concentrations (10 nM). Notably, the CD8 T cell clones did not recognize YopE66-80 of Y. enterocolitica strain WA:08 (MARSAIEFIKRMFSE), which contains multiple sequence substitutions between amino acids 69 and 77 (Figure 1D). Together, these results established that the CD8 T cell clones specifically recognized YopE69-77.
To identify and quantify YopE-specific CD8 T cells in vivo, we generated an MHC class I H2-Kb tetramer containing the Y. pestis YopE69-77 peptide (KbYopE69-77). While this tetramer stained very few cells in naïve mice, we detected a small but readily measurable increase in the percentage of tetramer-positive CD8 T cells in the lungs, spleens and peripheral blood of mice that had been immunized 3 months earlier with attenuated Y. pestis strain D27-pLpxL (Figure 2A). Moreover, the percentage and number of tetramer-positive CD8 T cells in the lung increased dramatically when the immunized mice were challenged intranasally with virulent strain D27. By day 4 after challenge, nearly 20% of pulmonary CD8 T cells stained positive for the tetramer (Figure 2A).
Figure 2. Detection of YopE69-77-specific CD8 T cells in vaccinated mice challenged with Y. pestis.

C57BL/6 mice were immunized twice with attenuated Y. pestis strain D27-pLpxL and then challenged intranasally with 200 MLD virulent Y. pestis strain D27 three months after the last immunization. At day 4 after challenge, cells were analyzed by flow cytometry. (A) Lung cells, peripheral blood leukocytes (PBL) and splenocytes were stained immediately with antibodies to CD4 and CD8 and an MHC class I H2-Kb tetramer loaded with YopE69-77 (KbYopE69-77). Plots are representative of five individual mice for each condition. The percentages of lymphocytes staining positive for CD8 and tetramer are shown, with parentheses depicting the percentage of tetramer-positive cells within the CD8 T cell population. YopE tetramer-positive CD8 T cells were detectable in the lungs, blood and spleens of vaccinated mice and their frequency increased dramatically in the lung after Y. pestis challenge. (B) Lung cells were stimulated ex vivo with plate-bound anti-CD3 mAb, Y. pestis YopE69-77 peptide or control peptide OVA257-264 and then stained for intracellular cytokines. Data depict results from one representative mouse that was vaccinated and challenged. The numbers depict the percentage of cells in the indicated quadrant. The YopE69-77 peptide specifically stimulated CD8 T cells to produce TNFα and IFNγ.
The specificity of CD8 T cells for YopE69-77 was further confirmed by intracellular cytokine staining. After challenging the immunized mice with virulent Y. pestis, 11% of CD8 cells in the lung could be induced to produce TNFα and IFNγ upon ex vivo stimulation with anti-CD3 mAb, a polyclonal stimulus for all effector T cells. Remarkably, nearly the same percentage of CD8 cells could be induced to produce these cytokines by culture with YopE69-77, whereas few CD8 T cells responded to a control peptide, OVA257-264 (Figure 2B). CD4 T cells did not respond to YopE69-77, further confirming the specificity of the CD8 T cell response to YopE. Together, these data indicate that YopE69-77 is a dominant antigen recognized by CD8 T cells in this C57BL/6 mouse model of pneumonic plague.
Immunization with YopE69-77 primes CD8 T cells that confer protection against pulmonary Y. pestis infection
To assess whether YopE-specific CD8 T cells could suffice to protect against Y. pestis infection, we vaccinated mice with the YopE69-77 epitope. Initially, we employed a vaccination strategy in which bone marrow-derived dendritic cells (DC) were pulsed with peptide and injected intravenously into mice. Specifically, C57BL/6 mice were vaccinated with DC that had been pulsed with either YopE69-77 or OVA257-264. Two weeks later, we confirmed that the DC vaccination primed peptide-specific T cells by measuring the frequency of cells in lung, spleen and peripheral blood that stained with MHC class I tetramers. After vaccination with OVA257- 264-pulsed DC, less than 1% of the lung CD8 T cells stained positive for the KbYopE69-77 tetramer (Figure 3A). In contrast, more than 20% of lung CD8 T cells stained with KbYopE69-77 tetramer after vaccination with YopE69-77-pulsed DC. Vaccination with YopE69-77-pulsed DC also increased the frequency of YopE69-77-specific CD8 T cells significantly in the spleen and peripheral blood, and increased the absolute number of these cells more than 1000-fold in the lung (Figure 3A). In parallel with the tetramer analysis, additional cohorts of vaccinated mice were challenged intranasally with 20 MLD of Y. pestis strain D27. In comparison with mice vaccinated with OVA257-264, mice vaccinated with YopE69-77 displayed modest but significant protection (p<0.0001), as evidenced by both a 2-day prolongation in median survival time and an increase in the overall survival rate to 25% (Figure 3B).
Figure 3. Vaccination with dendritic cells loaded with YopE69-77 peptide primes CD8 T cells and protects against lethal pulmonary Y. pestis challenge.

C57BL/6 mice were immunized intravenously with bone marrow-derived DC loaded with YopE69-77 (closed circles) or control OVA257-264 peptides (open circles). (A) On day 14 after DC transfer, lung cells were stained with antibodies to CD4 and CD8 and MHC class I tetramers KbYopE69-77. The left panel shows the percentages of lymphocytes staining positive for CD8 and KbYopE69-77 tetramer in lung (left axis), spleen (right axis) or PBL (right axis) and the right panel shows the numbers of these cells in lung and spleen at day 14 after DC transfer. The data depict the mean and SD of 5 mice per group. Vaccination significantly increased the frequency and number of CD8 T cells specific for YopE69-77 (* p<0.0001). (B) At day 14 after DC transfer, mice were challenged intranasally with 20 MLD Y. pestis strain D27. In comparison with mice immunized with DC loaded with OVA257-264 peptide, mice that received DC loaded with YopE69-77 peptide displayed significantly improved survival (p<0.0001 by log rank test; n=37 mice/group; data pooled from 3 independent experiments).
To investigate whether the protection conferred by immunization with YopE69-77 could be improved further, we immunized mice intranasally with 10 μg YopE69-77 and cholera toxin (CT), a mucosal adjuvant known to induce peptide-specific CD8 T cell responses (21). Control mice received CT adjuvant alone or CT and OVA257-264. Two weeks after the last of three immunizations, more than 50% of the lung CD8 T cells stained specifically with the KbYopE69-77 tetramer (Figure 4A). The absolute number of YopE69-77-specific CD8 T cells in the lung increased more than 1000-fold after immunization (Figure 4A). When challenged intranasally with 20 MLD Y. pestis strain D27, the YopE69-77-immunized mice demonstrated remarkably improved protection (p<0.0001), with 83% surviving the lethal challenge (Figure 4B). Parallel cohorts of mice vaccinated with 10-fold less YopE69-77 (i.e. 1 μg) showed fewer tetramer-positive cells but still displayed robust protection (75%, Figure 4B). When mice were challenged 35 days after the last immunization, lower percentages and numbers of YopE-specific CD8 T cells were measured (Figure 4C) but similar levels of protection were observed (Figure 4D), suggesting that a memory response to YopE69-77 can suffice to protect against Y. pestis infection.
Figure 4. Intranasal immunization with YopE69-77 confers protection against lethal pulmonary Y. pestis challenge.
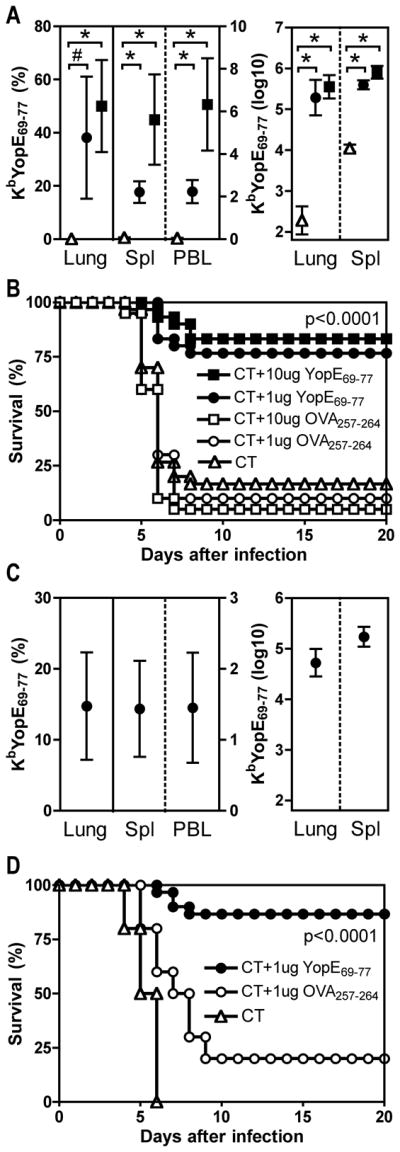
On days 0, 7 and 21, C57BL/6 mice were immunized intranasally with either 1 μg (closed circles) or 10 μg (closed squares) YopE69-77 mixed with cholera toxin (CT) adjuvant. Control mice received CT alone (open triangles), 1 μg (open circles) or 10 μg (open squares) OVA257-264 mixed with CT. Mice were either harvested without challenge (A and C) or challenged with Y. pestis (B and D) at day 37 (A and B) or day 56 (C and D). (A) The left panel shows the percentages of lymphocytes staining positive for CD8 and KbYopE69-77 tetramer in lung (left axis), spleen (right axis) or PBL (right axis) and the right panel shows the numbers of these cells in lung and spleen at day 37. The data depict the mean and SD of 6–8 mice per group. In comparison with CT alone, both doses of YopE69-77 significantly primed tetramer-positive cells (# p<0.01; * p<0.0001). (B) On day 37, parallel cohorts of mice were challenged intranasally with 20 MLD Y. pestis strain D27. In comparison with mice immunized with CT alone or CT and OVA257-264, mice immunized with CT and YopE69-77 displayed significantly increased survival (p<0.0001 for both 10 and for 1 μg by log rank test; n=30 mice for CT alone and YopE69-77 groups; n=20 for OVA257-264 groups). Data is pooled from 3 independent experiments. (C) The left panel shows the percentages of lymphocytes staining positive for CD8 and KbYopE69-77 tetramer in lung (left axis), spleen (right axis) or PBL (right axis) and the right panel shows the numbers of these cells in lung and spleen at day 56. The data depict the mean and SD of 10 mice per group. (D) On day 56, parallel cohorts of mice were challenged intranasally with 20 MLD Y. pestis strain D27. In comparison with mice immunized with CT alone or CT and OVA257-264, mice immunized with CT and YopE69-77 displayed significantly increased survival (p<0.0001 by log rank test; n=30 mice for YopE69-77 group; n=10 for CT alone and OVA257-264 groups). Data is pooled from 2 independent experiments.
The YopE-specific T cells in the mice immunized 35 days earlier with YopE69-77 and CT possessed a memory/effector phenotype; almost all of the YopE-specific CD8 T cells in the lung and spleen were CD44highCD62Llow and many of them also expressed CD43 (data not shown). In the spleen, very few of the YopE-specific cells expressed the classical early activation marker CD69, consistent with a resting memory phenotype. Interestingly, more than half of the YopE-specific cells in the lung expressed CD69 (data not shown), consistent with prior observations that lung airway memory CD8 T cells possess a highly activated phenotype (CD44highCD62LlowCD43+CD69+) long after viral clearance (22, 23).
Measurements of bacterial CFU revealed that YopE69-77 immunization helps mice control bacterial burden. Mice immunized with CT and YopE69-77 exhibited significantly reduced numbers of bacterial CFU in both lung and liver tissues at days 3, 4 and 5 after Y. pestis challenge, as compared with control mice immunized with CT alone or CT and OVA257-264 (Figure 5, p<0.05 for days 3 and 5 and p<0.01 for day 4). The burden had fallen below the detection limit of our assay in most of the mice immunized with YopE69-77 by day 7 after challenge (8 of 10 mice for lung; 6 of 10 mice for liver), whereas the control mice had succumbed to infection by that time.
Figure 5. Intranasal immunization with YopE69-77 decreases bacterial burden after pulmonary Y. pestis challenge.

On days 0, 7 and 21, C57BL/6 mice were immunized intranasally with 1 μg YopE69-77 mixed with CT adjuvant (closed circles). Control mice received CT alone (open triangles) or 1 μg OVA257-264 (open circles) mixed with CT. On day 37, mice were challenged intranasally with 20 MLD Y. pestis strain D27, and the bacterial burden in lung (A) and liver (B) tissues was measured on the indicated days after infection. In comparison with mice immunized with CT alone or CT and OVA257-264, mice immunized with CT and YopE69-77 exhibited significantly reduced bacterial burden in both tissues at days 3, 4 and 5 (*p<0.05; **p<0.01 by Kruskal-Wallis with Dunn’s multiple-comparison test). Data shown are median and interquartile range of 10 mice per group per time point, except many control animals had succumbed to infection by day 5 so only 3–4 mice per group were analyzed for CT alone and CT and OVA257-264 on day 5. The broken line depicts the limit of detection.
Depleting CD8 T cells at the time of challenge fully abrogated the protective effect of immunization (Figure 6A), further confirming that CD8 T cells mediated the protection. The protection conferred by immunization with YopE69-77 did not require antibody, as it was also evident in B cell-deficient μMT mice (Figure 6B). Taken together, these findings demonstrate that YopE is not only a dominant CD8 T cell epitope of natural Y. pestis infection but also a protective antigen in the C57BL/6 mouse model of pneumonic plague.
Figure 6. Protection conferred by intranasal immunization with YopE69-77 is mediated by CD8 T cells.

On days 0, 7 and 21, C57BL/6 wild type (A) or μMT (B) mice were immunized intranasally with either 10 μg (closed squares) or 1 μg (closed circles) YopE69-77 mixed with CT adjuvant. Control mice received CT alone (open triangles) or 10 μg OVA257-264 mixed with CT (open squares). On day 37, mice were challenged intranasally with 20 MLD Y. pestis strain D27. (A) At the time of challenge, the wild type mice that were immunized with CT and 10 μg YopE69-77 were subdivided into two groups. One group of mice was treated with depleting mAb specific for CD8 (aCD8, closed triangles) and the other was treated with an isotype-matched mAb (ctrl Ig, closed squares). Mice treated with mAb specific for CD8 displayed significantly reduced survival in compared with mice treated with control mAb (p<0.0001 by log rank test; n=20 mice/group). Data is pooled from 2 independent experiments. (B) In comparison with μMT mice immunized with CT alone, μMT mice immunized with CT and YopE69-77 displayed significantly increased survival (p<0.0001 for both 10 and for 1 μg by log rank test; n=20 mice/group). Data is pooled from 2 independent experiments.
Discussion
Y. pestis, Y. pseudotuberculosis, and Y. enterocolitica, the three Yersinia species that cause human disease, all descend from a common ancestor (8). The sequence of YopE is identical in most strains of Y. pestis and Y. pseudotuberculosis. Although the YopE69-77 sequence is altered in the 0:8 serotype of Y. enterocolitica, it is conserved in many other Y. enterocolitica strains. Thus, we anticipate that Y. pestis YopE69-77 will be a dominant and protective CD8 T cell epitope in many H2-Kb mouse models of disease caused by pathogenic Yersinia spp.
YopE was never previously shown to serve as a protective antigen for any Yersinia spp. One prior study of mutant Y. pseudotuberculosis strains suggested that YopE may be a target of rat CD8 T cells (24). However, subsequent mouse studies reported that immunization with YopE does not confer significant protection from plague (25, 26). Moreover, an ELISPOT-based screen using recombinant proteins did not identify YopE as an antigen recognized by T cells in BALB/c mice immunized with attenuated Y. pestis EV76 (27). The methods used in these prior studies differed significantly from those reported here, which focused specifically on antigens recognized by CD8 T cells and immunization strategies known to prime CD8 T cells. Further studies are required to determine whether Yersinia YopE is a dominant antigen recognized by CD8 T cells in all mammals, or only subsets thereof.
The extent to which the biology of Y. pestis YopE accounts for its immunodominance in the C57BL/6 mouse model of plague is presently unclear. Yersinia spp. use a plasmid-encoded type III secretion system to translocate YopE into the cytosol of eukaryotic cells (28), where it functions as a GTPase-activating protein (29). A series of studies have established that attenuated strains of Yersinia and Salmonella can translocate recombinant YopE-fusion proteins, thereby priming CD8 T cell responses to heterologous antigens (30-33). Although these prior studies used YopE as a molecular carrier to deliver heterologous antigens and did not report that YopE itself was an antigen, they suggest that translocation facilitates the presentation of antigens to CD8 T cells. However, our data indicate that heat-killed Y. pestis bacilli, which presumably lack the capacity to translocate YopE, also effectively deliver YopE69-77 to APC (Figure 1). In addition, several epitope prediction algorithms predicted that many peptides encoded by other translocated Yops should bind H2-Kb far more effectively than YopE69-77. Indeed, initially we attempted, but failed, to identify the CD8 T cell antigen by screening 90 high-scoring pCD1-encoded peptides predicted by the BIMAS and SYFPEITHI algorithms. Clearly, further studies are required to understand why YopE69-77 dominates the CD8 T cell response to plague.
Wang et al recently identified epitopes recognized by CD8 T cells in Balb/c mice immunized with a DNA vaccine encoding LcrV, a leading vaccine candidate (34). They also demonstrated that CD8 T cells may play a role in protection elicited by LcrV-based vaccines. However, Wang et al did not investigate whether LcrV is a dominant T cell antigen during Y. pestis infection. Our study decisively demonstrates that YopE69-77 is a dominant antigen recognized by Y. pestis-specific CD8 T cells in C57BL/6 mice immunized with live attenuated Y. pestis. Together, these two studies significantly increase the research utility of the mouse models of pneumonic plague; now, it is possible to apply sensitive, quantitative, pathogen-specific assays (e.g. tetramers, ELISPOTs, limiting dilution) to the study of anti-plague T cell responses and the development of vaccines incorporating cell-mediated defense. These T cell assays will complement existing tools and assays based on knowledge of the dominant antibody targets, F1 and LcrV. Moreover, the identification of YopE as an antigen recognized by CD8 T cells will permit direct assessments of the extent to which the efficacy of subunit vaccines can be improved by incorporating dominant targets for both antibodies and T cells.
We know remarkably little about the function of T cells during septic bacterial pneumonias and the capacities of vaccine-primed T cells to prevent or ameliorate disease in that setting. Protective epitopes recognized by CD8 T cells have been identified for a number of viral pulmonary pathogens. However, to our knowledge, this study reports the first protective CD8 epitope for any acutely lethal bacterial pneumonia. Given that bacterial infections of the lung are a leading cause of death worldwide, and given growing concerns about antibiotic resistant and weaponized bacteria, further studies of the mouse model of pneumonic plague could lead to improved vaccines and immunotherapeutics not only for Y. pestis but also for the many other bacterial pathogens that infect the lung.
Acknowledgments
We thank Drs. Eric Pamer, David Woodland, and Herman Staats for helpful discussions; Drs. Robert Brubaker and James Bliska for bacterial strains; Drs. Egil Lien and John Goguen for the pLpxL plasmid; Ms. Debra Duso for technical assistance; the Trudeau Institute Molecular Biology Core Facility for MHC class I tetramers, and the dedicated staff of the Trudeau Institute Animal facilities for the expert care of mice. The Gateway library was obtained through NIAID’s Pathogen Functional Genomics Resource Center, managed and funded by Division of Microbiology and Infectious Diseases, NIAID, NIH, DHHS and operated by the J. Craig Venter Institute.
Abbreviations
- CT
cholera toxin
- MLD
median lethal dose
Footnotes
This work was supported by funding from Trudeau Institute and Public Health Service grants R01-AI061577 and R01-AI071295.
References
- 1.Anevlavis S, Bouros D. Community acquired bacterial pneumonia. Expert Opin Pharmacother. 2010;11:361–374. doi: 10.1517/14656560903508770. [DOI] [PubMed] [Google Scholar]
- 2.Peleg AY, Hooper DC. Hospital-acquired infections due to gram-negative bacteria. N Engl J Med. 2010;362:1804–1813. doi: 10.1056/NEJMra0904124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casadevall A, Pirofski LA. A reappraisal of humoral immunity based on mechanisms of antibody-mediated protection against intracellular pathogens. Adv Immunol. 2006;91:1–44. doi: 10.1016/S0065-2776(06)91001-3. [DOI] [PubMed] [Google Scholar]
- 4.Perry RD, Fetherston JD. Yersinia pestis-etiologic agent of plague. Clin Microbiol Rev. 1997;10:35–66. doi: 10.1128/cmr.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brubaker RR. Yersinia pestis and bubonic plague. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandte E, editors. The prokaryotes, an evolving electronic resource for the microbiological community. Springer-Verlag; New York: 2000. [Google Scholar]
- 6.Inglesby TV, Dennis DT, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Koerner JF, Layton M, McDade J, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Schoch-Spana M, Tonat K. Plague as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA. 2000;283:2281–2290. doi: 10.1001/jama.283.17.2281. [DOI] [PubMed] [Google Scholar]
- 7.Galimand M, Guiyoule A, Gerbaud G, Rasoamanana B, Chanteau S, Carniel E, Courvalin P. Multidrug resistance in Yersinia pestis mediated by a transferable plasmid. N Engl J Med. 1997;337:677–680. doi: 10.1056/NEJM199709043371004. [DOI] [PubMed] [Google Scholar]
- 8.Wren BW. The yersiniae--a model genus to study the rapid evolution of bacterial pathogens. Nat Rev Microbiol. 2003;1:55–64. doi: 10.1038/nrmicro730. [DOI] [PubMed] [Google Scholar]
- 9.Russell P, Eley SM, Hibbs SE, Manchee RJ, Stagg AJ, Titball RW. A comparison of Plague vaccine, USP and EV76 vaccine induced protection against Yersinia pestis in a murine model. Vaccine. 1995;13:1551–1556. doi: 10.1016/0264-410x(95)00090-n. [DOI] [PubMed] [Google Scholar]
- 10.Szaba FM, Kummer LW, Wilhelm LB, Lin JS, Parent MA, Montminy-Paquette SW, Lien E, Johnson LL, Smiley ST. D27-pLpxL, an avirulent strain of Yersinia pestis, primes T cells that protect against pneumonic plague. Infect Immun. 2009;77:4295–4304. doi: 10.1128/IAI.00273-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feodorova VA, Corbel MJ. Prospects for new plague vaccines. Expert Rev Vaccines. 2009;8:1721–1738. doi: 10.1586/erv.09.129. [DOI] [PubMed] [Google Scholar]
- 12.Meyer KF, Cavanaugh DC, Bartelloni PJ, Marshall JD., Jr Plague immunization. I. Past and present trends. J Infect Dis. 1974;129(Suppl):S13–18. doi: 10.1093/infdis/129.supplement_1.s13. [DOI] [PubMed] [Google Scholar]
- 13.Welkos S, Pitt ML, Martinez M, Friedlander A, Vogel P, Tammariello R. Determination of the virulence of the pigmentation-deficient and pigmentation-/plasminogen activator-deficient strains of Yersinia pestis in non-human primate and mouse models of pneumonic plague. Vaccine. 2002;20:2206–2214. doi: 10.1016/s0264-410x(02)00119-6. [DOI] [PubMed] [Google Scholar]
- 14.Smiley ST. Current challenges in the development of vaccines for pneumonic plague. Expert Rev Vaccines. 2008;7:209–221. doi: 10.1586/14760584.7.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bashaw J, Norris S, Weeks S, Trevino S, Adamovicz JJ, Welkos S. Development of in vitro correlate assays of immunity to infection with Yersinia pestis. Clin Vaccine Immunol. 2007;14:605–616. doi: 10.1128/CVI.00398-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elvin SJ, Williamson ED. Stat 4 but not Stat 6 mediated immune mechanisms are essential in protection against plague. Microb Pathog. 2004;37:177–184. doi: 10.1016/j.micpath.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 17.Smiley ST. Immune defense against pneumonic plague. Immunol Rev. 2008;225:256–271. doi: 10.1111/j.1600-065X.2008.00674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin JS, Park S, Adamovicz JJ, Hill J, Bliska JB, Cote CK, Perlin DS, Amemiya K, Smiley ST. TNFalpha and IFNgamma contribute to F1/LcrV-targeted immune defense in mouse models of fully virulent pneumonic plague. Vaccine. 2010;29:357–362. doi: 10.1016/j.vaccine.2010.08.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin JS, Kummer LW, Szaba FM, Smiley ST. IL-17 contributes to cell-mediated defense against pulmonary Yersinia pestis infection. J Immunol. 2011;186:1675–1684. doi: 10.4049/jimmunol.1003303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motin VL, Georgescu AM, Fitch JP, Gu PP, Nelson DO, Mabery SL, Garnham JB, Sokhansanj BA, Ott LL, Coleman MA, Elliott JM, Kegelmeyer LM, Wyrobek AJ, Slezak TR, Brubaker RR, Garcia E. Temporal global changes in gene expression during temperature transition in Yersinia pestis. J Bacteriol. 2004;186:6298–6305. doi: 10.1128/JB.186.18.6298-6305.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porgador A, Staats HF, Faiola B, Gilboa E, Palker TJ. Intranasal immunization with CTL epitope peptides from HIV-1 or ovalbumin and the mucosal adjuvant cholera toxin induces peptide-specific CTLs and protection against tumor development in vivo. J Immunol. 1997;158:834–841. [PubMed] [Google Scholar]
- 22.Hikono H, Kohlmeier JE, Ely KH, Scott I, Roberts AD, Blackman MA, Woodland DL. T-cell memory and recall responses to respiratory virus infections. Immunol Rev. 2006;211:119–132. doi: 10.1111/j.0105-2896.2006.00385.x. [DOI] [PubMed] [Google Scholar]
- 23.Hogan RJ, Usherwood EJ, Zhong W, Roberts AA, Dutton RW, Harmsen AG, Woodland DL. Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J Immunol. 2001;166:1813–1822. doi: 10.4049/jimmunol.166.3.1813. [DOI] [PubMed] [Google Scholar]
- 24.Falgarone G, Blanchard HS, Virecoulon F, Simonet M, Breban M. Coordinate involvement of invasin and Yop proteins in a Yersinia pseudotuberculosis-specific class I-restricted cytotoxic T cell-mediated response. J Immunol. 1999;162:2875–2883. [PubMed] [Google Scholar]
- 25.Leary SE, Griffin KF, Galyov EE, Hewer J, Williamson ED, Holmstrom A, Forsberg A, Titball RW. Yersinia outer proteins (YOPS) E, K and N are antigenic but non-protective compared to V antigen, in a murine model of bubonic plague. Microb Pathog. 1999;26:159–169. doi: 10.1006/mpat.1998.0261. [DOI] [PubMed] [Google Scholar]
- 26.Andrews GP, Strachan ST, Benner GE, Sample AK, Anderson GW, Jr, Adamovicz JJ, Welkos SL, Pullen JK, Friedlander AM. Protective efficacy of recombinant Yersinia outer proteins against bubonic plague caused by encapsulated and nonencapsulated Yersinia pestis. Infect Immun. 1999;67:1533–1537. doi: 10.1128/iai.67.3.1533-1537.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li B, Zhou L, Guo J, Wang X, Ni B, Ke Y, Zhu Z, Guo Z, Yang R. High-throughput identification of new protective antigens from a Yersinia pestis live vaccine by enzyme-linked immunospot assay. Infect Immun. 2009;77:4356–4361. doi: 10.1128/IAI.00242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosqvist R, Magnusson KE, Wolf-Watz H. Target cell contact triggers expression and polarized transfer of Yersinia YopE cytotoxin into mammalian cells. EMBO J. 1994;13:964–972. doi: 10.1002/j.1460-2075.1994.tb06341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viboud GI, Bliska JB. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol. 2005;59:69–89. doi: 10.1146/annurev.micro.59.030804.121320. [DOI] [PubMed] [Google Scholar]
- 30.Russmann H, Weissmuller A, Geginat G, Igwe EI, Roggenkamp A, Bubert A, Goebel W, Hof H, Heesemann J. Yersinia enterocolitica-mediated translocation of defined fusion proteins to the cytosol of mammalian cells results in peptide-specific MHC class I-restricted antigen presentation. Eur J Immunol. 2000;30:1375–1384. doi: 10.1002/(SICI)1521-4141(200005)30:5<1375::AID-IMMU1375>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 31.Russmann H, Igwe EI, Sauer J, Hardt WD, Bubert A, Geginat G. Protection against murine listeriosis by oral vaccination with recombinant Salmonella expressing hybrid Yersinia type III proteins. J Immunol. 2001;167:357–365. doi: 10.4049/jimmunol.167.1.357. [DOI] [PubMed] [Google Scholar]
- 32.Wiedig CA, Kramer U, Garbom S, Wolf-Watz H, Autenrieth IB. Induction of CD8+ T cell responses by Yersinia vaccine carrier strains. Vaccine. 2005;23:4984– 4998. doi: 10.1016/j.vaccine.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 33.Trulzsch K, Sporleder T, Leibiger R, Russmann H, Heesemann J. Yersinia as oral live carrier vaccine: influence of Yersinia outer proteins (Yops) on the T-cell response. Int J Med Microbiol. 2008;298:59–67. doi: 10.1016/j.ijmm.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 34.Wang S, Goguen JD, Li F, Lu S. Involvement of CD8+ T cell-mediated immune responses in LcrV DNA vaccine induced protection against lethal Yersinia pestis challenge. Vaccine. 2011 doi: 10.1016/j.vaccine.2010.12.062. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]