

Abstract
A new series of 3-ethynyl-1H–indazoles has been synthesized and evaluated in both biochemical and cell-based assays as potential kinase inhibitors. Interestingly, a selected group of compounds identified from this series exhibited low micromolar inhibition against critical components of the PI3K pathway, targeting PI3K, PDK1 and mTOR kinases. Combination of computational modeling and structure-activity relationships studies reveal a possible novel mode for PI3K inhibition, resulting in a PI3Kα isoform specific compound. Hence, by targeting the most oncogenic mutant isoform of PI3K, the compound displays anti-proliferative activity both in monolayer human cancer cell cultures and in three-dimensional tumor models. Because of its favorable physicochemical, in vitro ADME and drug-like properties, we propose that this novel ATP mimetic scaffold could result useful in deriving novel selecting and multi-kinase inhibitors for clinical use.
Introduction
The PI3K/AKT/mTOR cascade is an important cellular signaling pathway that regulates intersecting biological processes such as cell growth and proliferation, cell survival, protein synthesis, and glycolysis metabolism.1–3 Physiological stimulation of this cascade occurs through the binding of growth factors (e.g., insulin or insulin-like growth factor; IGF-1) to receptor tyrosine kinases (RTK) on the cell surface. Subsequent activation of the lipid phosphatidylinositol 3-kinase (PI3K) leads to the phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2) to produce phosphatidylinositol-3,4,5-triphosphate (PIP3), which in turn interacts with the pleckstrin homology (PH) domain of AKT and recruits the kinase to the plasma membrane. Full activation of AKT requires phosphorylation of two distinct residues: Thr308 by the upstream kinase PDK1 and Ser473 by the mammalian target of rapamycin complex 2 (mTORC2).1–3 Activated AKT, among its wide array of downstream effects, increases protein synthesis rate by phosphorylation at Thr246 of the proline-rich substrate of 40 kDa (PRAS40). This substrate is further regulated through phosphorylation at Ser183 by the mTOR complex 1 (mTORC1). Downstream of mTORC1, signaling events can go through p70 S6 kinase (S6K1), which in turn phosphorylates the programmed cell death protein (PDCD4) at residue Ser457 (Figure 1).
Figure 1.

Schematic representation of the PI3K/mTOR signaling pathway highlighting the phosphorylation sites tested in our cellular and biochemical assays.
Because of its involvement in a variety of cellular events, such as survival, proliferation, cell motility and invasion,4 hyperactivation of the PI3K signaling pathway is one of the most common molecular events in nearly every form of human cancer.5–8 Indeed, aberrant activation of the PI3K cascade, including PTEN inactivation and PI3K activating mutations, are present in about 32 % of colon cancers, 30 % of breast cancers, 30% of melanomas,9 27 % of brain cancers, and 25 % of stomach cancers.10, 11 It is for these reasons that considerable drug discovery efforts are ongoing targeting several components of this signaling cascade, using either single and multiple target strategies.12–16 Interestingly, clinical data suggest that multikinase inhibitors (MKIs) produce greater benefit over single kinase inhibition, especially in solid tumors where different kinases and different pathways synergistically contribute to tumor proliferation.17–19 Indeed, emerging Phase II clinical trials data reveal that dual PI3K/mTOR inhibitors may be more effective having the advantage of being less susceptible to PI3K drug resistance owing their preserved activity against mTOR.16
In our drug discovery program we interrogated the PI3K signaling pathway and we report on the identification of a novel scaffold of the 3-ethynylindazole family as multiple PI3K/PDK1/mTOR inhibitor. Such scaffold represents a novel lead structure for the PI3K pathway, suitable for further functionalization and drug development.
Results and Discussion
Compounds 6–19 (Table 1) were prepared as shown in Scheme 1. Compound 1 was iodinated and Boc protection was performed according to the previously reported procedures.20 Compound 3 (Scheme 1) was coupled with the appropriate alkyne using the Sonogashira reaction conditions to give 4 and its analogs in moderate yield (45–65%). Compound 4 was treated with TFA to afford the final compound 5 and its analogs (6–19, Table 1) in good yields (89–95%). Similarly, compounds 20–21 (Table 1) were synthesized starting from 7-aza-indazole instead of indazole and following the same synthetic procedure reported in Scheme 1 for indazole derivatives.
Table 1.
Kinase and cell-based assay results for compounds 6–21a
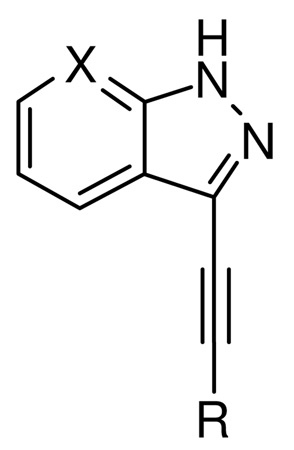 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compd ID |
R | X | Kinase assay IC50 (µM) |
Cell-based Assayd IC50 (µM) |
Cell-based Assay GI50 (µM) |
||||||
| PI3Kαb | mTORb | PDK1c | AKT [pT308] |
PRAS40 [pS183] |
PC3 | MDA- MB-231 |
MCF7 | Hela | |||
| 6 | 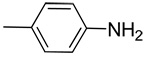 |
CH | 1.05 | 4.97 | 39.20 | 17.09 | 10.80 | 2.51 | 8.28 | 3.23 | 3.60 |
| 7 |  |
CH | >100 | >100 | >50 | >100 | >50 | ND | ND | ND | ND |
| 8 |  |
CH | >100 | >100 | >100 | >100 | >200 | ND | ND | ND | ND |
| 9 | 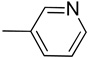 |
CH | 1.85 | >100 | 20.80 | 23.5 | >50 | 6.81 | 10.48 | 3.66 | 5.21 |
| 10 | 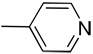 |
CH | 0.36 | 10.70 | 3.87 | 12.85 | 13.60 | 2.43 | 7.84 | 2.58 | 2.53 |
| 11 | CH | >50 | >100 | >100 | >100 | >50 | ND | ND | ND | ND | |
| 12 | 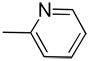 |
CH | >100 | >100 | >25 | >100 | >50 | ND | ND | ND | ND |
| 13 | 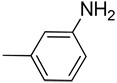 |
CH | 5.12 | >100 | 12.60 | 12.90 | 5.10 | 2.84 | 8.29 | 10.39 | 5.21 |
| 14 | 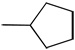 |
CH | >100 | >100 | >100 | >50 | >50 | ND | ND | ND | ND |
| 15 | CH | >100 | >100 | >100 | >50 | >50 | ND | ND | ND | ND | |
| 16 | 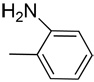 |
CH | >100 | >100 | >100 | >100 | >50 | ND | ND | ND | ND |
| 17 | 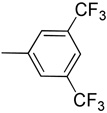 |
CH | >100 | >100 | >100 | >100 | >100 | ND | ND | ND | ND |
| 18 | 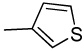 |
CH | >100 | >100 | >100 | >100 | >100 | ND | ND | ND | ND |
| 19 | 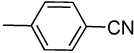 |
CH | >100 | >100 | >100 | >50 | >50 | ND | ND | ND | ND |
| 20 |  |
N | 3.05 | >10 | >50 | ND | 19.1 | ND | ND | ND | ND |
| 21 | 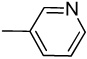 |
N | 9.0 | 5.0 | >50 | ND | >10 | ND | ND | ND | ND |
Values are means of at least three or more experiments with a typical standard deviation of less than ± 20%.
LanthaScreen Eu kinase binding assay.
Z’-LYTE kinase activity assay.
LanthaScreen cellular assays in HEK293E cell line; AKT [pThr308] for PI3K/PDK1 readout and PRAS40 [pSer183] for mTORC1 readout. ND: not determined (compound was not tested).
Scheme 1. Synthetic scheme for compounds 6–21.

R = aryl or alkyl, see Table 1
X= CH (1-19) or N (20-21)
Reagents and conditions. (a) Iodine, KOH, DMF, rt; (b) di-tert-butyl dicarbonate, Et3N, DMAP, CH3CN, rt; (c) aryl or aliphatic alkyne, Pd(Ph3P)2Cl2, CuI, Et3N, CH3CN, rt; (d) TFA, CH2Cl2, rt.
We chose to initially screen the synthesized compounds (shown in Table 1) in a cell-based format using a LanthaScreen cellular assay technology, recently reported and successfully applied to the analysis of the PI3K/AKT/mTOR pathway.21–23 This assay platform uses individual cell lines that stably express key kinase substrate within the cascade as GFP-fusion proteins. Cells are stimulated, in the presence or absence of an inhibitor, to trigger endogenous kinase activity and phosphorylation of the substrate. The phosphorylation status of the GFP-substrate is detected after cell lysis by addition of a terbium (Tb)-labeled phosphospecific antibody, via time-resolved Förster resonance energy transfer (TR-FRET) signal between Tb and GFP.21 We used three phosphorylation readouts to interrogate pathway signaling: AKT [pThr308] for PI3K/PDK1, PRAS40 [pThr246] for AKT, and PRAS40 [pSer183] for mTORC1 activity (Figure 1).
Compounds were incubated with the different cell lines in 10-point dose-response format for 60 minutes to generate IC50 curves. Among all newly synthesized series of 3-ethynyl-1H–indazoles (6 to 19), we found that compounds 6, 10, 13 inhibited both AKT and PRAS40 phosphorylation, while compound 9 inhibited only AKT phosphorylation, with potencies in the low micromolar range (Table 1).
To understand if the inhibition of AKT phosphorylation at Thr308 was a result of a direct inhibition of PDK1 or PI3K, we performed a cell-free kinase assay against both kinases. All the compounds which displayed inhibition in cell based assays (6, 9, 10, 13) resulted to be active against both kinases, with the highest potency for 10 (IC50 = 361 nM) against PI3Kα. Similarly, we tested the compounds against mTOR through a biochemical assay. Compounds 6 and 10 are multiple PI3Kα/PDK1/mTOR inhibitors, while compounds 9 and 13 are dual PI3Kα /PDK1 inhibitors (Table 1). All compounds tested were inactive against AKT as measured by a biochemical kinase assay for AKT1 (data not shown). Further in vitro displacement assays against the PI3K isoforms revealed that compounds 6, 10 and 13 are α-isoform specific and ATP competitive with about 100-fold selectivity over the β and γ isoforms (Table 2).
Table 2.
Biochemical Selectivity Profile against PI-3K isoforms, IC50 (µM)
These assays were conducted by Invitrogen’s SelectScreen Biochemical Kinase (SSBK) Profiling Service. a Values are means of at least three or more experiments with a typical standard deviation of less than ± 20%.
In order to rationalize the affinity observed for the lead structure 10, we studied its binding mode within the ATP binding site of PI3Kα by using computational docking studies. A target binding pocket was derived from the X-ray crystal structure of the ternary complex involving the most common mutant of PI3K catalytic domain p110α (H1047R), its regulatory subunit (p85α) and the drug wortmannin (PDB id: 3hhm). From the docked binding pose, compound 10 appears to be deeply inserted in the ATP-binding site (Figure 2A): the N-2 atom of compound 10 seems to be involved in hydrogen bonding interactions with the OH of Tyr836 and the indazole moiety further stabilized via hydrogen bond contacts between the NH-1 and the N-2 of the heterocycle with the backbone carbonyl of Asp810 and the backbone NH of Asp933, respectively (Figure 2B). Furthermore, the compound binds to the hinge region of the kinase via a hydrogen bond between the protonated NH of the pyridine ring and the backbone carbonyl of Val851 (Figure 2). From these studies it became apparent that compounds of this series capable of interacting with the hinge region are most effective against PI3Kα. Indeed, the isomer 9, whose protonated nitrogen could still constitutes a suitable hydrogen bonding donor group interacting with the hinge residue Val851, showed good potency (IC50 = 1.85 µM); in compounds 6 and 13, where the ethynyl-indazole moiety is linked to an aniline, is the amino group at the 3- or 4-position on the phenyl ring to be involved in the hydrogen bond with the backbone carbonyl of Val851. Indeed, they both showed a low micromolar inhibition (IC50 values of 1.05 µM and 5.12 µM respectively).
Figure 2.

Docking studies of compound 10 within the ATP-binding site of PI3Kα (H1047R) mutant (PDB id: 3hhm).24 (A) Surface representation of the active site of p110α with the ATP mimic compound 10. Surface generated with MOLCAD37 and color coded according to cavity depth (blue, shallow; yellow, deep). Protein residues involved in hydrogen bonds are highlighted. (B) Ribbon representation of the PI3Kα (H1047R) mutant in complex with compound 10 in the same binding pose of A). Protein residues involved in hydrogen bonds showed as capped sticks.
The shift of the amino group at the 2-position on the phenyl ring (16) doesn’t allow the compound to form the critical interaction with the hinge region, thus explaining its loss of activity (IC50 > 100 µM). As expected, the simple phenyl (7), fluorophenyl (8), trifluoromethylphenyl (17), thiophenyl (18), 4-cyanophenyl (19) substitutions on 3-ethynylindazole showed poor activity. Accordingly, aliphatic alkynes such as cyclopentyl (14), cyclopropyl (15), and N,N-dimethyl (11) were inactive both in the biochemical and cell-based assays (Table 1).
Keeping the pyridine group as the most efficient substituent, we explored possible modifications on the indazole ring (20–21). Isosteric replacement of the indazole with the pyrazolopyridine ring revealed to be detrimental for PI3Kα inhibition. Indeed, by comparing the most active compound of the 3-ethynylindazole series (10, IC50 = 361 nM) with the corresponding pyrazolopyridine analog (20, IC50 = 3.05 µM) a 10-fold loss of activity was observed and the same trend was followed by compound 9 (IC50 = 1.85 µM) versus compound 21 (IC50 = 9.0 µM). Such decrease in activity is expected given that the additional nitrogen atom is not engaged in hydrogen bonding interactions in the docked structures of compounds 20 and 21 in the binding pocket of PI3Kα (Supporting Information).
Since the ATP binding pocket of mTOR is highly homologous to that of PI3K, it is not surprising that some of the compounds inhibit both kinases (Table 1). Indeed, even previously reported selective PI3K inhibitors show some degree of activity against mTOR.16 However, noteworthy compound 10 in our cellular and biochemical assays inhibits also the less structurally related PDK-1. Inhibition of multiple kinases on the PI3K pathway may be highly desirable to suppress oncogenic transformations that result from the activation of the pathway in cancer cells.
To evaluate the drug-likeness of the proposed series on empirical ground25, 26, we gathered information about physicochemical properties of the compounds, such as solubility, cell permeability, ligand efficiency indices,27–29 as well as plasma and microsomal stability (Table 3 and Supporting Information). Hence, investigation of these parameters suggests that the compounds of this novel scaffold series may be deemed suitable as lead-candidates for further drug optimizations.30
Table 3.
Ligand efficiency indices and in vitro ADME properties of 10 and 13
| Parameter | 10 | 13 | |
|---|---|---|---|
| Ligand Efficiency | LE (Kcal mol−1) | 0.5 | 0.4 |
| Binding Efficiency Index | BEI | 29.4 | 22.7 |
| Surface Binding Efficiency Index | SEI | 17.5 | 10.5 |
| Lipophilic Ligand Efficiency | LLE | 4.2 | 2.5 |
| Microsomal stability | t ½ (min) | 35 | >60 |
| Plasma stability | % remaining after 60 min | 97 | 100 |
On the basis of their cellular efficacy and favorable biopharmaceutical properties, we further investigated the anti-proliferative activity of the most promising compounds (6, 9, 10, and 13) against human cancer cells lines including prostatic (PC3), breast (MD-MA-231 and MCF7), and cervical (HeLa) cancers. The results are summarized in Table 1. The anti-tumor activity of compound 10, which displayed the highest cellular potency inhibiting PI3Kα, mTOR and PDK1, has been further evaluated against growth/proliferation of the glioblastoma U87 cell line when tested in three-dimensional cultures. The three-dimensional cell culture system has been chosen because it better recapitulates real human tissue with respect to oxygen and nutrient levels, cell-cell contacts and cellular architecture compared with conventional two-dimensional cell culture systems.31 It has also been observed that spheroid cultures exhibit differential sensitivity to known chemotherapeutic agents, as reported for the wortmannin derivative PX-866 (22)32 which showed higher potency in multicellular 3D system then in monolayer.33 We chose U87 glioblastoma cells because they exhibit deregulated PI3K signaling due to the functional loss of the tumor suppressor PTEN (Figure 1). U87 spheroids, generated through the previously reported hanging drop method,34 were treated daily, for a total of 6 days, with compound 10 and 22 as control at different concentrations (Figure 3). Treatment with 10 µM of compound 10 every day strongly suppressed cell growth/proliferation, as proved by detected spheroid volumes, comparable to those produced by the potent PI3K inhibitor 22 (Figure 3).
Figure 3.

U87 cells grown as spheroids generated by the hanging drop method34 and treated with the indicated concentration of 22 or compound 10 every day, for a total of 6 days. Left panel: Phase-contrast photos representative of four independent experiments. Right panel: spheroid length and width (measured with an optical micrometer) were used to calculate spheroid volumes (µm3).
In summary, we successfully developed a useful synthetic route to obtain 3-ethynylindazole derivatives which lead to the identification of a multiple PI3K/PDK1/mTOR inhibitor, with a nanomolar inhibition against PI3Kα. Other indazoles were recently reported as possible inhibitors of other protein kinases,35 however the scaffolds reported here, in particular compound 10, represent a novel and valuable starting point for the development of inhibitors of the PI3K pathway and potentially other kinases (see below). Moreover, due to the observed drug-likeness and in vitro ADME properties of this novel scaffold (Table 3), we anticipate that further selective and/or multi-kinase inhibitors could arise from further derivatization of this novel ATP mimetics. In fact, preliminary selectivity panels with compound 10 against 314 protein kinases (Supporting Information) reveals that less than 10% of these proteins showed ≥ 50% inhibition at 10 µM (Table 4). Hence, based on these data, we envision that further elaborations of compound 10 could lead to further selective or multi-kinase inhibitors against a variety of drug targets.
Table 4.
Kinases which showed an inhibition higher than 50 % at 10µM concentration of compound 10.
| KINASES TESTED | |
|---|---|
| ABL1 T3151I | MARK4 |
| CDK5/p25 | MELK |
| CHUK (IKK alpha) | MINK1 |
| CLK2 | MUSK |
| DNA-PK | MYLK2 (skMLCK) |
| FLT3 | NTRK1 (TRKA) |
| FLT3 D835Y | NTRK2 (TRKB) |
| FLT4 (VEGFR3) | NTRK3 (TRKC) |
| GSG2 (Haspin) | NUAK1 (ARK5) |
| IRAK1 | PDGFRA T674I |
| JNK1 (MAPK8) | PDGFRA V561D |
| JNK2 (MAPK9) | RET V804L |
| KDR (VEGFR2) | RET Y791F |
| LRKK2 | SGK (SGK1) |
| LRRK2 G2019S | SYK |
| MAP4K4 (HGK) | |
Experimental Section
Chemistry
Unless otherwise indicated, all anhydrous solvents were commercially obtained and stored in Sure-seal bottles under nitrogen. All other reagents and solvents were purchased as the highest grade available and used without further purification. Thin-layer chromatography (TLC) analysis of reaction mixtures was performed using Merck silica gel 60 F254 TLC plates, and visualized using ultraviolet light. NMR spectra were recorded on Varian 300 or 500 MHz instruments. Chemical shifts (δ) are reported in parts per million (ppm) referenced to 1H (Me4Si at 0.00). Coupling constants (J) are reported in Hz throughout. Mass spectral data were acquired on Shimadzu LCMS-2010EV for low resolution, and on an Agilent ESI-TOF for either high or low resolution. Purity of all compounds was obtained in a HPLC Breeze from Waters Co. using an Atlantis T3 3µm 4.6×150 mm reverse phase column. The eluant was a linear gradient with a flow rate of 1 ml/min from 95% A and 5% B to 5% A and 95% B in 15 min followed by 5 min at 100% B (Solvent A: H2O with 0.1% TFA; Solvent B: acetonitrile with 0.1% TFA). The compounds were detected at λ=254 nm. Purity of key compounds was established by elemental analysis as performed on a Perkin Elmer series II-2400 and HPLC analysis and determined to be > 95%. Combustion analysis was performed by NuMega Resonance Labs, San Diego, CA, USA.
Synthesis of tert-butyl 3-(phenylethynyl)-1H-indazole-1-carboxylate (4, R = Ph)
To a solution of compound 3 (343 mg, 1 mmol) in CH3CN (5 mL) were added phenyl acetylene (0.13 mL, 1.2 mmol), CuI (20 mg, 0.10 mmol), Pd(Ph3P)2Cl2 (70 mg, 0.10 mmol), and Et3N (0.42 mL, 3 mmol) respectively at room temperature under nitrogen atmosphere. The reaction mixture was stirred at room temperature for 16 h then solvent was removed in vacuo. The residue was chromatographed over silica gel (2% ethyl acetate in hexane) to give a Boc-protected product 4 (270 mg, 85%).
Synthesis of 3-(phenylethynyl)-1H-indazole (7)
To a solution of 4 (150 mg, 0.47 mmol) in CH2Cl2 (3 mL) was added TFA (1 mL) at room temperature. The resulting mixture was stirred at room temperature for 3 h then solvent and TFA were removed in vacuo. The residue was extracted with CH2Cl2 (50 mL), washed with saturated NaHCO3 aqueous solution (3 × 30 mL), brine (30 mL), dried (MgSO4) and concentrated in vacuo. The residue was chromatographed over silica gel (40% ethyl acetate in hexane) to afford a white solid compound 7 (94 mg, 92%). 1H NMR (300 MHz, CDCl3) δ 7.26–7.34 (m, 1H), 7.40–7.50 (m, 5H), 7.66–7.76 (m, 2H), 7.95 (d, J = 7.8 Hz, 1H), 11.68 (br s, 1 H, NH); MS m/z 219 (M+H)+, 166, 155, 121, 88; HRMS calcd for C15H11N2 219.0917 (M+H), found 219.0918.
Following the above mentioned procedure and the use of the appropriate starting materials and reagents, compounds 6 to 16 were prepared. Yields refer to the final deprotection step.
Synthesis of 4-((1H-indazol-3-yl)ethynyl)aniline (6)
Yield: 65%; 1H NMR (300 MHz, CD3OD) δ 5.24 (br s, 2 H, NH2), 6.57–6.60 (m, 2 H), 7.12–7.15 (m, 1H), 7.24 (d, J = 7.5 Hz, 2 H), 7.28–7.46 (m, 2 H), 7.70 (d, J = 6.9 Hz, 1 H), 11.82 (br s, 1 H, NH); MS m/z 234 (M+H)+, 158, 149, 121, 102, 65; HRMS calcd for C15H12N3 234.1031 (M+H), found 234.1021.
Synthesis of 3-((3,5-difluorophenyl)ethynyl)-1H-indazole (8)
Yield: 89%; 1H NMR (300 MHz, DMSO) δ 7.27 (t, J = 7.5 Hz, 1H), 7.36–7.51 (m, 4H), 7.63 (d, J = 8.4 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 13.32 (br s, 1 H); MS m/z 255 (M+H)+, 149, 121; HRMS calcd for C15H9F2N2 255.0728 (M+H), found 255.0726.
Synthesis of 3-(pyridin-3-ylethynyl)-1H-indazole (9)
Yield: 92%; 1H NMR (300 MHz, CDCl3) δ 7.24–7.41 (m, 3H), 7.46 (m, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.87–7.99 (m, 2H), 8.63 (s, 1H), 11.32 (br s, 1 H, NH); MS m/z 220 (M+H)+, 192, 121, 102; HRMS calcd for C14H10N3 220.0875 (M+H), found 220.0877.
Synthesis of 3-(pyridin-4-ylethynyl)-1H-indazole (10)
Yield: 91%; 1H NMR (400 MHz, DMSO) δ 7.29 (t, J = 7.3 Hz, 1 H), 7.45 (t, J = 7.3 Hz, 1 H), 7.62–7.67 (m, 3 H), 7.89 (d, J = 7.9 Hz, 1 H), 8.65 (d, J = 6.1 Hz, 2 H), 13.68 (br s, 1 H, NH); HRMS calcd for C14H10N3 220.0875 (M+H), found 220.0878. Anal. calcd for C14H9N3: C, 76.70; H, 4.14; N, 19.17. Found: C, 76.58; H, 4.29; N, 19.01.
Synthesis of 3-(1H-indazol-3-yl)-N,N-dimethylprop-2-yn-1-amine (11)
Yield: 90%; 1H NMR (300 MHz, CDCl3) δ 2.47 (s, 6H), 3.67 (s, 2H), 7.18–7.27 (m, 1H), 7.40 (t, J = 7.6 Hz, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 7.8 Hz, 1H), 11.65 (br s, 1 H, NH); MS m/z 200 (M+H)+, 155, 127, 83; HRMS calcd for C12H14N3 200.1182 (M+H), found 200.1190.
Synthesis of 3-(pyridin-2-ylethynyl)-1H-indazole (12)
Yield: 91%; 1H NMR (300 MHz, CD3OD) δ 7.29 (t, J = 6.9 Hz, 1H), 7.40–7.55 (m, 2H), 7.60 (d, J = 8.1 Hz, 1H), 7.77 (d, J = 6.6 Hz, 2H), 7.93 (d, J = 6.9 Hz, 2H), 11.31 (br s, 1 H, NH); HRMS calcd for C14H10N3 220.0875 (M+H), found 220.0878.
Synthesis of 3-((1H-indazol-3-yl)ethynyl)aniline (13)
Yield: 91%; 1H NMR (300 MHz, CD3OD) δ 5.26 (br s, 2 H, NH2), 6.76 (d, J = 7.8 Hz, 1 H), 6.94 (d, J = 7.5 Hz, 1 H), 6.97 (s, 1 H), 7.13 (t, J = 8.1 Hz, 1H), 7.24 (t, J = 8.1 Hz, 1 H), 7.44 (t, J = 8.1 Hz, 1 H), 7.56 (d, J = 7.8 Hz, 1 H), 7.84 (d, J = 8.7 Hz, 1 H), 11.32 (br s,1 H, NH); HRMS calcd for C15H12N3 234.1031 (M+H), found 234.1025. Anal. calcd for C15H11N3: C, 77.23; H, 4.75; N, 18.01. Found: C, 77.01; H, 4.89; N, 17.89.
Synthesis of 3-(cyclopentylethynyl)-1H-indazol (14)
Yield: 92%; 1H NMR (300 MHz, CDCl3) δ 1.66–1.69 (m, 2H), 1.80–189 (m, 4H), 2.02–2.19 (m, 2H), 2.94–3.08 (m, 1H), 7.22 (t, J = 7.4 Hz, 1H), 7.41 (t, J = 7.4 Hz, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 11.84 (br s, 1 H, NH); HRMS calcd for C14H15N2 211.1235 (M+H), found 211.1233.
Synthesis of 3-(cyclopropylethynyl)-1H-indazole (15)
Yield: 90%; 1H NMR (300 MHz, CDCl3) δ 0.92–1.00 (m, 4H), 1.56–1.67 (m, 1H), 7.17–7.27 (m, 1H), 7.36–7.46 (m, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.82 (d, J = 8.1 Hz, 1H), 9.86 (br s, 1 H, NH); HRMS calcd for C12H11N2 183.0922 (M+H), found 183.0925.
Synthesis of 2-((1H-indazol-3-yl)ethynyl)aniline (16)
Yield: 81%; 1H NMR (300 MHz, CD3OD) δ 5.26 (br s, 2 H, NH2), 6.58–6.60 (m, 2 H), 7.12–7.15 (m, 1H), 7.24 (d, J = 7.5 Hz, 2 H), 7.28–7.46 (m, 2 H), 7.70 (d, J = 6.9 Hz, 1 H), 11.80 (br s, 1 H, NH); MS m/z 234 (M+H)+, 158, 149, 121, 102, 65; HRMS calcd for C15H12N3 234.1031 (M+H), found 234.1021.
Synthesis of 3-((3,5-bis(trifluoromethyl)phenyl)ethynyl)-1H-indazole (17)
Yield: 90%; 1H NMR (300 MHz, CDCl3) δ 7.32 (t, J = 8.5 Hz, 1H), 7.49 (t, J = 6.6 Hz, 1H), 7.85–7.94 (m, 2H), 8.07 (s, 2H), 9.56 (br s, 1 H, NH); MS m/z 355 (M+H)+, 275, 244, 149, 130, 127, 121, 118, 88; HRMS calcd for C17H9F6N2 355.0664 (M+H), found 355.0664.
Synthesis of 3-(thiophen-3-ylethynyl)-1H-indazole (18)
Yield: 92%; 1H NMR (300 MHz, CDCl3) δ 7.22–7.46 (m, 4H), 7.63–7.70 (m, 2H), 7.90 (d, J = 8.1 Hz, 1H), 9.52 (br s , 1 H, NH); MS m/z 225 (M+H)+, 130, 121, 102; HRMS calcd for C13H9N2S 225.0481 (M+H), found 225.0484.
Synthesis of 4-((1H-indazol-3-yl)ethynyl)benzonitrile (19)
Yield: 91%; 1H NMR (300 MHz, CD3OD) δ 7.28 (t, J = 7.5 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.59 (d, J = 8.1 Hz, 1H), 7.74–7.76 (m, 4H), 7.85 (d, J = 8.1 Hz, 1H), 10.89 (br s, 1 H, NH); MS m/z 244 (M+H)+, 217, 149, 141, 130, 127, 121, 102; HRMS calcd for C16H10N3 244.0869 (M+H), found 244.0875.
Synthesis of 3-(pyridin-4-ylethynyl)-1H-pyrazolo[3,4-b]pyridine (20)
Yield: 86%; 1H NMR (300 MHz, DMSO-d6) δ 7.37 (dd, J = 8.1 and 4.5 Hz, 1 H), 7.65–7.69 (m, 2 H), 8.42 (dd, J = 8.1 and 1.2 Hz, 1 H), 8.65 (dd, J = 4.5 and 1.2 Hz, 1 H), 8.67–8.71 (m, 2 H), 13.89 (br s, 1 H, NH); MS m/z 221 (M+H)+, 159, 130, 121, 102, 88, 85, 56; HRMS calcd for C13H9N4 (M+H) 221.0827, found 221.0822.
Synthesis of 3-(pyridin-2-ylethynyl)-1H-pyrazolo[3,4-b]pyridine (21)
Yield: 83%; 1H NMR (300 MHz, DMSO-d6) δ 7.35 (dd, J = 8.1 and 4.2 Hz, 1 H), 7.42 (t, J = 5.7 Hz, 1 H), 7.79 (d, J = 7.5 Hz, 1 H), 7.90 (t, J = 7.7 Hz, 1 H), 8.35 (d, J = 8.1 Hz, 1 H), 8.60–8.67 (m, 2 H), 13.87 (br s, 1 H, NH); HRMS calcd for C13H9N4 (M+H) 221.0827, found 221.0824.
Assays
LanthaScreen Cellular assay
Cell-based assays for both AKT and PRAS40 phosphorylation were carried out using LanthaScreen cellular assay technology from Invitrogen (Carlsbad, CA).21 Briefly, the assay protocol for compound screening is as follows. Cells were plated in white tissue culture-treated 384-well assay plates at a density of 20,000 cells per well in 32 µL of assay medium (low glucose DMEM + 0.1% bovine serum albumin, BSA). After overnight serum starvation, cells were pretreated with 4 µL of compound at the indicated concentrations (10-point dose-response in duplicate) for 60 minutes. The cells were then stimulated with insulin (EC80 concentration of ~5 ng/mL) for 30 minutes to activate PI3K/AKT/mTOR signaling. The assay medium was subsequently removed via aspiration and cells were lysed by the addition of 20 µL of LanthaScreen cellular assay lysis buffer supplemented with protease and phosphatase inhibitor cocktails (Sigma P8340 and P2850, respectively) as well as 2 nM of the Tb-labeled detection antibody, also from Invitrogen. Following 2 hour assay equilibration at room temperature, the TR-FRET emission ratios were acquired on a PerkinElmer EnVision fluorescence plate reader (Waltham, MA) with TRF laser excitation and emission wavelengths of 520 nm and 495 nm. Data analysis and curve-fitting was per formed using XLfit4 and GraphPad Prism4 software.
PI3Kα in vitro assay
The lipid kinase PI3K was assayed by using a LanthaScreen Eu Kinase Binding® Assay technology from Invitrogen. To a solution of the compounds diluted in assay buffer (25 mM TrisHCl, 5 mM MgCl2, 0.02% chaps) the following components were added: the kinase tracer (PV6088, 20 nM final concentration), the kinase PIK3CA/PIK3R1 (PV4788, 5 nM final concentration) and Eu-labeled anti-His tag antibody (PV6089 2 nM final concentration).
The assay was performed in Corning 3673 white 384-well assay plates. Following 1 hour assay equilibration at room temperature, the TR-FRET emission ratios were acquired on a PerkinElmer EnVision fluorescence plate reader (Waltham, MA) with TRF laser excitation and emission wavelengths of 665 nm and 615 nm. Data analysis and curve-fitting was per formed using XLfit4 and GraphPad Prism4 software.
mTOR in vitro assay
Mammalian target of rapamycin (mTOR) was assayed by using a LanthaScreen Eu Kinase Binding® Assay technology from Invitrogen. To a solution of the compound diluted in assay buffer (25 mM TrisHCl, 5 mM MgCl2, 0.02% chaps) the following components were added: the kinase tracer (PV6087, 20 nM final concentration), the kinase FRAP1 (PV4753, mTOR, 5nM final concentration) and Eu-labeled anti-GST tag antibody (PV5594, 2nM final concentration).
The assay was performed in Corning 3673 white 384-well assay plates. Following 1 hour assay equilibration at room temperature, the TR-FRET emission ratios were acquired on a PerkinElmer EnVision fluorescence plate reader (Waltham, MA) with TRF laser excitation and emission wavelengths of 665 nm and 615 nm. Data analysis and curve-fitting was per formed using XLfit4 and GraphPad Prism4 software.
PDK1 in vitro assay
The PDK1 kinase was tested by using the Z´-LYTE® biochemical assay technology from Invitrogen (PV 3180). The assay was performed in Corning 3676 black 384-well assay plate. The final 10 µL Kinase Reaction consisted of 9.75 – 49.4 ng PDK1 and 2 µM of the substarte Ser/Thr 07 in 50 mM of HEPES buffer at pH 8.0, containing 0.01% BRIJ-35, 10 mM MgCl2, 1 mM EGTA, 0.01% NaN3. ATP concentration at the Km value was used. After the 1 hour Kinase Reaction incubation, 5 µL of a 1:32768 dilution of Development Reagent A was added. After the development reaction, where a site-specific protease recognizes and cleaves the non-phosphorylated peptide, a ratiometric read-out of the donor emission (Coumarin, 445 nm) over the acceptor (Fluorescein, 520 nm) was detected by a PerkinElmer EnVision fluorescence plate reader (Waltham, MA). Data analysis and curve-fitting was per formed using XLfit4 and GraphPad Prism4 software.
Anti-proliferative Assay
All human cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA) and were maintained in 5% CO2 at 37°C. Human cervical carcinoma (Hela) and U87 glioblastoma cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Cellgro) supplemented with 10% fetal bovine serum (FBS, Omega Scientific) and 1% penicillin/streptomycin (Omega Scientific). MDA-MB-231 and MCF7 breast cancer cells and PC3 prostate cancer cells were cultured in RPMI + GlutaMAX Medium (Gibco) plus 10% FBS and 1% penicillin/streptomycin.
Approximately 3000 cells were seeded into individual wells of a 96-well tissue culture plate and incubated for 24hours. Cells were replenished with fresh medium (0.1ml/well) and exposed to triplicates of different concentration solutions (from 0.5 to 100µM) of test compounds. The analyzed inhibitors were dissolved in DMSO reaching a final DMSO concentration of 0.5%. After incubation for 72 hours at 37 °C and 5% CO2, cell viability was assessed using ATPlite assay from Perkin Elmer (Waltham, MA). Viability was normalized to control cells which were treated with the vehicle, DMSO. The reported IG50 values were calculated by PRISM 5 (Graphpad).
3D culture assay
U87 cells were induced to form spheroids via a hanging drop method.34 Cells were plated at 200 cells per well (20ul) of a Nunc-60 well microwell MiniTray (Polystyrene). The trays were covered, inverted and placed in a humidity chamber for 5 days till one spheroid formed in each well. The spheroids were then transferred into a 48 well plate coated in 1% low melting point agarose to prevent them from adhering. The spheroids were measured and DMSO or drug was added every 24 hours for 6 days. Spheroid length and width (measured with an optical micrometer) were used to calculate spheroid volumes (µm3).
Microsomal Stability Assay (Rat Liver Microsome assay)
Test compound solutions were incubated with rat liver microsomes (RLM) for 60 minutes at 37.5 °C. The final incubation solutions contained 4µM test compound, 2mM NADPH, 1 mg/ml (total protein) microsomes, and 50mM phosphate (pH 7.2). Compound solutions, protein, and phosphate were pre-incubated at 37.5 °C for 5 min and the reactions were initiated by the addition of NADPH and incubated for 1 hour at 37.5 °C. Aliquots were taken at 15 minute time-points and quenched with the addition of methanol containing internal standard. Following protein precipitation and centrifugation, the samples were analyzed by LC-MS. Test compounds were run in duplicate with 2 control compounds of known half life.
Plasma Stability Assay
Test compound solution was incubated (1 µM, 2.5% final DMSO concentration) with fresh rat plasma at 37 °C. The reactions were terminated at 0, 30, and 60 min by the addition of two volumes of methanol containing internal standard. Following protein precipitation and centrifugation, the samples were analyzed by LC-MS. The percentage of parent compound remaining at each time point relative to the 0 min sample is calculated from peak area ratios in relation to the internal standard. Compounds were run in duplicate with a positive control known to be degraded in plasma.
Molecular Modeling
Molecular modeling studies were conducted on a Linux workstation and a 64 3.2-GHz CPUs Linux cluster. Docking studies were performed using the X-ray coordinates of p110alpha (H1037R) mutant in complex with niSH2 of p85alpha and the drug wortmannin (PDB code: 3hhm).24 The PI3Kα crystal structure was extracted from the protein data bank and the complexed ligand was used to define the binding site for docking of small molecules. The genetic algorithm (GA) procedure in the GOLD docking software performed flexible docking of small molecules whereas the protein structure was static.36–38 For each compound, 20 solutions were generated and subsequently ranked according to GoldScore. Molecular surfaces were generated with MOLCAD 38 and docked structures analyzed with Sybyl (Tripos Inc., ST. Louis).
Supplementary Material
Acknowledgment
We gratefully acknowledge financial support from the NIH (grant P01CA128814 to MP).
Abbreviations list
- PI3K
Phosphatidylinositol 3-kinase
- PDK1
3-phosphoinositide dependent protein kinase-1
- RTK
receptor tyrosine kinases
- AKT
Protein Kinase B
- mTOR
mammalian target of rapamycin
- PTEN
phosphatase and tensin homolog protein
- PIP2
phosphatidylinositol-4,5-bisphosphate
- PIP3
phosphatidylinositol-3,4,5-triphosphate
- PDCD4
programmed cell death protein
- S6K1
S6 kinase beta-1
- GFP
Green fluorescent protein
Footnotes
Supporting Information Available: A list of 314 kinases used for the selectivity profile, graphs reporting plasma and microsomal stability of compounds 10 and 13, docking studies with compound 20, are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 2.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kandel ES, Skeen J, Majewski N, Di Cristofano A, Pandolfi PP, Feliciano CS, Gartel A, Hay N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol Cell Biol. 2002;22:7831–7841. doi: 10.1128/MCB.22.22.7831-7841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 5.Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JFM. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets. 2008;8:187–198. doi: 10.2174/156800908784293659. [DOI] [PubMed] [Google Scholar]
- 6.Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13:433–442. doi: 10.1016/j.molmed.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Cicenas J. The potential role of Akt phosphorylation in human cancers. Int J Biol Markers. 2008;23:1–9. doi: 10.1177/172460080802300101. [DOI] [PubMed] [Google Scholar]
- 8.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haluska FG, Tsao H, Wu H, Haluska FS, Lazar A, Goel V. Genetic alterations in signaling pathways in melanoma. Clin Cancer Res. 2006;12:2301s–2307s. doi: 10.1158/1078-0432.CCR-05-2518. [DOI] [PubMed] [Google Scholar]
- 10.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 11.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JKV, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 12.Maira S-M, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 13.Lindsley CW, Barnett SF, Layton ME, Bilodeau MT. The PI3K/Akt pathway: recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr Cancer Drug Targets. 2008;8:7–18. doi: 10.2174/156800908783497096. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27:5511–5526. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- 15.Fasolo A, Sessa C. mTOR inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2008;17:1717–1734. doi: 10.1517/13543784.17.11.1717. [DOI] [PubMed] [Google Scholar]
- 16.Brachmann S, Fritsch C, Maira S-M, Garcia-Echeverria C. PI3K and mTOR inhibitors: a new generation of targeted anticancer agents. Curr Opin Cell Biol. 2009;21:194–198. doi: 10.1016/j.ceb.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 17.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, Garcia-Echeverria C, Weissleder R, Mahmood U, Cantley LC, Wong K-K. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morphy R. Selectively nonselective kinase inhibition: striking the right balance. J Med Chem. 53:1413–1437. doi: 10.1021/jm901132v. [DOI] [PubMed] [Google Scholar]
- 19.Serra V, Markman B, Scaltriti M, Eichhorn PJA, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J, Baselga J. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–8030. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 20.Gaitonde S, De SK, Tcherpakov M, Dewing A, Yuan H, Riel-Mehan M, Krajewski S, Robertson G, Pellecchia M, Ronai Ze. BI-69A11-mediated inhibition of AKT leads to effective regression of xenograft melanoma. Pigment Cell Melanoma Res. 2009;22:187–195. doi: 10.1111/j.1755-148X.2009.00544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlson CB, Robers MB, Vogel KW, Machleidt T. Development of LanthaScreen cellular assays for key components within the PI3K/AKT/mTOR pathway. J Biomol Screen. 2009;14:121–132. doi: 10.1177/1087057108328132. [DOI] [PubMed] [Google Scholar]
- 22.De SK, Stebbins JL, Chen L-H, Riel-Mehan M, Machleidt T, Dahl R, Yuan H, Emdadi A, Barile E, Chen V, Murphy R, Pellecchia M. Design, synthesis, and structure-activity relationship of substrate competitive, selective, and in vivo active triazole and thiadiazole inhibitors of the c-Jun N-terminal kinase. J Med Chem. 2009;52:1943–1952. doi: 10.1021/jm801503n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stebbins JL, De SK, Machleidt T, Becattini B, Vazquez J, Kuntzen C, Chen L-H, Cellitti JF, Riel-Mehan M, Emdadi A, Solinas G, Karin M, Pellecchia M. Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc Natl Acad Sci U S A. 2008;105:16809–16813. doi: 10.1073/pnas.0805677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang C-H, Kinzler KW, Vogelstein B, Amzel LM. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A. 2009;106:16996–17001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hann MM, Oprea TI. Pursuing the leadlikeness concept in pharmaceutical research. Curr Opin Chem Biol. 2004;8:255–263. doi: 10.1016/j.cbpa.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 26.Oprea TI, Davis AM, Teague SJ, Leeson PD. Is there a difference between leads and drugs? A historical perspective. J Chem Inf Comput Sci. 2001;41:1308–1315. doi: 10.1021/ci010366a. [DOI] [PubMed] [Google Scholar]
- 27.Abad-Zapatero C, Metz JT. Ligand efficiency indices as guideposts for drug discovery. Drug Discov Today. 2005;10:464–469. doi: 10.1016/S1359-6446(05)03386-6. [DOI] [PubMed] [Google Scholar]
- 28.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 29.Perola E. An analysis of the binding efficiencies of drugs and their leads in successful drug discovery programs. J Med Chem. 53:2986–2997. doi: 10.1021/jm100118x. [DOI] [PubMed] [Google Scholar]
- 30.Hajduk PJ. Fragment-based drug design: how big is too big? J Med Chem. 2006;49:6972–6976. doi: 10.1021/jm060511h. [DOI] [PubMed] [Google Scholar]
- 31.Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol. 2006;7:211–224. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 32.Ihle NT, Williams R, Chow S, Chew W, Berggren MI, Paine-Murrieta G, Minion DJ, Halter RJ, Wipf P, Abraham R, Kirkpatrick L, Powis G. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther. 2004;3:763–772. [PubMed] [Google Scholar]
- 33.Howes AL, Chiang GG, Lang ES, Ho CB, Powis G, Vuori K, Abraham RT. The phosphatidylinositol 3-kinase inhibitor, PX-866, is a potent inhibitor of cancer cell motility and growth in three-dimensional cultures. Mol Cancer Ther. 2007;6:2505–2514. doi: 10.1158/1535-7163.MCT-06-0698. [DOI] [PubMed] [Google Scholar]
- 34.Kelm JM, Timmins NE, Brown CJ, Fussenegger M, Nielsen LK. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol Bioeng. 2003;83:173–180. doi: 10.1002/bit.10655. [DOI] [PubMed] [Google Scholar]
- 35.Poulsen A, William A, Lee A, Blanchard S, Teo E, Deng W, Tu N, Tan E, Sun E, Goh KL, Ong WC, Ng CP, Goh KC, Bonday Z. Structure-based design of Aurora A & B inhibitors. J Comput Aided Mol Des. 2008;22:897–906. doi: 10.1007/s10822-008-9224-5. [DOI] [PubMed] [Google Scholar]
- 36.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 37.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 38.Teschner M, Henn C, Vollhardt H, Reiling S, Brickmann J. Texture mapping: a new tool for molecular graphics. J Mol Graph. 1994;12:98–105. doi: 10.1016/0263-7855(94)80074-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.