

Abstract
G protein-coupled receptors (GPCRs) are the most numerous and diverse type of cell surface receptors, accounting for about 1% of the entire human genome and relaying signals from a variety of extracellular stimuli that range from lipid and peptide growth factors to ions and sensory inputs. Activated GPCRs regulate a multitude of target cell functions, including intermediary metabolism, growth and differentiation, and migration and invasion. The GPCRs contain a characteristic 7-transmembrane domain topology and their activation promotes complex formation with a variety of intracellular partner proteins, which form basis for initiation of distinct signaling networks as well as dictate fate of the receptor itself. Both termination of active GPCR signaling and removal from the plasma membrane are controlled by protein post-translational modifications of the receptor itself and its interacting partners. Phosphorylation, acylation and ubiquitination are the most studied post-translational modifications involved in GPCR signal transduction, subcellular trafficking and overall expression. Emerging evidence demonstrates that protein S-nitrosylation, the covalent attachment of a nitric oxide moiety to specified cysteine thiol groups, of GPCRs and/or their associated effectors also participates in the fine-tuning of receptor signaling and expression. This newly appreciated mode of GPCR system modification adds another set of controls to more precisely regulate the many cellular functions elicited by this large group of receptors.
Keywords: GPCR, G protein, GRK, beta-arrestin, dynamin, S-nitrosylation
Introduction
G protein-coupled receptors (GPCRs) are the founding members of the large family of 7-transmembrane (7TM) receptors, which constitute the most abundant type of cell surface receptors in mammals. The 7TM receptor superfamily, which comprises approximately 800 members in humans, is divided into six subgroups on the basis of sequence homology and ligand identity. The GPCRs form the largest portion of 7TM receptors and they regulate a wide spectrum of cellular functions that are elicited by multiple extracellular factors, including neurotransmitters, hormones, and sensory stimuli [1]. Activated GPCRs control essential cellular processes, including metabolic homeostasis, cell cycle progression, and cell migration and invasion, and serve as targets for majority of therapeutic drugs [2, 3].
Classical signaling unit of a GPCR consists of ligand-bound receptor, heterotrimeric αβγ G proteins, and plasma membrane-expressed effector [4]. Signal initiation by GPCRs commences with ligand agonist binding that promotes conformational change in the receptor [5], thereby allowing it to function as a guanine nucleotide exchange factor (GEF) to catalyze the replacement of GDP for GTP on the α subunit of the heterotrimeric G proteins [1–3]. The Gα-GTP and Gβγ subunits independently, but coordinately activate downstream effectors to produce specific cellular responses (Figure 1). Signaling by the Gα-GTP and Gβγ subunits is terminated upon the hydrolysis of GTP to GDP via intrinsic GTPase activity of the specific Gα subunit, which is promoted by interaction with Gα subtype-specific regulator of G protein signaling (RGS) proteins that exert GTPase activating protein (GAP) function [6]. The resultant Gα-GDP reassembles with available Gβγ subunits leading to the re-formation of inactive Gαβγ complex and signal termination.
Figure 1. Classical signaling by G protein-coupled receptors.

A, Binding of ligand agonist promotes coupling of the receptor to heterotrimeric G protein leading to the exchange of GDP for GTP on the α subunit. Both Gα-GTP and Gβγ subunits transduce signals affecting expression of soluble second messengers that, in turn, impact the cellular response. B, Phosphorylation of agonist-occupied GPCR by GRK. Free Gβγ subunits mediate recruitment of GRK2 to the plasma membrane into close proximity to activated receptor, prompting the receptor phosphorylation. In addition to Gβγ subunits, GRK2 also binds to plasma membrane-anchored Gαq-GTP and prevents its signaling by sequestration. C, Agonist-bound and GRK-phosphorylated GPCR forms high affinity binding site for βArrestins. The binding of βArrestin to receptor prevents further activation of G proteins and initiates the process of receptor internalization.
Ligand binding to GPCRs causes rearrangement of their transmembrane domains. In the case of the canonical GPCR β2 adrenergic receptor (β2AR), agonist binding induces rearrangement of helix 3 and 6 that engenders conformational changes in the intracellular domains with the consequent coupling to appropriate G proteins and controlled activation of downstream effectors [7, 8]. Evidence is accumulating that stimulated GPCRs do not necessarily activate their effectors to the same extent; distinct ligands (i.e. agonist or antagonist) may exhibit collateral efficacies [9]. An explanation for this phenomenon may lie in the various conformations that a ligand-occupied GPCR may adopt. A GPCR may have conformations that favor coupling to different subsets of G proteins, or other binding partners such as βArrestin (βArrestin1 and βArrestin2) proteins [10]. As a result, the binding of a specific ligand can induce the G protein signaling, the βArrestin signaling, or the blockade of one pathway and activation of the other. For example, the βAR ligand carvedilol functions as an inverse agonist for G protein-mediated adenylyl cyclase activation, but as an agonist for βArrestin-mediated ERK phosphorylation [11].
Heterotrimeric G proteins are typically divided into four groups based on sequence homology of the Gα subunit: Gαs, Gαi, Gαq and Gα12 [2, 3, 6]. The Gαs proteins (Gαs and olfactory Gαolf) stimulate adenylyl cyclases while Gαi proteins (Gαi, Gαo, Gαz, transducin and gustducin) are generally sensitive to pertussis toxin and are often inhibitors of adenylyl cyclases. Activated adenylyl cyclases produce cyclic adenosine monophosphate (cAMP), which activates protein kinase A (PKA). The Gαq (Gαq, Gα11, Gα14) proteins regulate the activity of phosphatidylinositol-specific phospholipases, thereby generating the second messengers inositol 1,4,5 trisphosphate and diacylglycerol. These two molecules increase levels of intracellular Ca+2 and subsequently cause the activation of protein kinase C (PKC). The remaining Gα12 (Gα12, Gα13) family members regulate activity of low molecular weight GTPase Rho through interaction with specific RhoGEFs [6, 12, 13]. In addition to the 16 characterized Gα subunits, there are 5 known Gβ and 12 Gγ subunits. Regulatory mechanisms controlling complex formation between Gα and specific combinations of Gβγ subunits, or preference of specific Gβγ isoforms to effectors, remain largely unexplored. Nonetheless, Gβγ subunits have been demonstrated to transduce signals, independent of Gα, to regulate the activity of several classical effector molecules, including adenylyl cyclases and phospholipase C.
Duration of the ligand agonist-activated GPCR signal is regulated principally by two groups of proteins (Figure 1): the GPCR kinases (GRKs) and βArrestins [1, 10]. The seven GRKs belong to the AGC (PKA, PKG and PKC) group of kinases [14] and can be categorized into three groups based on sequence homology and function [3, 15]. The visual GRKs include GRK1 and GRK7 and are strictly expressed in the retina. The GRK2 group (a.k.a. βARKs) contains ubiquitously expressed GRK2 and GRK3. Finally, the GRK4 group contains GRK4, GRK5 and GRK6 with GRK5 and GRK6 being ubiquitously expressed and GRK4 being expressed mainly in the testes, cerebellum and kidney [3, 4]. The GRKs share a common general structure that encompasses an amino-terminal domain with similarity to RGS proteins, a conserved central kinase domain and a variable regulatory membrane-targeting carboxyl-terminal domain [16]. Unlike majority of the AGC family kinases that are activated by phosphorylation of the activation motif, the GRKs are activated by conformational rearrangement as a result of interaction with appropriate activated GPCR [17]. Remarkably, GRKs seem to possess poor specificity for substrate GPCRs, and once activated by a receptor, they appear relatively promiscuous in the site(s) they phosphorylate [3].
Visual GRKs are stabilized on the plasma membrane as a result of post-translational modification of their carboxyl-terminal CAAX motif by prenylation, with GRK1 being farnesylated and GRK7 geranylated. GRK1 is regulated by Ca+2 through association with the Ca+2-binding protein recoverin, which inhibits kinase activity [18]. Partition of the GRK2 (and GRK3) to the plasma membrane is facilitated by their interaction, through a carboxyl-terminal pleckstrin-homology (PH) domain, with free Gβγ subunits and inositol phospholipids [19–21]. Binding to these partners increases the kinase activity of GRK2 to phosphorylate activated receptors [19, 20]. The transient GRK2 association with the plasma membrane is also facilitated through its RGS-like (RH) domain interaction with palmitoylated, plasma membrane-anchored Gαq-GTP protein (Figure 1). The GRK2 RH domain does not possess a GAP activity. Rather, the binding of GRK2 to Gαq-GTP inhibits the activated Gαq signaling most likely by sequestering it away from its effectors. Targeting of GRK5 to the plasma membrane is accomplished through a combination of an amino-terminal phospholipid binding domain and a carboxyl-terminal amphipathic helix membrane-binding domain [22]. Constitutive expression of GRK4 and GRK6 on the plasma membrane is accomplished through a post-translational palmitoylation modification of carboxyl-terminal cysteine residues [23, 24]. Palmitoylation of GRK6 was reported to increase its kinase activity [25, 26]. Like other GRKs, activation of GRK4 members is regulated by protein-protein interactions and phosphorylation. For example, GRK5 is activated by interaction with substrate GPCRs, but inhibited by binding to Ca+2-calmodulin, actin, or α-actinin [27]. GRK5 is also a substrate for PKC, and PKC phosphorylation inhibits the GRK5 kinase activity [28].
Rapid termination of GPCR signaling is brought about by binding of cytosolic βArrestin proteins to ligand agonist-occupied and GRK-phosphorylated receptors. The binding of βArrestin to receptors uncouples the receptor from its cognate G protein (Figure 1), resulting in a decreased responsiveness of the signaling system to agonist, termed desensitization [1, 29]. There are four βArrestin-like proteins. Arrestin1 and 4, like GRK1 and GRK7, are strictly expressed in the retinal rods and cones, and Arrestin2 and 3 (a.k.a. βArrestin1 and βArrestin2, respectively) are ubiquitously expressed in mammalian cells [1, 3, 4]. All four Arrestin proteins bind GRK-phosphorylated GPCRs leading to blockade of receptor-G protein coupling (Figure 1). The βArrestin1 and 2 proteins exhibit 78% identity in their amino acid sequences and they structurally differ mainly in their carboxyl termini. In distinction from Arrestin1 and 4, the βArrestin proteins form complexes with components of the vesicle trafficking machinery and, therefore, regulate the processes of GPCR endocytosis, intracellular trafficking, resensitization and downregulation [30]. Emerging evidence demonstrates that in addition to their classical roles as terminators of GPCR/G protein signaling, the βArrestins may function as bona fide adaptors and signal transducers from a variety of activated receptor types [31, 32]. Mainly, the βArrestins have been demonstrated to serve as scaffolds to transduce signals involved in mitogenesis, including the activation of JNK and ERK MAP kinases.
GPCR systems are subject to regulatory events involving post-translational modifications, such as phosphorylation, acylation and ubiquitination [33, 34]. These modifications regulate all facets of receptor biology, including receptor signaling, subcellular trafficking and expression. For instance, GRK-mediated phosphorylation of agonist-occupied GPCR converts the receptor to high affinity binding site for βArrestins that, in turn, begin the process of uncoupling the receptor from the G protein and targeting it to clathrin-coated pits for internalization [1, 2, 4, 10]. Likewise, both GRK and βArrestin proteins are subject to post-translational modifications that regulate their activities (vide infra). In this minireview, we will highlight recent discoveries of S-nitrosylation-dependent post-translational modifications of GPCR systems, and their emerging impact on target receptor signaling and expression. Exemplar GRK2, βArrestin2 and dynamin are highlighted as substrates of S-nitrosylated GPCR signaling intermediates.
Protein S-nitrosylation
The production of nitric oxide (NO) is catalyzed by NO synthase (NOS) family of enzymes, which utilize NADPH as an electron donor to oxidize one molecule of L-arginine at the guanidine nitrogen, producing the Nω-OH-L-arginine intermediate that is subsequently oxidized to yield L-citrulline and one molecule of NO [35, 36]. Currently, three distinct NOS isoforms have been cloned and characterized, and they are: nNOS (neuronal NOS or NOS1), iNOS (inducible NOS or NOS2), and eNOS (endothelial NOS or NOS3). In general, eNOS functions as a myristoylated membrane-bound enzyme whereas iNOS and nNOS are soluble proteins [35]. The eNOS and nNOS behave as Ca+2-dependent, constitutively expressed NOS enzymes that generate rapid, but brief, increases in NO synthesis. The Ca+2-independent iNOS is detectable (albeit at very low levels) in resting-states, and its expression can be induced by stimuli such as inflammatory cytokines and bacteria, leading to sustained but delayed increases in NO production. In addition to NOSs, NO may be generated from endogenous higher nitrogen oxides [37] as well as low-molecular weight nitrosothiols (such as S-nitrosoglutathione; GSNO).
In the classical paradigm, NO binds heme iron domain of soluble guanylate cyclase, leading to the synthesis of cyclic guanosine monophosphate (cGMP) that activates downstream mediators such as ion channels, phosphodiesterases and protein kinases [38]. Hence, at physiological concentrations, NO is a vital intracellular signaling molecule. However, prolonged NO overproduction is associated with pathogenesis of a variety of diseases, including inflammation and cancer [39]. Although NO has a relatively short half life (millisec to sec) and is thought to be highly reactive, recent results demonstrate it regulates target cells by cGMP-independent mechanisms. The reaction of NO with superoxide anion to form reactive peroxynitrite, or other NO metabolites, is presumed to also mediate some NO effects in target cells.
Compelling evidence demonstrates that NO participates in S-nitrosylation of cysteine residues in target proteins (to regulate the cellular response) independent of cGMP signaling. NO reversibly modifies the thiol (-SH) group of cysteine residues to nitrosothiols (S-NO) [35, 40, 41]. The precise mechanism of protein S-nitrosylation remains elusive. For example, it is not clear whether direct association with NOS is required for S-nitrosylation, or whether NOS or another enzyme catalyzes the nitrosylation reaction, similar to the actions of protein kinases. Nevertheless, it is well established now that S-nitrosylation occurs as a result of NOS-mediated generation of NO [35, 40, 41]. Evidence linking NOS activity to S-nitrosylation was first demonstrated by Stamler who showed that eNOS-dependent S-nitrosylation endowed proteins with vasodilatory activity [42]. Follow-up work from the same group evidenced the S-nitrosylation of hemoglobin by eNOS [43], ryanodine receptor by nNOS [44], and caspase by iNOS [45]. The Snyder group showed in vivo S-nitrosylation of neuronal proteins, which co-localize with nNOS, was abolished in nNOS−/− mice [46]. Hence, NOS-driven S-nitrosylation seems to be a critical redox-based method of protein post-translational modification, analogous to phosphorylation.
S-nitrosylation reactions are specific, i.e. not every cysteine-containing protein is S-nitrosylated and not every cysteine residue in a protein becomes S-nitrosylated. For example, the ryanodine receptor tetramer contains 84 cysteine residues, but only 12 (i.e. 3 per subunit) are subject to reversible S-nitrosylation [44]. An S-nitrosylation consensus sequence, a sequential 4 amino acid motif consisting of a polar amino acid, an acidic or basic amino acid, a cysteine and an acidic amino acid, has been proposed [42]. A second motif containing adjacent hydrophobic side chains has been suggested in the catalysis of S-nitrosylation reaction [40]. Nonetheless, more work is needed to better establish identity of S-nitrosylation protein motifs. Protein S-nitrosylation may affect various cellular functions, such as enzymatic activity, protein-protein interactions and subcellular localization [35, 40, 41]. Some proteins are activated by physiological S-nitrosylation, including dynamin [47], ryanodine receptor [48], βArrestin2 [49] and small GTPase Ras [50]. Examples of protein functions that are inhibited by physiological S-nitrosylation include eNOS [51] and GRK2 [52].
To date, about a 1000 proteins have been demonstrated to undergo S-nitrosylation modification. However, unlike the case of protein phosphorylation that is carried out by 100s of protein kinases, there exist only three NOSs, casting doubt that the NOSs themselves mediate the overwhelming number of substrate S-nitrosylation reactions. Emerging evidence indicates NO groups may be transferred, in multi-protein complex situations, from a donor S-nitrosylated protein to an acceptor S-nitrosylation substrate [53, 54]. This new finding may explain temporal and spatial requirements of protein S-nitrosylation as participants (and transducers) of signaling networks, and it suggests that protein S-nitrosylation may be carried out not only by the mere 3 characterized NOSs and nitrosothiols, but also by the 100s of S-nitrosylated proteins. Removal of NO from thiol groups (i.e. denitrosylation) is mainly catalyzed by two enzymatic systems: the S-nitrosoglutathione reductase (GSNOR) and the thioredoxin (Trx)/Trx reductase systems (for review see [37]).
GPCR-induced NO production
Under physiologic conditions, NO is produced by eNOS and is maximally activated following the binding to calmodulin. The binding of eNOS to calmodulin serves two functions: to disrupt the inhibitory association with caveolin and to facilitate transfer of electrons between the reductase and oxygenase domains of the enzyme [35]. Several signaling networks converge on Ca+2 release from intracellular stores, thereby providing rapid and regulated mechanisms to activate eNOS through calmodulin. Stimulation of a large number of GPCRs that couple mainly to Gq increases levels of intracellular Ca+2 and consequent eNOS activation. For example, stimulation with angiotensin, thrombin, or bradykinin promotes the Gq-dependent increase in intracellular Ca+2 levels, eNOS activation and NO production.
In addition to calmodulin, eNOS is activated by phosphorylation, and several kinases have been demonstrated to use eNOS as a substrate. Both activating and inactivating eNOS phosphorylation reactions have been demonstrated: phosphorylation of Ser-1177, Ser-635 and Ser-617 residues stimulate while phosphorylation of Thr-495 and Ser-116 inhibit the eNOS activity [55]. In the case of Ser-1177, it may be phosphorylated by several kinases including PKA, Akt and Ca+2/calmodulin-dependent kinase II (CaM kinase II; [56–58]). As we discussed, Gs- and Gq-coupled receptors can activate PKA and CaM kinase II via increased accumulation of intracellular cAMP and Ca+2, respectively. GPCRs can also transduce signals to activate Akt, and it has been reported that Gs-, Gq- and Gi-coupled receptors can activate the phosphatidylinositol 3-OH kinase (PI3K)/Akt module through transactivated receptor tyrosine kinases [2], principally the epidermal growth factor receptor (EGFR). In this pathway, stimulation of the GPCR leads to the release of Gα-GTP and Gβγ subunits, which through incomplete intermediaries induce the tyrosine phosphorylation of EGFR. Subsequent to EGFR phosphorylation, the steps involved in GPCR-mediated and EGFR-mediated Akt activation are indistinguishable. Activated EGFR recruits PI3K leading to its phosphorylation-dependent activation. Other studies have shown that free Gβγ subunits directly bind to PI3Kγ and activate it [59]. Hence, GPCRs may transduce the multiple signaling networks to activate eNOS leading to the production of NO that mediates protein S-nitrosylation.
GPCR S-nitrosylation
One prominent feature of GPCRs signaling is their ability to instantiate feedback loops to inhibit the signal that initiated the original process. For instance, in addition to being phosphorylated by GRKs, GPCRs may be phosphorylated by second messenger-activated protein kinases, such as PKA or PKC [1, 29]. Once activated, these kinases phosphorylate GPCRs regardless of their agonist occupancy status, resulting in a so-called heterologous desensitization. In addition to receptor desensitization, phosphorylation of GPCRs by second messenger-dependent PKA/PKC may lead to a switch in the coupling preference of the GPCR to a different heterotrimeric G protein, thereby altering the multitude of signaling networks initiated by the single agonist-receptor pair. For example, phosphorylation of β2AR by PKA decreases the receptor coupling to Gs (thereby reducing cAMP production) and simultaneously enhances the receptor coupling to Gi that actively inhibits the Gs-mediated adenylyl cyclase signal [60]. The regulated coupling of β2AR to Gi also initiates new signaling networks, such as βArrestin-mediated ERK activation [61].
Chemical NO-donors, including endogenous GSNO, have also been demonstrated to control GPCR activity, albeit the mechanisms of NO action appear to vary depending upon receptor subtype. For example, in situ treatment with NO-donor nitrosothiols disrupted the M2 muscarinic [62] and bradykinin [63] receptor coupling to target G proteins. The treatment with nitrosothiols decreased β2AR-mediated cAMP accumulation, albeit by inhibiting receptor palmitoylation and consequent Gs-coupling [64]. In the case of α1-adrenergic receptor, treatment with GSNO inhibited the catecholamine-induced lung vessel vasoconstriction as a result of decreased ligand binding and increased receptor S-nitrosylation [65]. Likewise, treatment with NO-donor sodium nitroprusside decreased binding affinity of angiotensin II to its AT1 receptor by S-nitrosylation of the receptor on a single Cys-289 residue [66]. In animals, the knockout of GSNOR resulted in increased expression of β2AR in lung and heart tissues, consistent with the observation that GSNO inhibits agonist-induced receptor downregulation [52]. Hence, in parallel to GPCR signal regulation by feedback loops as a result of receptor phosphorylation, NO, which can be produced in response to multiple GPCRs activation, may initiate regulatory feedback loops to control the GPCR signal as a result of receptor S-nitrosylation and inhibition of coupling to effector G proteins.
GRK2 S-nitrosylation
Agonist binding to GPCRs is followed by recruitment of GRKs into close proximity to the activated receptor and, for example, GRK2 is catalytically activated by interaction with activated GPCRs [3, 4, 16]. GRK2 translocation from the cytosol to the plasma membrane is accomplished through binding to activated receptor, acidic inositol phospholipid groups, prenylated Gβγ and palmitoylated Gαq subunits (Figure 2). The GRK2 phosphorylates specific Serine and Threonine residues in the third intracellular loop and carboxyl terminus of agonist-occupied receptor. GRK2 phosphorylation exerts little impact on receptor-G protein coupling, but substantially increases the receptor’s affinity to cytosolic βArrestins that, in turn, interdict the receptor-mediated activation of G proteins [1, 4]. In addition to regulation of its function by subcellular distribution, the activation of GRK2 is also regulated by protein-protein interactions and phosphorylation. For instance, GRK2 activity is reduced by interaction with Ca+2-bound calmodulin [67, 68], caveolin [69], or α-actinin [27]. Phosphorylation of GRK2 by PKC or tyrosine kinase c-Src increases its activity, whilst phosphorylation of GRK2 by ERK decreases the GRK2 kinase activity and promotes its redistribution from the plasma membrane to the cytosol.
Figure 2. S-nitrosylation of GRK2 by eNOS.
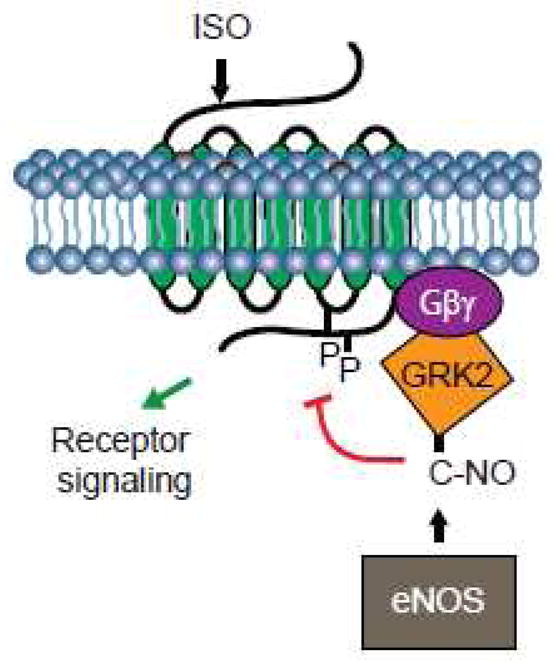
Stimulation of β2 adrenergic receptor with isoproterenol (ISO) promotes the GRK2 S-nitrosylation by eNOS on a single cysteine residue leading to the inhibition of kinase activity. As a result, the receptor escapes phosphorylation and consequent desensitization and down-regulation.
Being mindful that only a handful of GRKs regulate activity of 100s of GPCR and non-GPCR substrates, it is highly likely that multiple regulatory mechanisms exist to control the GRKs subcellular distribution and activity. While it is well accepted that GRKs activity is also regulated by interaction with protein and lipid partners as well as through feedback regulatory phosphorylation modifications, a recent report demonstrated that GRK2 kinase activity is also regulated by β2AR agonist-induced S-nitrosylation [52]. Previous work demonstrated eNOS activation following stimulation of β2AR with isoproterenol through Ca+2 influx- [70, 71] and PKA- and Akt-dependent mechanisms [70]. Isoproterenol induced the eNOS-mediated GRK2 S-nitrosylation, mainly on a single Cys-340 residue [52]. The GRK2 S-nitrosylation inhibited kinase activity as measured by a reduction of agonist-induced β2AR phosphorylation, thereby preventing receptor desensitization and downregulation (Figure 2). It is noteworthy mentioning that Cys-340 is also present in GRK3 but not in other GRKs or related AGC kinases. The Cys-340 is located on a β-sheet that borders the activation loop of the GRK2 catalytic domain and, in member AGC kinases, the loop contains a phosphorylated threonine residue that is required for kinase activity [17, 72]. S-nitrosylation may, therefore, perturb the structure of the GRK2 activation loop resulting in inhibition of kinase activity. By inhibiting β2AR phosphorylation (Figure 2) and consequent downregulation, GRK2 S-nitrosylation may restore receptor function that is dysregulated in many disease states, including cardiovascular and asthma. Notably, levels of S-nitrosylated GRK2 are increased in GSNOR knockout mice [52].
βArrestin S-nitrosylation
Cytosolic βArrestin proteins are enriched on the plasma membrane by binding to ligand-occupied and GRK-phosphorylated GPCR (Figure 3). The βArrestins were functionally characterized based on their ability to sterically hinder activated GPCR interaction with effector G protein, hence instantiating signal termination [1, 29]. Emerging evidence shows additional βArrestin functions (Figure 3). For example, termination of cAMP signal, which is initiated following activation of Gs-coupled receptors, is mediated by its degradation through the phosphodiesterase (PDE) enzymes, and it was recently shown that βArrestins form a complex with PDE4Ds in the cytosol and co-translocate with PDE4Ds to the plasma membrane upon activation of β2AR [73]. Hence, βArrestins can limit cAMP-dependent signals through active degradation of already synthesized cAMP. In the case of Gq-coupled M1 muscarinic receptor, the βArrestins bind to and recruit diacylglycerol kinase (DGK) to the plasma membrane. The DGK uses diacylglycerol as a substrate to generate phosphatidic acid, thereby turning off signaling through diacylglycerol, such as activation of PKC [74]. Lastly, activated D2 dopaminergic receptor recruits βArrestin2 that forms a complex with Akt and the phosphatase PP2A [75]. Dephosphorylation of Akt by PP2A results in the D2 receptor-mediated inactivation of Akt that contributes to the locomoter activity of dopaminergic neurotransmission. These recent observations provide new ways wherein βArrestins limit second messenger-dependent signals not only by interdicting receptor-G protein coupling, but also through active degradation of already synthesized second messengers (Figure 3).
Figure 3. βArrestin-mediated signaling.
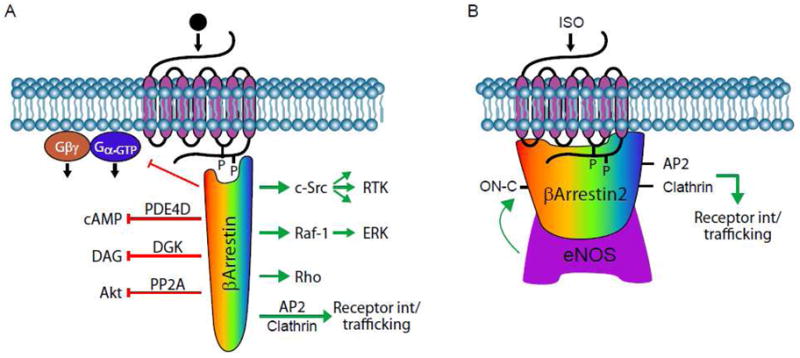
A, Stimulation of a GPCR with ligand agonist promotes the GRK-mediated receptor phosphorylation and consequent recruitment of cytosolic βArrestin. Depending upon the nature of ligand-receptor pair and post-translational modification of βArrestin itself, the βArrestin can adopt a specific conformation that allows it to select an interacting partner(s) that, in turn, dictate the cellular response. B, S-nitrosylation of βArrestin2 by eNOS following β2 adrenergic receptor stimulation with isoproterenol (ISO). The S-nitrosylated βArrestin2 favors interaction with endocytic machinery components clathrin and AP-2, leading to receptor internalization through clathrin-coated endocytic portals.
Evidence that βArrestins directly impact signal transduction (other than to affect receptor desensitization) came, unexpectedly, from studies aimed at determining the cellular response to modulating vesicle trafficking [76–78]. Pharmacologic inhibitors of vesicle fission [77] or forced over-expression of dominant negative forms of βArrestin1 [76, 78] resulted in diminished receptor-mediated activation of the ERK MAP kinases. Support for the idea that βArrestins play a role in GPCR-mediated ERK activation came with the observation that βArrestin1 served as a binding partner for activated c-Src [61], and mutations in βArrestin1 that prevent its interaction with receptor or c-Src inhibited β2AR stimulation of ERK (Figure 3). Soon thereafter, activation of the neurokinin1 receptor with substance P was shown to attenuate cell apoptosis in a mechanism that depended on βArrestin-mediated recruitment of c-Src [79]. These studies provided confidence that βArrestins initiate a ‘second-wave’ signaling from the same receptors that they desensitize. DeFea and colleagues demonstrated that agonist stimulation resulted in formation of a complex containing activated PAR2, βArrestin1, Raf-1, and phosphorylated ERK [80], and the Lefkowitz group showed that stimulation with angiotensin II induced the βArrestin2, Raf-1, MEK1, and phosphorylated ERK complex formation [81]. Collectively, these studies identified the additional function of βArrestins to act as scaffolds bringing into close proximity activated receptors and (ERK MAP kinase) signaling intermediates.
A cardinal requirement for GPCR signal initiation is ligand binding-induced conformation changes in the receptor structure [5] that allows it to couple to target G proteins and to exert a GEF function. Likewise, the binding of βArrestin to receptor promotes conformational changes in βArrestin, permitting its interaction with downstream effectors [82]. Choice of binding partner may be controlled by nuanced changes in βArrestin structure that may result from binding to activated GPCR, or undergoing post-translational modifications. Indeed, βArrestins have been demonstrated to undergo the regulated phosphorylation [83, 84], ubiquitination [85] and more recently S-nitrosylation [49]. For example, βArrestin1 is phosphorylated on Ser-412 by ERK and βArrestin2 is phosphorylated mainly on Thr-383 by casein kinase II [83, 84, 86]. Phosphorylation of βArrestins selectively regulates their function: while the binding of βArrestins to GPCRs and consequent GPCR desensitization is not affected, their binding to clathrin and subsequent receptor internalization requires dephosphorylation, albeit by as yet unidentified phosphatase(s). The βArrestins also serve as substrates for ubiquitination [85], the addition of a 76 amino acid polypeptide ubiquitin. Upon receptor activation by agonist, βArrestins are polyubiquitinated by RING finger E3 ligase MDM2 that, in turn, regulates GPCR internalization and trafficking. For example, activation of β2AR, which dissociates from βArrestin before internalization, results in transient ubiquitination of the βArrestin. On the other hand, activation of V2 vasopressin receptor, which internalizes together with βArrestin, induces prolonged ubiquitination of the βArrestin [87], suggesting that the ubiquitination status of βArrestin determines stability of the receptor-βArrestin complex and the trafficking pattern of the receptor. The enzyme ubiquitin-specific protease 33 was recently shown to catalyze the deubiquitination of βArrestin [88], establishing feedback regulatory mechanisms to control the βArrestin function. In addition to serving as substrates for ubiquitination, the βArrestins also serve as adaptors that bring the E3 ligase into close proximity to target GPCRs, leading to receptor ubiquitination that regulates the receptors’ endocytic trafficking and lysosomal targeting.
Recently, βArrestin2, but not βArrestin1, was shown to be S-nitrosylated on Cys-410 that provided an additional regulatory step in the life cycle of GPCR [49]. The βArrestin2 was shown to form a complex with eNOS, and stimulation of β2AR, which activates eNOS, promoted the dynamic βArrestin2-eNOS complex formation and βArrestin2 S-nitrosylation (Figure 3). Active β2AR-induced S-nitrosylation of βArrestin2 resulted in the dissociation of βArrestin2 from eNOS and association with the endocytic machinery components clathrin and AP-2, thereby facilitating an increase in the rate of β2AR trafficking from the plasma membrane. The βArrestin2 also forms a complex with iNOS leading to increased NO production [89]. Here, stimulation of bradykinin receptor 1 (B1R) initiates a βArrestin2 (but not βArrestin1) →ERK → iNOS signal that promotes the iNOS phosphorylation and consequent NO synthesis [89]. Of note, EGF-induced activation of ERK was shown to be insufficient to activate iNOS, suggesting pools of βArrestin- and EGF-activated ERK are distinct, or βArrestin2 recruits additional mediators involved in the B1R-regulated iNOS activation.
Functionally, it appears that dephosphorylation of βArrestin1 on Ser-412 exerts similar effects as S-nitrosylation of Cys-410 on βArrestin2 (as assayed with binding to clathrin/AP-2 and β2AR internalization), and it is interesting to note that Ser-412 (in βArrestin1) and Cys-410 (in βArrestin2) are not conserved in βArrestin2 and βArrestin1, respectively. Hence, βArrestin2 S-nitrosylation mirrors βArrestin1 dephosphoryation in the context of β2AR internalization, supporting the idea that S-nitrosylation and phosphorylation reactions may operate in unison to regulate GPCR trafficking. Remarkably, comprehensive proteomic analyses revealed βArrestin2 interacts with over 250 partner proteins [90] that are involved in cellular signaling, cytoskeletal organization, and nucleic acid binding. However, the functional consequence of S-nitrosylation on these interactions awaits further investigation, and it will not be surprising if some of these interactions are dictated by βArrestin2 S-nitrosylation status.
Dynamin S-nitrosylation
GPCRs undergo constitutive and agonist-stimulated endocytosis and the internalized receptors can recycle back to the plasma membrane for reuse, or can be targeted to lysosomes for degradation. Internalization of GPCRs, in particular, and cell surface receptors, in general, is typically mediated by invaginations of the plasma membrane regions that are coated with clathrin (i.e. clathrin coated pits), or enriched in caveolin (i.e. caveolae) proteins [91]. Notably, late-stage “pinching off” and internalization of either clathrin-coated pits or caveolae are dependent upon large GTPase dynamin that facilitates fission of these endocytic vesicles from the plasma membrane [92]. Unlike classical small GTPases and α subunits of heterotrimeric G proteins, dynamin has a relatively low affinity for GTP binding (Km, 10–100 μM), high intrinsic rate of GTP hydrolysis (Kcat, 2–100 min−1), and its GTPase activity is increased (5–10 fold over basal) as a result of self assembly [93, 94]. In the native state, dynamin exists as homotetramer [92, 95]. When diluted into low ionic strength conditions in vitro, dynamin tetramers can spontaneously self assemble into a supermolecular structure consisting of spiral stacks of rings (Figure 4), similar to the structure observed in vivo in which dynamin appears as an electron dense collar around the necks of endocytic pits [96, 97]. Upon GTP binding, dynamin self-assembles into spiral collars at the necks of invaginations, which is thought to stimulate its GTPase activity, thereby allowing it to mediate scission of budding vesicles from the plasma membrane [92].
Figure 4. Dynamin S-nitrosylation promotes the G protein-coupled receptor internalization.
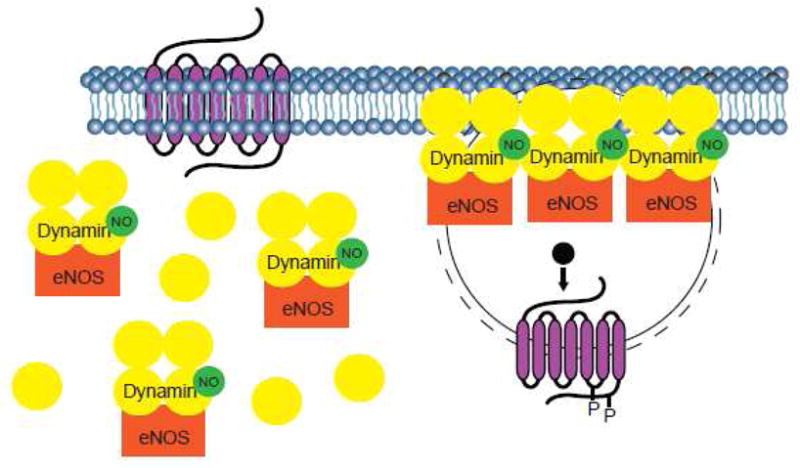
In the absence of agonist, dynamin tetramers may be bound to eNOS and present in the cytosol. Upon stimulation with agonist, eNOS induces the S-nitrosylation of dynamin that, in turn, promotes the dynamin recruitment to the neck of invaginated pits on the plasma membrane. The S-nitrosylation also increases dynamin self-assembly and its GTPase activity, thereby enhancing rate of vesicle scission from the plasma membrane and internalization of activated receptor.
Three distinct mammalian dynamin genes have been recognized: neuronal dynamin1, ubiquitously expressed dynamin2, and dynamin3, which is detected only in brain, testis and lung [92]. All three genes encode at least four alternatively spliced variants that could result in expression of at least twelve different isoforms. All dynamin isoforms consist of four distinct structural domains: a highly conserved N-terminal GTPase domain represents the catalytic workhorse of dynamin; mutations within this domain (i.e. K44A) result in abolishment of GTP binding and hydrolysis, leading to the inhibition of vesicle detachment from the plasma membrane. A central PH domain binds phosphoinositides and other proteins and is thought to mediate dynamin’s recruitment to the plasma membrane [93]. A GTPase effector domain (GED) encodes for GAP activity, which interacts with the GTPase domain of the same protein monomer, or of neighboring dynamin monomers [94]. Finally, a C-terminal proline-arginine rich domain (PRD) is thought to bind other proteins, such as the tyrosine kinase c-Src, and we demonstrated that dynamin1 is a c-Src substrate [98] and that c-Src-mediated phosphorylation regulates the dynamin1 function in receptor endocytosis [99]. Follow-up reports demonstrated the c-Src-mediated tyrosine phosphorylation of dynamin2 and its requirement to mediate the vesicle fission [100, 101].
Dynamin GTPase activity is controlled by self oligomerization that is regulated by dynamin membrane recruitment to coated pits, and PH domain of dynamin interacts with membrane lipids [92]. However, the affinity of the PH domain to membrane lipids is low (1 mM for inositol-1,4,5-trisphosphate), suggesting that dynamin may employ alternate mechanisms to translocate to membranes. Indeed, βγ subunits of heterotrimeric G proteins have been implicated in the recruitment of dynamin to the plasma membrane following GPCR activation [102]. Dynamin membrane recruitment may also occur via interaction of its PRD with SH3-domain containing proteins, such as amphiphysin. Another PRD-interacting protein that may control dynamin membrane recruitment is eNOS (Figure 4). Accordingly, dynamin and eNOS colocalize within membrane compartments and immunoprecipitation of eNOS from endothelial cells coprecipitates dynamin [103]. In vitro, dynamin binds directly to eNOS with high affinity (Kd ~ 30 nM) and overexpression of dynamin2 increases the eNOS activity [104].
The binding of eNOS to dynamin exerts a reciprocal activation of dynamin [47] and related protein Drp1 [105] through a mechanism that involves S-nitrosylation (Figure 4). In vitro and in situ studies show that dynamin1 [47], dynamin2 [106] and Drp1 [105] are subject to S-nitrosylation modifications. Dynamin1 is S-nitrosylated on Cys-607 [47], dynamin2 on Cys-86 and Cys-607 [106], and Drp1 on Cys-644 [105]. In all cases, S-nitrosylation appears to be required for optimal dynamin function. Forced overexpression of dynamin1 C607A, for example, reduces rate of agonist-induced β2AR internalization [47]. Similarly, overexpression of dynamin2 C86/607A decreases the bacteria invasion of bladder epithelial cells [107]. Drp1 S-nitrosylation triggers mitochondrial fission, synaptic loss, and neuronal damage that are key mediators of Alzheimer’s disease [105]. The S-nitrosylation promotes dynamin self-assembly and increases GTPase activity (Figure 4), thus accelerating the process of vesicle fission from the plasma membrane (for dynamin1 and dynamin2) and mitochondrial fragmentation (for Drp1). Hence, the binding of dynamin to eNOS may serve as an anchor for dynamin, thereby stabilizing dynamin at the plasma membrane (Figure 4). The S-nitrosylation-dependent interactions between dynamin and eNOS may regulate subcellular localization as well as multimerization of dynamin and its assembly-linked activities.
Conclusions and perspectives
Protein modification by S-nitrosylation, like phosphorylation, is now recognized as a regulatory mechanism involved in propagating as well as fine-tuning signaling networks from a variety of receptor types (including G protein-coupled, tyrosine kinase and nuclear receptors). It is evident that GPCR systems undergo multiple specific modifications during their life cycle, and it will be important to elucidate effects of these modifications in combination and not in isolation. Integrative post-translational modifications approach of GPCR systems that incorporate S-nitrosylation may provide a more comprehensive understanding of mechanisms controlling receptor expression and function. Also important is the establishment of possible existence of cross-talk between the different types of post-translational modifications, which is predicted to control the temporal and spatial GPCR signaling networks. Future studies on the molecular mechanisms by which GPCR system S-nitrosylation modifications regulate various cellular processes will likely improve our understanding of some human diseases and may provide rationale for discovery of effective drugs to improve their clinical outcomes.
Acknowledgments
This work was supported, in part, by grants CA129155 and AI079014 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 2.Daaka Y. G proteins in cancer: The prostate cancer paradigm. Science STKE. 2004;re2:1–10. doi: 10.1126/stke.2162004re2. [DOI] [PubMed] [Google Scholar]
- 3.Premont RT. Once and future signaling - G protein-coupled receptor kinase control of neuronal sensitivity. Neuromol Med. 2005;7:129–147. doi: 10.1385/NMM:7:1-2:129. [DOI] [PubMed] [Google Scholar]
- 4.Moore CAC, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- 5.Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelly P, Casey PJ, Meigs TE. Biologic functions of the G12 subfamily of heterotrimeric G proteins: Growth, migration, and Metastasis. Biochemistry. 2007;46:6677–6687. doi: 10.1021/bi700235f. [DOI] [PubMed] [Google Scholar]
- 7.Rasmussen SGF, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VRP, Sanishvili R, Fischetti RF, Schertler GFX, Weis WI, Kobilka BK. Crystal structure of the human beta(2) adrenergic G-protein-coupled receptor. Nature. 2007;450:383–U384. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 8.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into β2 adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 9.Kenakin T. New concepts in drug discovery: Collateral efficacy and permissive antagonism. Nat Rev Drug Discov. 2005;4:919–927. doi: 10.1038/nrd1875. [DOI] [PubMed] [Google Scholar]
- 10.Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, Shenoy SK, Lefkowitz RJ. A unique mechanism of β-blocker action: carvedilol stimulates βArrestin signaling. Proc Natl Acad Sci US A. 2007;104:16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marinissen MJ, Gutkind JS. Scaffold proteins dictate Rho GTPase-signaling specificity. Trends Biochem Sci. 2005;30:423–426. doi: 10.1016/j.tibs.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 15.Penela P, Murga C, Ribas C, Lafarga V, Mayor F. The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol. 2010;160:821–832. doi: 10.1111/j.1476-5381.2010.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lodowski DT, Pitcher JA, Capel WD, Lefkowitz RJ, Tesmer JJG. Keeping G proteins at bay: A complex between G protein-coupled receptor kinase 2 and Gβγ. Science. 2003;300:1256–1262. doi: 10.1126/science.1082348. [DOI] [PubMed] [Google Scholar]
- 17.Tobin AB, Butcher AJ, Kong KC. Location, location, location … site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci. 2008;29:413–420. doi: 10.1016/j.tips.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorodovikova EN, Senin II, Philippov PP. Calcium-sensitive control of rhodopsin phosphorylation in the reconstituted system consisting of photoreceptor - membranes, rhodopsin kinase and recoverin. FEBS Lett. 1994;353:171–172. doi: 10.1016/0014-5793(94)01030-7. [DOI] [PubMed] [Google Scholar]
- 19.Pitcher JA, Inglese J, Higgins JB, Arriza JL, Casey PJ, Kim C, Benovic JL, Kwatra MM, Caron MG, Lefkowitz RJ. Role of βγ-subunits of G proteins in targeting the βAdrenergic receptor kinase to membrane-bound receptors. Science. 1992;257:1264–1267. doi: 10.1126/science.1325672. [DOI] [PubMed] [Google Scholar]
- 20.Pitcher JA, Fredericks ZL, Stone WC, Premont RT, Stoffel RH, Koch WJ, Lefkowitz RJ. Phosphatidylinositol 4,5-bisphosphate (PIP2)-enhanced G protein-coupled receptor kinase (GRK) activity - Location, structure, and regulation of the PIP2 binding site distinguishes the GRK subfamilies. J Biol Chem. 1996;271:24907–24913. doi: 10.1074/jbc.271.40.24907. [DOI] [PubMed] [Google Scholar]
- 21.Daaka Y, Pitcher JA, Richardson M, Stoffel RH, Robishaw JD, Lefkowitz RJ. Receptor and Gβγ isoform-specific interactions with G protein-coupled receptor kinases. Proc Natl Acad Sci US A. 1997;94:2180–2185. doi: 10.1073/pnas.94.6.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Premont RT, Koch WJ, Inglese J, Lefkowitz RJ. Identification, purification, and characterization of GRK5, a member of the family of G protein-coupled receptor kinases. J Biol Chem. 1994;269:6832–6841. [PubMed] [Google Scholar]
- 23.Stoffel RH, Randall RR, Premont RT, Lefkowitz RJ, Inglese J. Palmitoylation of G protein-coupled receptor kinase, GRK6 - lipid modification diversity in the GRK family. J Biol Chem. 1994;269:27791–27794. [PubMed] [Google Scholar]
- 24.Premont RT, Macrae AD, Stoffel RH, Chung NJ, Pitcher JA, Ambrose C, Inglese J, MacDonald ME, Lefkowitz RJ. Characterization of the G protein-coupled receptor kinase GRK4 - Identification of four splice variants. J Biol Chem. 1996;271:6403–6410. doi: 10.1074/jbc.271.11.6403. [DOI] [PubMed] [Google Scholar]
- 25.Loudon RP, Benovic JL. Altered activity of palmitoylation-deficient and isoprenylated forms of the G protein-coupled receptor kinase GRK6. J Biol Chem. 1997;272:27422–27427. doi: 10.1074/jbc.272.43.27422. [DOI] [PubMed] [Google Scholar]
- 26.Stoffel RH, Inglese J, Macrae AD, Lefkowitz RJ, Premont RT. Palmitoylation increases the kinase activity of the G protein-coupled receptor kinase, GRK6. Biochemistry. 1998;37:16053–16059. doi: 10.1021/bi981432d. [DOI] [PubMed] [Google Scholar]
- 27.Freeman JLR, Pitcher JA, Li XL, Bennett V, Lefkowitz RJ. αActinin is a potent regulator of G protein-coupled receptor kinase activity and substrate specificity in vitro. FEBS Lett. 2000;473:280–284. doi: 10.1016/s0014-5793(00)01543-x. [DOI] [PubMed] [Google Scholar]
- 28.Pronin AN, Benovic JL. Regulation of the G protein-coupled receptor kinase GRK5 by protein kinase C. J Biol Chem. 1997;272:3806–3812. doi: 10.1074/jbc.272.6.3806. [DOI] [PubMed] [Google Scholar]
- 29.Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Annu Rev Biochem. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- 30.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. βArrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 31.DeFea KA. Stop that cell! beta-arrestin-dependent chemotaxis: A tale of localized actin assembly and receptor desensitization. Annu Rev of Physiol. 2007;69:535–560. doi: 10.1146/annurev.physiol.69.022405.154804. [DOI] [PubMed] [Google Scholar]
- 32.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by βArrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 33.Torrecilla I, Tobin AB. Co-ordinated covalent modification of G protein-coupled receptors. Current Pharm Design. 2006;12:1797–1808. doi: 10.2174/138161206776873716. [DOI] [PubMed] [Google Scholar]
- 34.Tobin AB. G protein-coupled receptor phosphorylation: where, when and by whom. Br J Pharmacol. 2008;153:S167–S176. doi: 10.1038/sj.bjp.0707662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dudzinski DM, Igarashi J, Greif D, Michel T. The regulation and pharmacology of endothelial nitric oxide synthase. Annu Rev Pharmacol Toxicol. 2006;46:235–276. doi: 10.1146/annurev.pharmtox.44.101802.121844. [DOI] [PubMed] [Google Scholar]
- 36.Luiking YC, Engelen MP, Deutz NE. Regulation of nitric oxide production in health and disease. Current Opin Clin Nut Metab Care. 2010;13:97–104. doi: 10.1097/MCO.0b013e328332f99d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10:721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 38.Murad F. Shattuck Lecture: Nitric oxide and cyclic GMP in cell signaling and drug development. New England J Med. 2006;355:2003–2011. doi: 10.1056/NEJMsa063904. [DOI] [PubMed] [Google Scholar]
- 39.Pervin S, Chaudhuri G, Singh R. NO to Breast: When, Why and Why Not? Current Pharm Design. 2010;16:451–462. doi: 10.2174/138161210790232130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: Purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 41.Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S)NO signals: Translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 43.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: A dynamic activity of blood involved in vascular control. Nature. 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 44.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 45.Eu JP, Liu LM, Zeng M, Stamler JS. An apoptotic model for nitrosative stress. Biochemistry. 2000;39:1040–1047. doi: 10.1021/bi992046e. [DOI] [PubMed] [Google Scholar]
- 46.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 47.Wang GF, Moniri NH, Ozawa K, Stamler JS, Daaka Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc Natl Acad Sci US A. 2006;103:1295–1300. doi: 10.1073/pnas.0508354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonzalez DR, Beigi F, Treuer AV, Hare JM. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci US A. 2007;104:20612–20617. doi: 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ozawa K, Whalen EJ, Nelson CD, Mu YY, Hess DT, Lefkowitz RJ, Stamler JS. S-nitrosylation of βArrestin regulates βAdrenergic receptor trafficking. Mol Cell. 2008;31:395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lim KH, Ancrile BB, Kashatus DF, Counter CM. Tumour maintenance is mediated by eNOS. Nature. 2008;452:646–U611. doi: 10.1038/nature06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Erwin PA, Lin AJ, Golan DE, Michel T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2005;280:19888–19894. doi: 10.1074/jbc.M413058200. [DOI] [PubMed] [Google Scholar]
- 52.Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, Koch WJ, Daaka Y, Lefkowitz RJ, Stamler JS. Regulation of βAdrenergic receptor signaling by S-nitrosylation of G protein-coupled receptor kinase 2. Cell. 2007;129:511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 53.Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JVK, Snowman AM, Law L, Hester LD, Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol. 2010;12:1094–U1089. doi: 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakamura T, Wang L, Wong CCL, Scott FL, Eckelman BP, Han XM, Tzitzilonis C, Meng FJ, Gu ZZ, Holland EA, Clemente AT, Okamoto S, Salvesen GS, Riek R, Yates JR, Lipton SA. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol Cell. 2010;39:184–195. doi: 10.1016/j.molcel.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kolluru GK, Siamwala JH, Chatterjee S. eNOS phosphorylation in health and disease. Biochimie. 2010;92:1186–1198. doi: 10.1016/j.biochi.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 56.Boo YC, Sorescu G, Boyd N, Shiojima L, Walsh K, Du J, Jo HJ. Shear stress stimulates phosphorylation of endothelial nitric oxide synthase at Ser(1179) by Akt-independent mechanisms - Role of protein kinase A. J Biol Chem. 2002;277:3388–3396. doi: 10.1074/jbc.M108789200. [DOI] [PubMed] [Google Scholar]
- 57.Boo YC, Hwang J, Sykes M, Michell BJ, Kemp BE, Lum H, Jo H. Shear stress stimulates phosphorylation of eNOS at Ser(635) by a protein kinase A-dependent mechanism. Am J Physiol Heart Circ Physiol. 2002;283:H1819–H1828. doi: 10.1152/ajpheart.00214.2002. [DOI] [PubMed] [Google Scholar]
- 58.Michell BJ, Harris MB, Chen ZP, Ju H, Venema VJ, Blackstone MA, Huang W, Venema RC, Kemp BE. Identification of regulatory sites of phosphorylation of the bovine endothelial nitric oxide synthase at serine 617 and serine 635. J Biol Chem. 2002;277:42344–42351. doi: 10.1074/jbc.M205144200. [DOI] [PubMed] [Google Scholar]
- 59.LopezIlasaca M, Crespo P, Pellici PG, Gutkind JS, Wetzker R. Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase γ. Science. 1997;275:394–397. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- 60.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the β2 adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 61.Luttrell LM, Ferguson SSG, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin FT, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. beta-arrestin-dependent formation of β2 adrenergic receptor Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 62.Aronstam RS, Martin DC, Dennison RL, Cooley HG. S-nitrosylation of M2 muscarinic receptor thiols disrupts receptor-G protein coupling. In: Abood LG, Lajtha A, editors. Diversity of Interacting Receptors. Vol. 757. 1995. pp. 215–217. [DOI] [PubMed] [Google Scholar]
- 63.Miyamoto A, Laufs U, Pardo C, Liao JK. Modulation of bradykinin receptor ligand binding affinity and its coupled G proteins by nitric oxide. J Biol Chem. 1997;272:19601–19608. doi: 10.1074/jbc.272.31.19601. [DOI] [PubMed] [Google Scholar]
- 64.Adam L, Bouvier M, Jones TLZ. Nitric oxide modulates β2 adrenergic receptor palmitoylation and signaling. J Biol Chem. 1999;274:26337–26343. doi: 10.1074/jbc.274.37.26337. [DOI] [PubMed] [Google Scholar]
- 65.Nozik-Grayck E, Whalen EJ, Stamler JS, McMahon TJ, Chitano P, Piantadosi CA. S-nitrosoglutathione inhibits α1 adrenergic receptor-mediated vasoconstriction and ligand binding in pulmonary artery. Am J Physiol Lung Cell Mol Physiol. 2006;290:L136–L143. doi: 10.1152/ajplung.00230.2005. [DOI] [PubMed] [Google Scholar]
- 66.Leclerc PC, Lanctot PM, Auger-Messier M, Escher E, Leduc R, Guillemette G. S-nitrosylation of cysteine 289 of the AT(1) receptor decreases its binding affinity for angiotensin II. Br J Pharmacol. 2006;148:306–313. doi: 10.1038/sj.bjp.0706725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chuang TT, Paolucci L, DeBlasi A. Inhibition of G protein-coupled receptor kinase subtypes by Ca2+/calmodulin. J Biol Chem. 1996;271:28691–28696. doi: 10.1074/jbc.271.45.28691. [DOI] [PubMed] [Google Scholar]
- 68.Pronin AN, Satpaev DK, Slepak VZ, Benovic JL. Regulation of G protein-coupled receptor kinases by calmodulin and localization of the calmodulin binding domain. J Biol Chem. 1997;272:18273–18280. doi: 10.1074/jbc.272.29.18273. [DOI] [PubMed] [Google Scholar]
- 69.Carman CV, Lisanti MP, Benovic JL. Regulation of G protein-coupled receptor kinases by caveolin. J Biol Chem. 1999;274:8858–8864. doi: 10.1074/jbc.274.13.8858. [DOI] [PubMed] [Google Scholar]
- 70.Ferro A, Queen LR, Priest RM, Xu BA, Ritter JM, Poston L, Ward JPT. Activation of nitric oxide synthase by β2 adrenoceptors in human umbilical vein endothelium in vitro. Br J Pharmacol. 1999;126:1872–1880. doi: 10.1038/sj.bjp.0702512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Figueroa XF, Poblete I, Fernandez R, Pedemonte C, Cortes V, Huidobro-Toro JP. NO production and eNOS phosphorylation induced by epinephrine through the activation of β-adrenoceptors. Am J Physiol Heart Circ Physiol. 2009;297:H134–H143. doi: 10.1152/ajpheart.00023.2009. [DOI] [PubMed] [Google Scholar]
- 72.Lima B, Forrester MT, Hess DT, Stamler JS. S-Nitrosylation in Cardiovascular Signaling. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ. Targeting of cyclic AMP degradation to β2 adrenergic receptors by beta-arrestins. Science. 2002;298:834–836. doi: 10.1126/science.1074683. [DOI] [PubMed] [Google Scholar]
- 74.Nelson CD, Perry SJ, Regier DS, Prescott SM, Topham MK, Lefkowitz RJ. Targeting of diacylglycerol degradation to M1 muscarinic receptors by βArrestins. Science. 2007;315:663–666. doi: 10.1126/science.1134562. [DOI] [PubMed] [Google Scholar]
- 75.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/βArrestin2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 76.Daaka Y, Luttrell LM, Ahn S, Della Rocca GJ, Ferguson SSG, Caron MG, Lefkowitz RJ. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998;273:685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- 77.Luttrell LM, Daaka Y, DellaRocca GJ, Lefkowitz RJ. G protein-coupled receptors mediate two functionally distinct pathways of tyrosine phosphorylation in rat 1a fibroblasts - Shc phosphorylation and receptor endocytosis correlate with activation of Erk kinases. J Biol Chem. 1997;272:31648–31656. doi: 10.1074/jbc.272.50.31648. [DOI] [PubMed] [Google Scholar]
- 78.Pierce KL, Maudsley S, Daaka Y, Luttrell LM, Lefkowitz RJ. Role of endocytosis in the activation of the extracellular signal-regulated kinase cascade by sequestering and nonsequestering G protein-coupled receptors. Proc Natl Acad Sci US A. 2000;97:1489–1494. doi: 10.1073/pnas.97.4.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.DeFea KA, Vaughn ZD, O’Bryan EM, Nishijima D, Dery O, Bunnett NW. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a βArrestin-dependent scaffolding complex. Proc Natl Acad Sci US A. 2000;97:11086–11091. doi: 10.1073/pnas.190276697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ge L, Ly Y, Hollenberg M, DeFea K. A βArrestin-dependent scaffold is associated with prolonged MAPK activation in pseudopodia during protease-activated receptor-2-induced chemotaxis. J Biol Chem. 2003;278:34418–34426. doi: 10.1074/jbc.M300573200. [DOI] [PubMed] [Google Scholar]
- 81.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Activation and targeting of extracellular signal-regulated kinases by βArrestin scaffolds. Proc Natl Acad Sci US A. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin - sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- 83.Lin FT, Krueger KM, Kendall HE, Daaka Y, Fredericks ZL, Pitcher JA, Lefkowitz RJ. Clathrin-mediated endocytosis of the βAdrenergic receptor is regulated by phosphorylation/dephosphorylation of βArrestin1. J Biol Chem. 1997;272:31051–31057. doi: 10.1074/jbc.272.49.31051. [DOI] [PubMed] [Google Scholar]
- 84.Lin FT, Miller WE, Luttrell LM, Lefkowitz RJ. Feedback regulation of βArrestin1 function by extracellular signal-regulated kinases. J Biol Chem. 1999;274:15971–15974. doi: 10.1074/jbc.274.23.15971. [DOI] [PubMed] [Google Scholar]
- 85.Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated β2 adrenergic receptor and βArrestin. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- 86.Kim YM, Barak LS, Caron MG, Benovic JL. Regulation of arrestin3 phosphorylation by casein kinase II. J Biol Chem. 2002;277:16837–16846. doi: 10.1074/jbc.M201379200. [DOI] [PubMed] [Google Scholar]
- 87.Martin NP, Lefkowitz RJ, Shenoy SK. Regulation of V2 vasopressin receptor degradation by agonist-promoted ubiquitination. J Biol Chem. 2003;278:45954–45959. doi: 10.1074/jbc.M308285200. [DOI] [PubMed] [Google Scholar]
- 88.Shenoy SK, Modi AS, Shukla AK, Xiao KH, Berthouze M, Ahn S, Wilkinson KD, Miller WE, Lefkowitz RJ. βArrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc Natl Acad Sci US A. 2009;106:6650–6655. doi: 10.1073/pnas.0901083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kuhr FK, Zhang YK, Brovkovych V, Skidgel RA. beta-Arrestin 2 is required for B1 receptor-dependent post-translational activation of inducible nitric oxide synthase. FASEB J. 2010;24:2475–2483. doi: 10.1096/fj.09-148783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, Yates JR, Lefkowitz RJ. Functional specialization of βArrestin interactions revealed by proteornic analysis. Proc Natl Acad Sci US A. 2007;104:12011–12016. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lajoie P, Nabi IR. Lipid rafts, caveolae, and their endocytosis. Int Rev Cell Mol Biol. 2010;282:135–163. doi: 10.1016/S1937-6448(10)82003-9. [DOI] [PubMed] [Google Scholar]
- 92.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 93.Warnock DE, Hinshaw JE, Schmid SL. Dynamin self-assembly stimulates its GTPase activity. J Biol Chem. 1996;271:22310–22314. doi: 10.1074/jbc.271.37.22310. [DOI] [PubMed] [Google Scholar]
- 94.Sever S, Muhlberg AB, Schmid SL. Impairment of dynamin’s GAP domain stimulates receptor-mediated endocytosis. Nature. 1999;398:481–486. doi: 10.1038/19024. [DOI] [PubMed] [Google Scholar]
- 95.Muhlberg AB, Warnock DE, Schmid SL. Domain structure and intramolecular regulation of dynamin GTPase. EMBO J. 1997;16:6676–6683. doi: 10.1093/emboj/16.22.6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kosaka T, Ikeda K. Reversible blockage of membrane retrieval and endocytosis in the garland cell of the temperature-sensitive mutant of drosophila melanogaster, shibire ts1. J Cell Biol. 1983;97:499–507. doi: 10.1083/jcb.97.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Takei K, McPherson PS, Schmid SL, Decamilli P. Tubular membrane invaginations coated by dynamin rings are induced by GTPγS in nerve terminals. Nature. 1995;374:186–190. doi: 10.1038/374186a0. [DOI] [PubMed] [Google Scholar]
- 98.Ahn S, Maudsley S, Luttrell LM, Lefkowitz RJ, Daaka Y. Src-mediated tyrosine phosphorylation of dynamin is required for β2 adrenergic receptor internalization and mitogen-activated protein kinase signaling. J Biol Chem. 1999;274:1185–1188. doi: 10.1074/jbc.274.3.1185. [DOI] [PubMed] [Google Scholar]
- 99.Ahn S, Kim J, Lucaveche CL, Reedy MC, Luttrell LM, Lefkowitz RJ, Daaka Y. Src-dependent tyrosine phosphorylation regulates dynamin self-assembly and ligand-induced endocytosis of the epidermal growth factor receptor. J Biol Chem. 2002;277:26642–26651. doi: 10.1074/jbc.M201499200. [DOI] [PubMed] [Google Scholar]
- 100.Shajahan AN, Timblin BK, Sandoval R, Tiruppathi C, Malik AB, Minshall RD. Role of Src-induced dynamin2 phosphorylation in caveolae-mediated endocytosis in endothelial cells. J Biol Chem. 2004;279:20392–20400. doi: 10.1074/jbc.M308710200. [DOI] [PubMed] [Google Scholar]
- 101.Tosoni D, Puri C, Confalonieri S, Salcini AE, De Camilli P, Tacchetti C, Di Fiore PP. TTP specifically regulates the internalization of the transferrin receptor. Cell. 2005;123:875–888. doi: 10.1016/j.cell.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 102.Lin HC, Gilman AG. Regulation of dynamin I GTPase activity by G protein βγ subunits and phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 1996;271:27979–27982. doi: 10.1074/jbc.271.45.27979. [DOI] [PubMed] [Google Scholar]
- 103.Cao S, Yao J, McCabe TJ, Yao Q, Katusic ZS, Sessa WC, Shah V. Direct interaction between endothelial nitric oxide synthase and dynamin2 - Implications for nitric oxide synthase function. J Biol Chem. 2001;276:14249–14256. doi: 10.1074/jbc.M006258200. [DOI] [PubMed] [Google Scholar]
- 104.Cao S, Yao J, Shah V. The proline-rich domain of dynamin2 is responsible for dynamin-dependent in vitro potentiation of endothelial nitric oxide synthase activity via selective effects on reductase domain function. J Biol Chem. 2003;278:5894–5901. doi: 10.1074/jbc.M212546200. [DOI] [PubMed] [Google Scholar]
- 105.Cho DH, Nakamura T, Fang JG, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates βAmyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V. Nitric oxide promotes endothelial cell survival signaling through S-nitrosylation and activation of dynamin2. J Cell Sci. 2007;120:492–501. doi: 10.1242/jcs.03361. [DOI] [PubMed] [Google Scholar]
- 107.Wang Z, Humphrey C, Frilot N, Wang G, Nie Z, Moniri NH, Daaka Y. Dynamin2- and endothelial nitric oxide synthase-regulated invasion of uropathogenic Escherichia coli into bladder epithelial cells. J Cell Biol. 2010;192:101–110. doi: 10.1083/jcb.201003027. [DOI] [PMC free article] [PubMed] [Google Scholar]