

Abstract
Exposure of alcohols 2a-2j to 2-silyl-butadienes in the presence of ruthenium complexes modified by (R)-SEGPHOS or (R)-DM-SEGPHOS results in redox-triggered generation of allylruthenium-aldehyde pairs, which combine to form products of carbonyl crotylation 4a-4j in the absence of stoichiometric byproducts and with high levels of syn-diastereo- and enantioselectivity. In the presence of isopropanol under otherwise identical conditions, aldehydes 3a-3j are converted to an equivalent set of adducts 4a-4j. Whereas reactions conducted using conventional heating require 48 hours, microwave irradiation enables full conversion in only 4 hours. Finally, as illustrated in the conversion of adduct 4a to compounds 6a and 6b, diastereoselective hydroboration-Suzuki cross-coupling with aryl and vinyl halides followed by Fleming-Tamao oxidation enables generation of anti,syn-stereotriads found in numerous polyketide natural products.
Introduction
Roughly 20% of top-selling small molecule therapeutic agents are polyketides,1,2 and it is estimated that polyketides are five times more likely to possess useful drug activity compared to other families of natural products.3 Among methods for polyketide construction,4 the addition of chirally modified crotylmetal reagents to carbonyl compounds ranks as one of the foremost strategies used for the generation of polypropionate substructures.5-8 However, the most broadly utilized protocol of this type, Brown's crotylation,5b,c generates superstoichiometric quantities of a secondary alcohol byproduct, isopinocampheol, which frequently complicates product isolation and has prevented implementation of this technology at the process level.9 Consequently, efforts toward asymmetric carbonyl crotylation protocols continue unabated.6-8
Recently, we reported a catalytic method for anti-diastereo- and enantioselective carbonyl crotylation from the alcohol or aldehyde oxidation level employing α-methyl allyl acetate as the crotyl donor.10c,d This transformation represents one among a broad, new class of catalytic C-C couplings in which hydrogen exchange between alcohols and π-unsaturated reactants triggers generation of electrophile-nucleophile pairs that combine to form products of carbonyl addition.11,12 Whereas chirally modified iridium complexes promote such transformations with excellent control of diastereo- and enantioselectivity,10,11c,d enantioselective ruthenium catalyzed processes have proven elusive.13 For example, in ruthenium catalyzed butadiene-alcohol C-C couplings, products of carbonyl crotylation appear as mixtures of syn- and anti-diastereomers.13a
The low levels of diastereoselectivity associated with ruthenium catalyzed crotylations are attributed to incomplete partitioning of (Z)- and (E)-σ-crotylruthenium intermediates, which react stereospecifically through closed transition structures to deliver syn- and anti-diastereomers, respectively. It was reasoned that 2-silyl-substituted butadienes, readily prepared from chloroprene, would enforce generation of “pseudo-(Z)-σ-crotylruthenium” isomers, potentially generating products of carbonyl syncrotylation.14 Here, we report that upon exposure of primary alcohols 2a-2j to 2-silyl-butadiene 1 in the presence of chirally modified ruthenium catalysts, products of carbonyl crotylation 4a-4j are formed with high levels of syn-diastereo- and enantioselectivity. Under related transfer hydrogenation conditions employing isopropanol as terminal reductant, aldehydes 3a-3j are converted to an equivalent set of carbonyl crotylation products 4a-4j with comparable levels of stereoselectivity (Figure 1).
Figure 1.

syn-Diastereo- and Enantioselective carbonyl crotylation in the absence of stoichiometric byproducts via ruthenium catalyzed hydrohydroxyalkylation of 2-silyl-butadienes
Results and Discussion
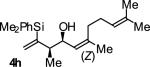
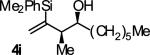
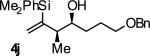
In an initial set of experiments, a range of 2-silyl substituted dienes were assayed for their ability to engage in efficient, syn- diastereoselective carbonyl crotylation from the alcohol oxidation level. Such dienes are conveniently prepared from the Grignard reagent derived from chloroprene, which itself is generated in situ from 3,4-dichloro-1-butene.15 It was found that upon exposure of the 2-silyl substituted butadiene 1 to aliphatic alcohol 2j in the presence of RuHCl(CO)(PPh3)3, rac-BINAP, in THF solvent at 95 °C, the desired product of syn-crotylation 4j was obtained as a single diastereomer in 25% yield. It should be noted that large alkyl substituents (tert-butyl and mesityl) at the 2-position of butadiene also enforce syn-diastereoselectivity. Encouraged by these results an assay of chiral ligands was undertaken. Remarkably, although a racemic background reaction is catalyzed by RuHCl(CO)(PPh3)3, the catalyst modified by (R)-DM-SEGPHOS [(R)-(+)-5,5-bis(di[3,5-xylyl]phosphino)-4,4-bi-1,3-benzodioxole] promotes the reaction in 34% yield, >20:1 dr and 84% ee. In an effort to exclude triphenylphosphine, and potentially eliminate a competing background reaction, the phosphine-free precatalyst RuCl2(CO)(cymene) was assayed in combination with (R)-DM-SEGPHOS. However, conversion was low. In toluene, a less Lewis basic solvent, using RuHCl(CO)(PPh3)3 as precatalyst, the yield of 2j was significantly improved (66% yield, >20:1 dr, 86% ee). Under these conditions, diene 1 was coupled to a diverse range of alcohols 2a-2j. The products of crotylation 4a-4j were formed in good yields and with excellent control of syn-diastereo- and enantioselectivity (Table 1).
Table 1.
syn-Diastereo- and enantioselective carbonyl crotylation from the alcohol oxidation level.a
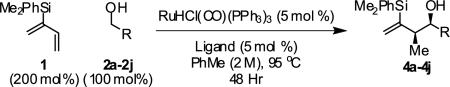 | |||||
|---|---|---|---|---|---|
| Entry | Product | Ligand | Yield [%] | ee [%] | syn:anti |
| 1 |
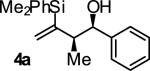
|
A | 81 | 87 | 13:1 |
| 2 |
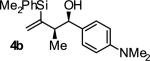
|
A | 87 | 90 | ≥20:1 |
| 3 |
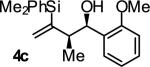
|
A | 84 | 90 | ≥20:1 |
| 4 |
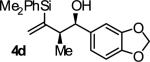
|
B | 85 | 90 | ≥20:1 |
| 5 |
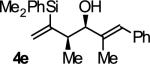
|
B | 70b | 90 | 11:1 |
| 6 |
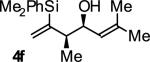
|
A | 67b,c | 90 | ≥20:1 |
| 7 |
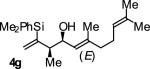
|
A | 70b | 92 | ≥20:1 |
| 8 |
|
A | 64b,c | 86 | ≥20:1 |
| 9 |
|
A | 65b,c,d | 88 | ≥20:1 |
| 10 |
|
A | 66 | 86 | ≥20:1 |
Ligand A = (R)-DM-SEGPHOS, Ligand B = (R)- SEGPHOS. Yields are of isolated material. Diastereoselectivity was determined through 1H NMR analysis of crude reaction mixtures. Enantiomeric excess was determined by chiral stationary phase HPLC analysis. See Supporting Information for details.
250 mol% 1.
THF
7 mol% catalyst.
Reactions conducted using conventional heating typically require 48 hours to reach full conversion. However, microwave irradiation promotes a dramatic increase in rate. For example, in a microwave reactor under otherwise standard conditions, the coupling of 2-silyl-butadiene 1 to alcohol 2d is complete in only 4 hours. The reaction product 4d is isolated in 88% yield with complete syn-diastereoselectivity (≥20:1 dr) and exceptional levels of enantioselectivity (93% ee) (eqn. 1).
 |
(eqn. 1) |
The reductive coupling of 2-silyl-butadiene 1 to aldehydes was attempted next. Toward this end, formic acid, 1,4-butanediol and isopropanol were assayed as terminal reductants under conditions otherwise identical to those established for reactions conducted from the alcohol oxidation level. Using isopropanol (200 mol%) as terminal reductant, diene 1 participates in highly regio-, diastereo- and enantioselective couplings to aldehydes 3a-3j to furnish an identical set of homoallylic alcohols 4a-4j in roughly equivalent isolated yields (Table 2).
Table 2.
syn-Diastereo- and enantioselective carbonyl crotylation from the aldehyde oxidation level.a
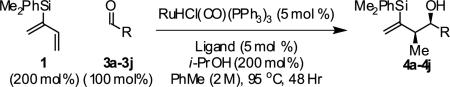 | |||||
|---|---|---|---|---|---|
| Entry | Product | Ligand | Yield [%] | ee [%] | syn:anti |
| 1 |
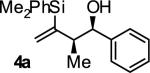
|
A | 76 | 88 | 12:1 |
| 2 |
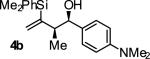
|
A | 80 | 93 | 19:1 |
| 3 |
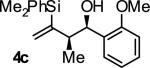
|
A | 71 | 88 | ≥20:1 |
| 4 |
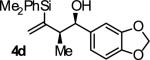
|
B | 91 | 90 | ≥20:1 |
| 5 |
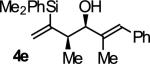
|
B | 75b | 93 | 11:1 |
| 6 |
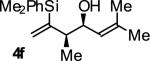
|
A | 56b,c | 90 | ≥20:1 |
| 7 |
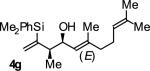
|
A | 71b | 91 | ≥20:1 |
| 8 |
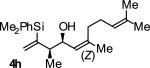
|
A | 66 | 84 | ≥20:1 |
| 9 |
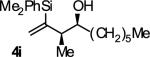
|
A | 53b,c,d,e | 84 | ≥20:1 |
| 10 |
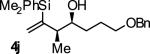
|
A | 50b,d,e | 84 | ≥20:1 |
Ligand A = (R)-DM-SEGPHOS, Ligand B = (R)- SEGPHOS. Yields are of isolated material. Diastereoselectivity was determined through 1H NMR analysis of crude reaction mixtures. Enantiomeric excess was determined by chiral stationary phase HPLC analysis. See Supporting Information for details.
250 mol% 1.
THF
7 mol% catalyst.
72 hours.
To illustrate the utility of the coupling products, compound 4a was converted to the silyl ether and subjected to diastereoselective hydroboration-Suzuki cross-coupling with aryl halides and vinyl halides to furnish adducts 5a and 5b, respectively.16 Protection of the hydroxyl moiety of 4a as the TBS is required to enforce high levels of diastereoselectivity (10:1 dr) in the hydroboration event. The benzyl ether derived from 4a displays low levels of diastereoselectivity (2:1 dr) upon hydroboration under identical conditions. Fleming-Tamao oxidation of the C-Si bond was especially challenging. However, upon exposure of the Suzuki coupling products 5a and 5b to Woerpel's modified conditions for oxidative C-Si bond cleavage,17 the diols 6a and 6b, which possess anti,syn-stereotriads found in numerous polyketide natural products, could be isolated in good yield (Scheme 1). Finally, if products of conventional syn-crotylation are desired, direct addition of TBAF to the reaction mixture enables protodesilylation in a convenient one-pot procedure, as demonstrated by the formation of 7a (eqn. 2).
 |
(eqn. 2) |
Scheme 1.

Diastereoselective hydroboration enables formation of anti,syn-stereotriads.
With regard to mechanism, the stoichiometric reaction of RuHCl(CO)(PPh3)3 with 1,2- and 1,3-dienes to form π-allyl complexes that have been characterized by single crystal x-ray diffraction and NMR, respectively, is known (Figure 2).18 Carbonyl addition by way of the σ-bound allylruthenium haptomer through a closed transition structure delivers the homoallylic ruthenium alkoxide, which upon substitution with a reactant alcohol provides a pentacoordinate ruthenium alkoxide. The vacant coordination site at this stage enables dehydrogenation to form an aldehyde and regenerate the ruthenium hydride to close the catalytic cycle (Scheme 2). The stereochemical outcome of the reaction may be predicted on the basis of the indicated model (Figure 2). Absolute and relative stereochemistry of adducts 4a-4j were assigned in analogy to 7a, which was compared to an authentic sample.7c
Figure 2.

Stereochemical model for 2-silyl-butadiene mediated syn-crotylation employing a (R)-DM-SEGPHOS modified ruthenium catalyst.a
Scheme 2.

Proposed catalytic mechanism for ruthenium catalyzed diene hydrohydroxymethylation as supported by established stoichiometric transformations.
Conclusion
In summary, exposure of alcohols 2a-2j to 2-silyl-butadiene 1 in the presence of the ruthenium catalyst obtained upon the combination of RuHCl(CO)(PPh3)3 and (R)-SEGPHOS or (R)-DM-SEGPHOS provides products of hydrohydroxyalkylation 4a-4j with complete regioselectivity and with good to excellent levels of diastereo- and enantioselectivity. In the presence of isopropanol, but under otherwise identical conditions, an equivalent set of adducts 4a-4j are generated in an equally selective fashion from aldehydes 3a-3j. In this way, catalytic syn-diastereo- and enantioselective carbonyl crotylation is achieved from the alcohol or aldehyde oxidation level. This carbonyl crotylation protocol circumvents stoichiometric byproducts and cryogenic conditions and does not require glove-box techniques, thus representing an important step toward the development of scalable methods for the construction of polyketide natural products. However, many unmet challenges remain, including the development of second generation catalysts that promote efficient, stereoselective couplings to α-chiral alcohols and aldehydes, and which enable related imine additions from the amine oxidation level. Future studies will address these goals.
Supplementary Material
Acknowledgment
The Robert A. Welch Foundation (F-0038), the NIH-NIGMS (RO1-GM069445) and the UT Austin, Center for Green Chemistry and Catalysis are acknowledged for financial support. The Natural Sciences and Engineering Research Council of Canada (NSERC) is acknowledged for generous postdoctoral support (J. M.).
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (1H NMR, 13C NMR, IR, HRMS). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected reviews on polyketide natural products, see: O'Hagan D. The Polyketide Metabolites. Ellis Horwood; Chichester, U.K.: 1991. Rimando AM, Baerson SR, editors. Polyketides Biosynthesis, Biological Activity, and Genetic Engineering. American Chemical Society; Washington, DC: 2007. (ACS Symposium Series 955).
- 2.a Newman DJ, Cragg GM. J. Nat. Prod. 2007;70:461. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]; b Newman DJ, Grothaus PG, Cragg GM. Chem. Rev. 2009;109:3012. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 3.Rohr J. Angew. Chem., Int. Ed. 2000;39:2847. doi: 10.1002/1521-3773(20000818)39:16<2847::aid-anie2847>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 4.For selected reviews on synthetic methods for polyketide construction, see: Paterson I, Doughty VA, Florence G, Gerlach K, McLeod MD, Scott JP, Trieselmann T. ACS Symp. Ser. 2001;783:195.Koskinen AMP, Karisalmi K. Chem. Soc. Rev. 2005;34:677. doi: 10.1039/b417466f.Yeung K-S, Paterson I. Chem. Rev. 2005;105:4237. doi: 10.1021/cr040614c.Schetter B, Mahrwald R. Angew. Chem., Int. Ed. 2006;45:7506. doi: 10.1002/anie.200602780.Morris JC, Nicholas GM, Phillips AJ. Nat. Prod. Rep. 2007;24:87. doi: 10.1039/b602832m.Paterson I. Total Synthesis of Polyketides using Asymmetric Aldol Reactions. In: Christmann M, Bräse S, editors. Asymmetric Synthesis. 2nd Edition Wiley-VCH Verlag GmbH & Co; Weinheim, Germany: 2008. pp. 293–298.Paterson I, Findlay AD. Aust. J. Chem. 2009;62:624.
- 5.For enantioselective carbonyl additions of chirally modified crotylmetal reagents, see: Hoffmann RW, Ladner W. Tetrahedron Lett. 1979;20:4653.Brown HC, Bhat KS. J. Am. Chem. Soc. 1986;108:293. doi: 10.1021/ja00279a042.Brown HC, Bhat KS. J. Am. Chem. Soc. 1986;108:5919. doi: 10.1021/ja00279a042.Roush WR, Halterman RL. J. Am. Chem. Soc. 1986;108:294.Roush WR, Ando K, Powers DB, Palkowitz AD, Halterman RL. J. Am. Chem. Soc. 1990;112:6339.Garcia J, Kim B-M, Masamune S. J. Org. Chem. 1987;52:4831.Burgos CH, Canales E, Matos K, Soderquist JA. J. Am. Chem. Soc. 2005;127:8044. doi: 10.1021/ja043612i.Riediker M, Duthaler RO. Angew. Chem., Int. Ed. Engl. 1989;28:494.Hafner A, Duthaler RO, Marti R, Rihs G, Rothe-Streit P, Schwarzenbach F. J. Am. Chem. Soc. 1992;114:2321.Panek JS, Yang M. J. Am. Chem. Soc. 1991;113:6594.Hu T, Takenaka N, Panek JS. J. Am. Chem. Soc. 2002;124:12806. doi: 10.1021/ja020853m.Hackman BM, Lombardi PJ, Leighton JL. Org. Lett. 2004;6:4375. doi: 10.1021/ol0480731.Kim H, Ho S, Leighton JL. J. Am. Chem. Soc. 2011;133:6517. doi: 10.1021/ja200712f.
- 6.For enantioselective Lewis acid catalyzed carbonyl additions of crotylmetal reagents, see: Furuta K, Mouri M, Yamamoto H. Synlett. 1991:561.Yanagasawa A, Ishiba A, Nakashima H, Yamamoto H. Synlett. 1997:88.Yanagasawa A, Kageyama H, Nakatsuka Y, Asakawa K, Matsumoto Y, Yamamoto H. Angew. Chem., Int. Ed. 1999;38:3701. doi: 10.1002/(sici)1521-3773(19991216)38:24<3701::aid-anie3701>3.0.co;2-d.Aoki S, Mikami K, Terada M, Nakai T. Tetrahedron Lett. 1993;49:1783.Motoyama Y, Okano M, Narusawa H, Makihara N, Aoki K, Nishiyama H. Organometallics. 2001;20:1580.Evans DA, Aye Y, Wu J. Org. Lett. 2006;8:2071. doi: 10.1021/ol0604771.Rauniyar V, Hall DG. Angew. Chem., Int. Ed. 2006;45:2426. doi: 10.1002/anie.200504432.Rauniyar V, Zhai H, Hall DG. J. Am. Chem. Soc. 2008;130:8481. doi: 10.1021/ja8016076.
- 7.For enantioselective Lewis base catalyzed carbonyl additions of crotylmetal reagents, see: Denmark SE, Coe DM, Pratt NE, Griedel BD. J. Org. Chem. 1994;59:6161. doi: 10.1021/jo052202p.Denmark SE, Coe DM, Pratt NE, Griedel BD. J. Org. Chem. 2001;59:6161. doi: 10.1021/jo052202p.Denmark SE, Fu J. J. Am. Chem. Soc. 2001;123:9488. doi: 10.1021/ja016552e.Iseki K, Kuroki Y, Takahashi M, Kishimoto S, Kobayashi Y. Tetrahedron. 1997;53:3513.Nakajima M, Saito M, Shiro M, Hashimoto S-I. J. Am. Chem. Soc. 1998;120:6419.Malkov AV, Dufková L, Farrugia L, Kočovsky P. Angew. Chem., Int. Ed. 2003;42:3674. doi: 10.1002/anie.200351737.
- 8.For enantioselective catalytic carbonyl crotylation via Nozaki-Hiyama coupling, see: Bandini M, Cozzi PG, Umani-Ronchi A. Polyhedron. 2000;19:537.Bandini M, Cozzi PG, Umani-Ronchi A. Tetrahedron. 2001;57:835.Bandini M, Cozzi PG, Umani-Ronchi A. Angew. Chem., Int. Ed. 2000;39:2327. doi: 10.1002/1521-3773(20000703)39:13<2327::aid-anie2327>3.0.co;2-9.Inoue M, Suzuki T, Nakada M. J. Am. Chem. Soc. 2003;125:1140. doi: 10.1021/ja021243p.Lee J-Y, Miller JJ, Hamilton SS, Sigman MS. Org. Lett. 2005;7:1837. doi: 10.1021/ol050528e.McManus HA, Cozzi PG, Guiry PJ. Adv. Synth. Catal. 2006;348:551.Xia G, Yamamoto H. J. Am. Chem. Soc. 2006;128:2554. doi: 10.1021/ja058454p.
- 9.In Brown's allylation protocol, the stoichiometric generation of isopinocampheol frequently complicates product isolation: Ireland RE, Armstrong JD, III, Lebreton J, Meissner RS, Rizzacasa MA. J. Am. Chem. Soc. 1993;115:7152.Burova SA, McDonald FE. J. Am. Chem. Soc. 2004;126:2495. doi: 10.1021/ja039618+.Ramachandran PV, Prabhudas B, Chandra JS, Reddy MVR. J. Org. Chem. 2004;69:6294. doi: 10.1021/jo0492416.White JD, Hansen JD. J. Org. Chem. 2005;70:1963. doi: 10.1021/jo0486387.Gao D, O'Doherty GA. Org. Lett. 2005;7:1069. doi: 10.1021/ol047322i.Gao D, O'Doherty GA. J. Org. Chem. 2005;70:9932. doi: 10.1021/jo051681p.Liu D, Xue J, Xie Z, Wei L, Zhang X, Li Y. Synlett. 2008:1526.
- 10.For enantioselective allylation, anti-crotylation and tert-prenylation via iridium catalyzed alcohol-mediated C-C couplings, see: Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:6340. doi: 10.1021/ja802001b.Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:14891. doi: 10.1021/ja805722e.Kim IS, Han S-B, Krische MJ. J. Am. Chem. Soc. 2009;131:2514. doi: 10.1021/ja808857w.Gao X, Townsend IA, Krische MJ. J. Org. Chem. 2011;76:2350. doi: 10.1021/jo200068q.Han SB, Kim I-S, Han H, Krische MJ. J. Am. Chem. Soc. 2009;131:6916. doi: 10.1021/ja902437k.
- 11.For recent reviews on C-C bond forming hydrogenation and transfer hydrogenation, see: Patman RL, Bower JF, Kim IS, Krische MJ. Aldrichim. Acta. 2008;41:95.Bower JF, Kim IS, Patman RL, Krische MJ. Angew. Chem. Int. Ed. 2009;48:34. doi: 10.1002/anie.200802938.Han SB, Kim IS, Krische MJ. Chem. Commun. 2009:7278. doi: 10.1039/b917243m.Bower JF, Krische MJ. Top. Organomet. Chem. 2011;43:107. doi: 10.1007/978-3-642-15334-1_5.
- 12.In related “hydrogen auto-transfer” or “borrowing hydrogen” processes, alcohol dehydrogenation and nucleophile generation occur independently. Hence, conventional pre-activated nucleophiles are required. Such processes deliver products of formal alcohol substitution rather than carbonyl addition. For selected reviews, see: Guillena G, Ramón DJ, Yus M. Angew. Chem. Int. Ed. 2007;46:2358. doi: 10.1002/anie.200603794.Hamid MHSA, Slatford PA, Williams JMJ. Adv. Synth. Catal. 2007;349:1555.Nixon TD, Whittlesey MK, Williams JMJ. Dalton Trans. 2009:753. doi: 10.1039/b813383b.Dobereiner GE, Crabtree RH. Chem. Rev. 2010;110:681. doi: 10.1021/cr900202j.Guillena G, Ramón DJ, Yus M. Chem. Rev. 2010;110:1611. doi: 10.1021/cr9002159. Related dehydrogenative couplings of amines also require pre-activated nucleophiles, see: Li C-J. Acc. Chem. Res. 2009;42:335. doi: 10.1021/ar800164n.
- 13.For ruthenium catalyzed alcohol-mediated C-C coupling, see: Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338. doi: 10.1021/ja801213x. Dienes.Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:14120. doi: 10.1021/ja805356j.Han H, Krische MJ. Org. Lett. 2010;12:2844. doi: 10.1021/ol101077v.Smejkal T, Han H, Breit B, Krische MJ. J. Am. Chem. Soc. 2009;131:10366. doi: 10.1021/ja904124b.Patman RL, Chaulagain MR, Williams VM, Krische MJ. J. Am. Chem. Soc. 2009;131:2066. doi: 10.1021/ja809456u. Alkynes.Williams VM, Leung JC, Patman RL, Krische MJ. Tetrahedron. 2009;65:5024. doi: 10.1016/j.tet.2009.03.068.Skucas E, Zbieg JR, Krische MJ. J. Am. Chem. Soc. 2009;131:5054. doi: 10.1021/ja900827p. Allenes.Zbieg JR, McInturff EL, Krische MJ. Org. Lett. 2010;12:2514. doi: 10.1021/ol1007235.Zbieg JR, McInturff EL, Leung JC, Krische MJ. J. Am. Chem. Soc. 2011;133:1141. doi: 10.1021/ja1104156.
- 14.Related Me3Si-substituted crotylmagnesium and crotylzinc reagents react with aldehydes to give racemic syn-adducts: Sato F, Kusakabe M, Kobayashi Y. J. Chem. Soc., Chem. Comm. 1984:1130.Helm MD, Mayer P, Knochel P. Chem. Comm. 2008:1916. doi: 10.1039/b802157k.
- 15.As practiced industrially, chloroprene is generated in situ from 3,4-dichloro-1-butene, which, in turn, derives from 1,3-butadiene. For conversion of chloroprene to the 2-silyl substituted by way of the Grignard reagent, see: Pidaparthi RR, Junker CS, Welker ME, Day CS, Wright MW. J. Org. Chem. 2009;74:8290. doi: 10.1021/jo901919m. See Supporting Information for experimental details related to the preparation of silyl diene 1.
- 16.For a related diastereoselective hydroboration-Suzuki cross-coupling sequence, see: Coleman RS, Gurrala SR. Org. Lett. 2005;7:1849. doi: 10.1021/ol050476t.
- 17.Smitrovich JH, Woerpel KA. J. Org. Chem. 1996;61:6044. doi: 10.1021/jo991312r. [DOI] [PubMed] [Google Scholar]
- 18.a Hiraki K, Ochi N, Sasada Y, Hayashida H, Fuchita Y, Yamanaka S. J. Chem. Soc. Dalton Trans. 1985:873. [Google Scholar]; b Xue P, Bi S, Sung HHY, Williams ID, Lin Z, Jia G. Organometallics. 2004;23:4735. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.