

Abstract
For the last decade, worldwide efforts for the treatment of anthrax infection have focused on developing effective vaccines. Patients that are already infected are still treated traditionally, using different types of standard antimicrobial agents. The most popular are antibiotics such as tetracyclines and fluoroquinolones. While aminoglycosides appear to be less effective antimicrobial agents than other antibiotics, synthetic aminoglycosides have been shown to act as potent inhibitors of anthrax lethal factor and may have potential application as antitoxins. Here, we present a structural analysis of the BA2930 protein, a putative aminoglycoside acetyltransferase, which may be a component of the bacterium’s aminoglycoside resistance mechanism. The determined structures revealed details of a fold characteristic only for one other protein structure in the PDB, namely YokD from Bacillus subtilis. Both BA2930 and YokD are members of the Antibiotic_NAT superfamily (PF02522). Sequential and structural analysis showed that residues conserved throughout the Antibiotic_NAT superfamily are responsible for the binding of the cofactor acetyl coenzyme A. The interaction of BA2930 with cofactors was characterized by both crystallographic and binding studies.
Keywords: Antibiotics resistance, Aminoglycoside N-acetyltransferase, Bacillus anthracis, AcCoA, crystal structure
1. Introduction
Widespread antibiotic use has caused evolutionary pressure for pathogenic bacteria to develop or acquire various mechanisms of resistance1 and some bacterial isolates are resistant to nearly all clinically used antibiotics2. Since aminoglycoside antibiotics have been widely used for over 50 years, resistance to this class of chemicals was one of the first kinds of antibiotic resistance developed by pathogenic bacteria3. This has led to great interest in the molecular mechanisms of bacterial aminoglycoside resistance.4
Aminoglycosides (e.g., streptomycin, gentamicin, and kanamycin) constitute a large, structurally diverse family of water-soluble polycationic amino sugars5. They are used to treat serious infections caused mostly by aerobic, Gram-negative bacteria and are used together with other antibiotics against certain Gram-positive bacteria. Aminoglycosides are mostly naturally occurring substances produced by actinomycetes of genus Streptomycetes or Micromonospor,6 but can be altered (or synthesized) chemically, resulting in extension or modification of their activities. In bacteria, aminoglycosides cause errors in the synthesis of proteins by binding the 16S rRNA of the 30S ribosomal fragment, and also disrupt the bacterial outer membrane and disturb the initiation of DNA replication.
Several bacterial aminoglycoside resistance mechanisms have been identified, including development of aminoglycoside-modifying enzymes, decreased membrane permeability, structural alteration of the ribosome, and removal of the compounds from the cell by efflux pumps.5 Among these mechanisms, chemical modification by aminoglycoside-modifying enzymes is the predominant one.7 All aminoglycoside-modifying enzymes function by attaching an additional chemical moiety to the antibiotic. This causes decreased affinity of aminoglycosides for the bacterial ribosome, and abrogates their antibiotic activity.8 Depending on which chemical moiety is attached, modifying enzymes are divided into three groups: O-phosphotransferases (APHs), which phosphorylate specific aminoglycoside hydroxyl groups in an ATP-dependent manner; N-acetyltransferases (AACs), which acetylate (primarily) amino groups in an acetyl-coenzyme-A-dependent manner; and O-nucleotidyltransferases (ANTs), which transfer nucleotide triphosphates to the substrate4. Of these three groups, N-acetyltransferases are most frequently found in clinical isolates.9 N-acetyltransferases can be further subdivided into four subgroups, based on the site of acyl transfer: 1-, 2’-, 3-, and 6’- acetyltransferases.3,10 N6’-acetylation is the most common modification.9
Here, we report the structure of the apo-form of a putative aminoglycoside N-acetyltransferase BA2930 from Bacillus anthracis str. Ames, as well as the structure of its complex with coenzyme A (CoA). Primary sequence analysis shows that BA2930 belongs to the Antibiotic_NAT Pfam family (PF02522). The structure of only one other member of the Antibiotic_NAT superfamily, namely the putative N-acetyltransferase YokD from Bacillus subtilis, is known. We determined that BA2930 deacetylates acetyl coenzyme A (AcCoA) in the presence of some aminoglycoside antibiotics, and by thin-layer chromatography (TLC), that some antibiotics are chemically modified by BA2930. In addition, we characterized two mutants of BA2930 that are presumed to interfere with the enzyme function, namely H183A and H183G. Analysis of crystals soaked or co-crystallized with acetyl AcCoA strongly suggests that His183 participates in the probable acetyl group transfer. Inhibition of this process is important not only for many strains of pathogenic bacteria that have already developed resistance to aminoglycosides, but could be of critical importance for the future treatment of anthrax, particularly if neomycin B and other synthetic aminoglycosides prove useful clinically as antitoxic agents.
2. Results and discussion
2.1 Overall structure
Both native BA2930 and its complex with CoA crystallized in the orthorhombic crystal system (space group P212121) with a dimer in the asymmetric unit (Table 1). Mutants of BA2930 also crystallized in the monoclinic system (space group P21), with two dimers in the asymmetric unit. CoA binding did not cause noticeable structural rearrangements in the BA2930 subunit. Comparison of different protein chains, both within the same asymmetric unit as well as across different crystal forms, showed that changes in main chain conformations are small with average Cα root mean square deviation (r.m.s.d.) values around 0.3–0.5 Å.
Table 1.
Summary of data collection, phasing, and refinement statistics. Data for the highest resolution shell are given in parentheses. Ramachandran plot statistics were calculated by MOLPROBITY.
| PDB code | 3E4F (wt+citric acid) |
3IJW (wt+CoA) |
3KZL (H183G +AcCoA) |
3N0S (H183A +AcCoA) |
3N0M (H183G +AcCoA) |
|---|---|---|---|---|---|
| Crystal | |||||
| Space group | P212121 | P212121 | P21 | P21 | P212121 |
| Unit cell | a=36.3 Å | a=36.5 Å | a=71.9 Å | a=72.0 Å | a=36.8 Å |
| b=108.0 Å | b=110.6 Å | b=109.2 Å | b=109.4 Å | b=110.6 Å | |
| c=132.8 Å | c=132.9 Å | c=73.8 Å | c=74.0 Å | c=132.6 Å | |
| α=β=γ=90° | α=β=γ=90° | β=112.0° | β=111.9° | α=β=γ=90° | |
| Solvent content (%) | 44 | 46 | 45 | 46 | 46 |
| Matthews coefficient (Å3/Da) | 2.2 | 2.3 | 2.2 | 2.3 | 2.3 |
| No. of molecules in asymmetric unit | 2 | 2 | 4 | 4 | 2 |
| Data collection | |||||
| Diffraction protocol | SAD | MR | MR | MR | MR |
| Wavelength (Å) | 0.9793 | 0.9793 | 0.9792 | 0.9786 | 0.9792 |
| Resolution (Å) | 50.00–2.00 | 50.00–1.90 | 50.00–2.10 | 50.00–2.15 | 50.00–2.40 |
| The highest resolution shell (Å) | 2.03–2.00 | 1.93–1.90 | 2.14–2.10 | 2.19–2.15 | 2.44–2.40 |
| Unique reflections | 36337 | 44002 | 61626 | 57925 | 21408 |
| Redundancy | 8.1(8.1) | 7.2(6.2) | 4.0(2.8) | 3.3(2.8) | 3.9(3.9) |
| Completeness (%) | 100(100) | 99.7(99.2) | 98.9(87.8) | 99.1(95.9) | 97.5(96.6) |
| I/σ(I) | 38.2(4.5) | 28.4(3.3) | 18.1(2.80) | 16.0(1.9) | 23.1(2.1) |
| Rmerge (%)- Bijvoet pairs merged | 8.2(49.6) | 9.0(55.5) | 11.7(57.1) | 9.0(48.7) | 8.4(54.6) |
| SAD analysis | |||||
| No. of Se sites located/theoretical | 17/18 | - | - | - | - |
| FOMMLPHARE | 0.27 | - | - | - | - |
| FOMDM | 0.74 | - | - | - | - |
| Refinement | |||||
| Reflections used for refinement | 34404 | 41511 | 57645 | 54459 | 20256 |
| Reflections in Rfree set | 1806 | 2203 | 3081 | 2907 | 1094 |
| Rwork (%) | 17.2 | 18.8 | 18.0 | 17.4 | 21.2 |
| Rfree (%) | 22.6 | 23.1 | 23.2 | 22.8 | 25.8 |
| RMSD bond length (Å) | 0.017 | 0.022 | 0.014 | 0.021 | 0.017 |
| RMSD bond angles (°) | 1.7 | 1.7 | 1.5 | 1.7 | 1.6 |
| Number of protein atoms | 4082 | 4139 | 8172 | 8260 | 4095 |
| Number of water molecules | 293 | 336 | 455 | 374 | 43 |
| Average B factor (Å2) | 32.3 | 32.7 | 17.9 | 15.6 | 57.1 |
| Average B factor - protein (Å2) | 32.0 | 32.2 | 17.4 | 14.2 | 57.4 |
| Average B factor - waters (Å2) | 42.2 | 37.8 | 20.8 | 35.7 | 43.1 |
| Average B factor – ligands (Å2) | 51.8 | 39.0 | 30.6 | 33.4 | 50.3 |
| B from Wilson plot (Å2) | 27.3 | 28.4 | 27.8 | 31.6 | 54.1 |
| Ramachandran plot | |||||
| Favored regions (%) | 96.8 | 97.2 | 97.3 | 97.2 | 95.4 |
| Additional allowed regions (%) | 3.2 | 2.8 | 2.7 | 2.8 | 4.6 |
Analysis of the structures of the apo- and holo-forms of BA2930 with DynDom11 showed that a subunit of BA2930 can be divided into two distinct fragments (Fig. 1a and 2). The larger fragment is comprised of a three-layer α-β-α sandwich with a central β-sheet surrounded by nine helices. The strand topology in the β-sheet is 6-7-5-1-2-4-3, with strands 4 and 7 antiparallel to the other five. The larger fragment includes the complete cofactor binding site. The smaller, lobe-like fragment (residues 73–101 and 193–216) comprises two β-stands and one α-helix and may accommodate a substrate binding site.
Fig. 1.

(a) Overall fold and dimeric structure of BA2930. The part of the protein containing the cofactor (CoA) binding site is shown in green, while the part thought to be involved in putative ligand binding is in red.
(b) The molecular surface of the BA2930 dimer, with the electrostatic charge distribution as calculated by APBS41 and rendered by PyMOL.42
Fig. 2.

Different properties of residues in the Ba2390 structures plotted as a function of residue number. (a) A plot of the atom contacts between molecules in crystal lattice: red - residues forming close contacts (from 2.0 to 3.5 Å), green - residues forming hydrogen bonds, pale blue – residues bridged by water molecule, grey - residues forming other contacts (from 3.5 to 4.5 Å). The top graph represents contacts in the structure of the apo-form of BA2930, and the bottom in the CoA-bound structure. Contacts and distances were calculated using the CONTACT program from the CCP4 suite. (b) Plot of the RMSD and distance profiles comparing the apo and CoA-bound structures. Blue lines - RMSD values between corresponding residues. Red lines - distances between corresponding Cα atoms. The pale, thin lines represent pairwise chain comparisons between structures. Averaged results using a window of 4 comparisons are plotted with bold lines. (c) Cα distances between superposed A and B chains from one structure. The structure of the apo-form of BA2930 is represented in orange and the CoA-bound structure in green. (d) Percent deviation of the Cα temperature factors from the average value. All calculations were performed with the BioShell package.43 Superposition of two BA2930 subunits yields Cα RMSD values of 0.4 Å for both apo- and CoA-bound forms of BA2930. Superposition of each chain of the apo-form on the corresponding chain of the CoA-bound form yields Cα RMSD values ranging from 0.3 to 0.4 Å. These differences can be attributed to mentioned before putative substrate binding lobe. We also observe a similar degree of flexibility in the crystallized mutants (data not shown).
A structural similarity search of the PDB reveals that only one protein (YokD from Bacillus subtilis; PDB code 2NYG) shares the same fold as BA2930 (Fig. 3). The sequence identity between these proteins is 52%. The subunit superposition of YokD and BA2930 yields a coordinate r.m.s.d. of 0.9 Å over 264 Cα atoms.
Fig. 3.

Comparison of the BA2930 (dark gray) and Bacillus subtilis YokD (light gray) structures. Coenzyme A (purple) was co-crystallized with YokD, and CoA (blue) with BA2930. The largest structural variations between the two structures are colored blue in BA2930 and purple in YokD.
Both BA2930 and YokD belong to the Antibiotic_NAT superfamily (PF02522) in the Pfam database. This family consists of 280 aminoglycoside-3-N-transferases (E.C. 2.3.1.81) from 106 species, almost exclusively bacterial. Figure 4 shows a multiple sequence alignment of BA2930, YokD and 11 other representative members of the Antibiotic_NAT family. The structure shows that the highly conserved residues Thr180, His183 and Glu186 are located in the cofactor and putative substrate binding sites (see Supplementary Materials and Fig. 5).
Fig. 4.

Multiple sequence alignment (MSA) for selected members of the Antibiotic_NAT family. An MSA of the 56 Antibiotic_NAT (PFAM2522) family seed sequences was prepared with PCMA44 followed by manual adjustments. The presented MSA shows a subset of the seed sequences, which represents the variability of the family alignment. Sequences are labeled with the name of the organism. The NCBI gene identification (gi) numbers of each sequence are: Vibrio shilonii – 149187267; Stigmatella aurantiaca - 115375112; Pseudomonas aeruginosa - 150956; Streptomyces fradiae – 62857296; Escheriachia coli - 134047089; Listeria monocytogenes - 167454283; Lactococcus lactis - 116326639; Clostridium butiricum - 182417486; Bacillus coagulans -124521550; Streptomyces coelicolor - 21220413; Salinispora arenicola – 159038228; Bacillus anthracis – 30257522; and Bacillus subtilis – 122921181. Amino acid residues responsible for binding of CoA/AcCoA by BA2930 are marked with red dots. Positions conserved among all sequences are highlighted in yellow and positions conserved in 80% of sequences are colored orange.
Fig. 5.

Surface representation and schematics of binding of BA2930 with ligands bound (citric acid (in the apo-form; (a), (aa)) or CoA ((b), (bb))), or docked (AcCoA and neomycin ((c), (cc))). The distances shown are calculated between non-hydrogen atoms. Atoms from the residue main chain are labeled next the box containing residue name and atoms from side chain are separated from this box by a solid line.
Gel filtration studies suggest that BA2930 is a dimer (data not shown). It is probable that the dimer found in the asymmetric unit represents the oligomeric state of BA2930 in solution. Both the PITA12 and PISA servers13 results are consistent with these findings. The decreases in solvent-accessible surface area of apo-BA2930 and the BA2930-CoA complex upon dimerization are 1320 Å2 and 1440 Å2, respectively. Both lay well above the cut-off value of 856 Å2 proposed for discrimination between homodimeric and monomeric proteins.14 The dimer interface comprises residues 5–15, 43–53, 73, 74 and 80–121. Hydrogen bonds (H-bonds) stabilizing the BA2930 dimer are limited to an interaction between the main chain O of Gln9 and Nε1 of Trp89 as well as between bridging water molecules and Ile104, Arg118 and Thr119. The residues involved in dimer formation are not conserved between YokD and BA2930.
2.2 Ligand binding
While several different derivatives of coenzyme A, including the short chain acetyl- and butyryl-CoA as well as the longer chain myristyl- and stearyl-CoA, were used in crystallization experiments of wild type BA2930, only the electron density corresponding to CoA was observed. This suggests that the CoA derivatives are hydrolyzed in the conditions of the crystallization experiment, though the possibility that the extra moieties were present but disordered cannot be ruled out.
The coenzyme binding site is formed by a single polypeptide chain (Fig. 5b). Both hydrogen bonds and hydrophobic interactions play a role in the binding of CoA by BA2930 (Figure 5bb). The adenine ring of CoA is buried in a hydrophobic pocket and is oriented parallel to the side chain of Trp43. In addition, two H-bonds are formed between the adenine ring and the main chain nitrogen and oxygen atoms of Ile44. The sugar moiety of CoA is not directly coordinated by the protein, but its diphosphate group forms four H-bonds with the Oγ atoms of Ser37 and Ser39 and nitrogen atoms of Leu38 and Lys112. The pantothenate fragment of CoA is anchored by two hydrogen bonds with the side chain of His35 and the main chain of Ser36. The interactions between CoA and BA2930 are similar to those observed in the structure of the YokD-CoA complex (Fig. 3). Although the conformation of CoA observed in BA2930 and YokD differ completely from those observed for GNAT family acetyltransferases, several common features can be observed. For example in one of the GNAT acetyltransferases from Bacillus anthracis (Fig. S1), the diphosphate moiety is bounded mostly by main chain nitrogens with one additional serine side chain, the pantothenate fragment is anchored by hydrogen bonds formed between both side chain and main chain atoms, and the ribose moiety does not form direct interactions with the protein. The most notable difference is that CoA in BA2930 is bound parallel to the protein and its binding site forms a continuous cleft with the putative ligand binding site, whereas in GNAT acetyltransferases, the panthothenate fragment is buried inside the protein and cofactor and ligand binding sites are located on the opposite sides of the protein.
In the crystal structure, the subunit of apo-BA2930 binds two ordered molecules of citric acid, a component of the crystallization solution. One molecule is bound at the protein surface near Ser148 and Ser150. The second molecule is located in the CoA binding site (Fig. 5a). This molecule of citric acid mimics the diphosphate moiety of the coenzyme, and forms an analogous pattern of H-bonds as diphosphate (Fig. 5aa). The CoA diphosphate/citric acid binding site of BA2930 carries a significant positive charge, consistent with an affinity for negatively charged ligands (Fig. 1b).
We used isothermal calorimetry to investigate the binding of BA2930 with CoA, AcCoA, and putative substrates, namely aminoglycoside antibiotics. Initially we tested the binding of CoA and AcCoA and shown that BA2930 binds both compounds. However, the affinity for CoA is 5-fold higher than that for AcCoA. This observation is consistent with a substrate activation mechanism for the presumed enzyme, where after the putative acetyl transfer and release of the acetylated product, binding of a subsequent substrate induces the release of CoA. This mechanism was proposed to describe kinetic data for some substrates of the aminoglycoside N-acetyltransferase AAC(6’)-Iy from Salmonella enteric.15 Subsequently, we investigated binding of these substrates to apo-BA2930 by ITC, as well as to BA2930 saturated with either CoA or AcCoA. In all cases, the ITC measurements did not detect a heat effect that could be associated with substrate binding and/or an enzymatic reaction.
2.3 Determination of possible BA2930 substrates
To determine possible substrates for the presumed enzyme, the structure of BA2930 in complex with CoA was screened in silico for docking to a set of ~1200 compounds in the FDA database (February 2008). The ten best docking solutions found in the screening experiment are summarized in Table 2 and are ranked by their calculated GScore, which mimics binding energies. Nine of the ten best solutions obtained were aminoglycoside antibiotics.
Table 2.
List of the 10 top hits obtained after docking of ~1200 compounds from the FDA approved list of drugs (February 2008) to BA2930. Compounds are ranked by their 'XP Gscore'. Extended list containing the first one hundred hits is provided in the Supplementary Materials.
| Compound name | Therapeutic Category |
Structure | XP GScore |
|
|---|---|---|---|---|
| 1 | Paromomycin | Antibacterial; Antiamebic | 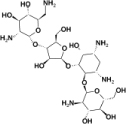 |
−13.3 |
| 2 | Kanamycin C | Antibacterial | 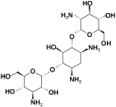 |
−12.4 |
| 3 | Amikacin | Antibacterial |  |
−12.1 |
| 4 | Streptomycin B | Antibacterial |  |
−12.1 |
| 5 | Streptomycin A | Antibacterial |  |
−11.0 |
| 6 | Lividomycin | Antibacterial |  |
−11.0 |
| 7 | Deferoxamine | Parenteral chelating agent |  |
−10.6 |
| 8 | Dihydrostreptomycin | Antibacterial |  |
−10.5 |
| 9 | Neomycin B | Antibacterial |  |
−10.1 |
| 10 | Kanamycin A | Antibacterial |  |
−10.1 |
To verify the results from the docking studies and determine putative substrates of BA2930 we employed various approaches. Co-crystallization and soaking of the protein with top-scoring compounds from the docking studies did not show any binding. The interaction with these compounds was not detected in solution as measured by ITC. BA2930 was also screened by glycan array by the Functional Glycomics Gateway (www.functionalglycomics.org). Two different methods of binding (sequential addition of the enzyme followed by anti-His, as well as precomplexing the enzyme with anti-His and anti-mouse IgG to increase the affinity and valency of binding) was applied. There was no binding to the glycan array detected using either method. Numerous aminoglycoside acetyltransferases have been shown to catalyze protein acetylation.16 To order to test this possibility, we conducted phage display assays (New England Biolabs), but no binding of putative peptide substrate was detected in the conditions recommended by the manufacturer.
To search for possible BA2930 substrates, we also used thin layer chromatography (TLC). This method showed that the enzyme is able to chemically modify several aminoglycoside antibiotics (Fig. S2). However, the rate of modification for these substrates seems to be relatively low. The antibiotics modified by the enzyme include tobramycin, amikacin, and kanamycin, while several other aminoglycoside antibiotics were not observed to be modified (see Supplementary Materials). These results indicate that half of the first 10 hits from the docking studies are indeed modified by BA2930. In addition, we measured the susceptibility of E. coli cells overexpressing BA2930 to a variety of aminoglycoside antibiotics, but the test did not demonstrate acquired resistance of cells against any of the antibiotics (Fig. S3).
However, BA2930 does deacetylate AcCoA in the presence of some aminoglycosides, as monitored by spectrophotometry (Supplementary Table 1). The kinetic rates vary depending upon the aminoglycoside present in the reaction mixture, with Km values ranging from 0.34 to 13.3 mM, and kcat values of 0.29 to 2.90 mM sec−1 M−1.
2.4 BA2930 structure-function studies
Along with the search for presumed BA2930 substrates, studies of the putative mechanism of action using site-directed mutagenesis of BA2930 were carried out. His183 is the most conserved residue among BA2930 homologs. It is located near the thiol group of CoA and the reactive amino group of docked putative substrates, such as neomycin B (Fig. 5cc). It is possible that the conserved His183 residue (Fig. 4) is needed for the putative enzymatic mechanism, together with two additional amino acids (Thr180 and Glu186) which may be responsible for the transfer of the acetyl group. These three highly conserved residues form a strong hydrogen bond network with the thiol group of coenzyme A. The bond formed between atom Oγ of Thr180 and the sulfur atom of CoA is weak (with a distance of 3.0 Å), while the H-bond formed between Glu186 and His183 is relatively strong (with an Oε2-Nδ distance of 2.3 Å).
Both the H183A and H183G mutants crystallized in the P21 space group, with four protein subunits in the asymmetric unit. In addition, the H183G mutant crystallized similarly to the wild-type protein in the orthorhombic crystal system (space group P212121) with two subunits in the asymmetric unit (Table 1). Analysis of the structures of the H183G (PDB codes 3KZL and 3N0M) and H183A (PDB code 3N0S) showed that they share a very similar overall structure and dimer assembly as the wild type, demonstrating that mutation did not alter the overall structure of the enzyme.
Unlike wild type BA2930, ordered electron density was observed for the complete AcCoA in the structures of crystals of both the H183G and H183A mutants grown in the presence of AcCoA. This suggests that replacement of His183 has altered the presumed ability of the protein to hydrolyze AcCoA.
ITC results measuring the binding of CoA and AcCoA to the H183G and H183A mutants (Table 3) showed that for both mutants, the replacement of His183 only slightly alters the affinity of BA2930 for CoA but significantly improves AcCoA binding. This suggests that the mechanism used by BA2930 to differentiate between AcCoA and CoA involves His183. The ambiguous stoichiometry observed for both mutants in the ITC experiments may be caused by the lower stability of the mutant protein (lower protein solubility).
Table 3.
Thermodynamic parameters obtained from titration of wild type (wt) BA2930 and two mutants H183G and H183A with CoA or AcCoA.
| Protein | Ligand | KD (µM) | ΔH (kcal·mol−1) | −TΔS (kcal·mol−1) | ΔG (kcal·mol−1) | N |
|---|---|---|---|---|---|---|
| wt | AcCoA | 33.35±2.53 | −21.71±0.46 | −15.61 | −37.32 | 1.140±0.013 |
| CoA | 6.23±1.81 | −12.47±0.53 | −5.36 | −17.83 | 1.000±0.031 | |
| H183G | AcCoA | 6.10±0.24 | −14.83±0.10 | −7.72 | −22.55 | 0.750±0.004 |
| CoA | 10.27±1.53 | −14.48±0.52 | −7.66 | −22.14 | 0.670±0.017 | |
| H183A | AcCoA | 3.72±0.29 | −17.40±0.20 | −9.98 | −27.39 | 0.610±0.005 |
| CoA | 8.69±0.69 | −14.79±0.25 | −7.87 | −22.66 | 0.670±0.008 |
In conclusion, sequence homology analysis suggests that the BA2930 protein from Bacillus anthracis str. Ames is an aminoglycoside N-acetyltransferase that belongs to the Antibiotic_NAT PFAM superfamily (PF02522). Currently, BA2930 and YokD are the only representatives of this family with structures reported in the PDB. They share not only a unique overall molecular architecture, but also form very similar dimeric assemblies. In both proteins two distinguishable cavities can be identified. One is the CoA/AcCoA binding site, while the other may be a binding site for an additional molecule. Docking experiments show that BA2930 may bind relatively large molecules like aminoglycoside antibiotics. This theoretical prediction was validated by the TLC experiments which shown that several antibiotics are modified by the enzyme, although the substrates identified have a relatively high apparent Michaelis-Menten constant (Table S1). ITC studies did not detect binding of any of the antibiotics tested. It is possible that in the conditions used during the binding studies, the interactions of the protein and antibiotics-- presuming the antibiotics are ligands of the protein-- are weak. Although the putative physiological substrate of BA2930 and the mechanism of its presumed action are still undefined, the presence of AcCoA in the structures of both the H183A and H183G mutant indicates that His183 may be needed for the possible acetyl group transfer.
3. Materials and methods
3.1 Protein cloning and expression
Ba2930 was cloned using a protocol previously described.17,18 Briefly, the DNA of the open reading frame of the ba2930 gene was extracted by PCR amplification using Bacillus anthracis str. Ames genomic DNA, gene-specific primers with 5’ LIC cloning tags appended, and Platinum® PCR SuperMix High Fidelity polymerase (Invitrogen). The quality of the PCR products was verified using the Caliper LC90 capillary electrophoresis platform. The gene was cloned into the pMCSG7 vector,18 a derivative of pET21a (Novagen), using a ligation-independent cloning (LIC) protocol. The pMCSG7 vector encodes for a 6×His-tag followed by a spacer and a TEV protease cleavage site on the N-terminus of the expressed protein. The amino acid triplet Ser-Asn-Ala remains on the N-terminus of the protein after cleavage of the tag with TEV protease.
The subsequent clone was transformed into DH10B chemically competent cells and selected on 2xYT agar + ampicillin (100 µg mL−1). Initially the protein was expressed in E. coli strain BL21(DE3)pLysS by growing 150 µL cultures in both M9 and 2xYT media with shaking at 37°C. When the optical density (at 600 nm) reached ~0.8 the cultures were induced with 1 mM IPTG, and grown overnight (~19 hours) with shaking at 25°C. The cell pellet was lysed using sonication, and the levels of protein expression and solubility levels of the both the crude lysate and the supernatant were determined using the Caliper LC90.
3.2 Site-Directed mutagenesis of Ba2930
To investigate the role of His183 in the presumed acetyl transfer, two single-site mutants were generated using the QuickChange site-directed mutagenesis kit (Stratagene) according to the manufacturer’s guidelines. The wild type Ba2930 gene cloned into the pMCSG7 vector was used as a template for all PCR amplifications. For each mutant, specific compliment primers conferring the site mutations were designed (forward: 5’-GATTCCAATACTTCTGTAGCTTTATCAGAAGTACGTTCTGG-3’ and reverse 5’-CCAGAACGTACTTCTGATAAAGCTACAGAAGTATTGGAATC-3’ for H183A and forward: 5’- GATTCCAATACTTCTGTAGGTTTATCAGAAGTACGTTCTGG-3’ and reverse 5’-CCAGAACGTACTTCTGATAAACCTACAGAAGTATTGGAATC-3’ for H183G). The presence of the desired mutations was confirmed by sequencing.
3.3 Large- scale proteins’ expression
For large-scale production, all three variants of BA2930--wild-type (wt), H183A and H183G--were expressed in E. coli strain BL21-CodonPlus(DE3), which harbors an extra plasmid encoding for three rare tRNAs (AGG and AGA for Arg, and ATA for Ile; Stratagene). The cells were grown in 4 flasks containing 1 L of M9 SeMET High-Yield growth media (Shanghai Medicilon) at 37°C to an optical density (at 600 nm) of approximately 1.2 and 0.4 mM IPTG was added to induce protein expression. After induction, the cells were incubated overnight with shaking at 25°C (wt for apo-enzyme crystallization) or at 18°C (wt, H183A, H183G for co-crystallization with AcCoA).
3.4 Protein purification
Cells expressing BA2930 wt (for apo-enzyme crystallization) were harvested and resuspended in a solubilization buffer (250 mM NaCl, 5% glycerol, 100 mM amonium sulfate, 50 mM phosphate-citrate buffer pH 7.5, 0.5 mM TCEP, 0.08% DDM and 5 mM imidazole), flash-frozen in liquid N2, and stored at −80°C. The thawed cells were lysed by sonication after the addition of 1 mM each of PMSF and benzamidine. The lysate was clarified by centrifugation (30 min at 17,000×g) and the liquid fraction was applied to a metal chelate affinity column charged with Ni2+ and pre-equilibrated with binding buffer (300 mM NaCl, 50 mM HEPES pH 7.5, 5% glycerol, 5 mM imidazole). The resin with bound protein was washed with wash buffer (300 mM NaCl, 50 mM HEPES pH 7.5, 5% glycerol, 30 mM imidazole) to remove weakly binding contaminants and then eluted from the column with elution buffer (300 mM NaCl, 5% glycerol, 50 mM HEPES pH 7.5, and 250 mM imidazole). The metal affinity tag was cleaved from the protein in the elution by treatment with recombinant His-tagged TEV protease19 followed by dialysis to remove the imidazole. The cleaved protein was then separated from the cleaved His-tag and the His-tagged protease by passing the mixture through a second Ni2+-chelate affinity column.
For crystallization of all variants of BA2930 with AcCoA, the protocol described above was modified as follows: all buffers used for purification contained 500mM NaCl. Harvested cells were resuspended directly in the binding buffer and immediately lysed by sonication after the addition of 1 mM each of PMSF and benzamidine.
3.5 Protein crystallization
After purification, protein was dialyzed in buffer containing 300 mM NaCl (BA2930 wt for apo-enzyme crystallization) or 500 mM NaCl (wt, H183A and H183G for co-crystallization with AcCoA), 10 mM HEPES pH 7.5, 0.5 mM TCEP, and concentrated using a BioMax concentrator (Millipore). Before crystallization, particulate matter was removed from the sample by passing it through a 0.2 µm Ultrafree-MC centrifugal filter (Millipore). All proteins were crystallized by hanging-drop vapor diffusion by mixing 2 µL of the protein solution (8 mg mL−1) with 2 µL of a precipitant solution. For apo-enzyme crystallization the precipitant solution contained 1.4 M sodium citrate and 0.1 M HEPES pH=7.5. For co-crystallization of BA2930 wt, H183A and H183G with CoA derivatives, the precipitant solution contained 0.1 M HEPES pH=7.5, 0.2 M MgCl2, 15% w/v PEG3350, or 0.1 M HEPES pH=7.5, 0.18 M MgCl2, 17% w/v PEG3350, 1% Chaps, or 0.1 M HEPES pH=7.5, 0.16 M MgCl2, 17% w/v PEG3350, 1% Chaps, respectively and either 5 mM AcCoA or 5 mM butyryl-CoA. In the case of co-crystallization with myristyl-CoA or arachidonyl-CoA, those compounds were added directly to the drop in solid form. Prior to data collection, crystals were transferred into Paratone-N (apo-enzyme crystals) and then flash cooled in a steam of cold nitrogen or cooled without cryoprotectants (wt, H183A and H183G crystals).
3.6 Data collection and structure solution
Data were collected at a temperature of 100 K on beamlines 19-ID (BA2930 apo-enzyme) and 19-BM (BA2930 wt and H183A mutant co-crystallization with CoA) of the Structural Biology Center,20 and on beamline 21-ID-G of the Life Sciences Collaborative Access Team at the Advanced Photon Source (APS). Data collection, structure determination, and refinement statistics are summarized in Table 1. Diffraction data from crystals of Se-Met substituted protein were processed with HKL-2000.21 The structure of the apo-protein was solved by Se single-wavelength anomalous diffraction (SAD), and initial models were built with HKL-3000.22 HKL-3000 is integrated with SHELXD, SHELXE,23 MLPHARE,24 DM,25 ARP/wARP,26 CCP4,27 SOLVE,28 and RESOLVE.29,30 The resulting model was further refined with REFMAC5,31 and COOT.32 MOLPROBITY33 and ADIT34 were used for structure validation. The structure of BA2930 wt complexed with CoA was solved by using initial phases calculated from the structure of the apo-protein, as implemented in HKL-3000. The structure of H183A and H183G mutants were solved by MR with HKL-3000 and MOLPREP35 using wt structure as model. Refinement and validation of the complex structures were done similarly.
3.7 Molecular docking study
All docking experiments were done with Glide (Schrodinger Suite 2008) in XP (Extreme Precision) docking mode36. LigPrep and Epik were used for preparing both protein and ligands, which were docked flexibly. Receptor grids were generated and centered on the residues defining the putative active site, near the CoA binding site. All Glide default parameters were used and no binding constraints were defined for docking.
Chain A of the BA2930 structure and a modeled AcCoA molecule was used in all calculations. In the BA2930 model, all solvent molecules were removed and selenomethionines were altered to methionines. The residues for which side chains were not observed in the electron density map were modeled assuming their most probable conformations. The protonation states and tautomers of all ligands were generated by Epik. Hydrogens were added and the protein structure was minimized using the OPLS2005 force field.37 A database of Federal Drug Administration (FDA) structures as of February 2008 was used for docking.
3.8 Isothermal titration calorimetry (ITC)
ITC measurements were performed at 25°C using an iTC200 isothermal titration calorimeter (MicroCal). Protein preparations were dialyzed against a 100 mM Tris pH=7.5 and 50 mM NaCl buffer. All cofactors and ligands injected were also dissolved in the same buffer. The concentrations of wild-type BA2930 and the H183G and H183A mutants were 132 µM, 125 µM, and 129 µM respectively. All three proteins were titrated by seventeen injections of 1.2 mM CoA or AcCoA ligand solutions (for wt 1.4 mM CoA). The first injection of each ligand solution had a volume of 0.4 µL, followed by sixteen 2.4 µL injections at 320 s intervals. The experiment was performed in high gain mode with the syringe rotating at 700 rpm. The same titration protocol was used to characterize antibiotic binding with 1.5 mM solutions of antibiotics (either kanamycin, amikacin, paromomycin, neomycin, streptomycin, dihydrostreptomycin, or deferoxamine) added to solutions containing either 132 µM protein solution (wt) alone or a mixture of 132 µM wt protein with 1.4 mM AcCoA.
3.9 Thin layer chromatography (TLC)
Antibiotic acetylation was carried out in 20 µl reaction mixtures containing 50 µg/ml of the protein, 2 mM AcCoA and 2 mM antibiotic in a buffer of 50 mM HEPES pH 7.5. Buffer and AcCoA alone, as well as mixtures of enzyme and antibiotic without AcCoA, were used as a negative controls. Reaction mixtures were spotted on 60 F254 silica gel plates (Merck) and developed with 5% potassium phosphate. The antibiotics and their reaction products were detected by spraying ninhydrin reagent (0.5% ninhydrin in acetone). The following compounds were used for the initial overnight test: kanamycin, paromomycin, neomycin, amikacin, streptomycin, dihydrostreptomycin, gentamicin, tobramycin, deferoxamine, moxalactam, and cefmetazole. All were obtained from Sigma, save kanamycin, which was obtained from Fisher Scientific. Antibiotics that proved promising following the initial overnight test were subsequently tested with different incubation times (1h, 3h and over night); specifically kanamycin, paromomycin, neomycin, amikacin, getamicin, and tobramycin.
3.10 Susceptibility tests
Susceptibility for aminoglycosides were determined with E. coli BL21-CodonPlus(DE3) cells harboring an pMCSG7 plasmid with the Ba2930 gene. Cells were induced with 0.4 mM IPTG for 30 min., then approximately 109 cells were spotted on agar plates containing LB Broth Lennox (RPI) with 100 µg/ml ampicillin, 34 µg/ml chloramphenicol and 0.5 mM IPTG. Then Antimicrobial Susceptibility Test discs (BD BBL™), containing 30 µg of amikacin, 30 µg of neomycin, 30 µg of kanamycin, 10 µg of streptomycin, 10 µg of tobramycin or 10 µg of gentamicin, were applied to each plate. As a control 109 of uninduced bacteria was spotted on the agar plate without IPTG. After overnight incubation at 37°C, the zones of inhibition around each spotted culture were assessed.
3.11 Measurement of enzyme activity
Activity was monitored spectrophotometrically by measuring absorbance at 324 nm caused by the reaction of the free sulfhydryl group of enzymatically deacetylated CoA with 4,4’-dithopiridine.38,39,40 Reactions were monitored continuously using a SPECTRA MAX190 spectrophotometer. Enzyme activities were calculated using a molar absorption coefficient of reaction product 4-thiopyridone (ε324=19,800 M−1 cm−1). Reaction mixtures contained: 50mM Tris, pH 7.5, 1mM EDTA, 0.8 mM Aldrithiol™-4 (SIGMA), 100 µM AcCoA, 20 µM enzyme, and seven concentrations (50µM- 1.5 mM) of different antibiotics (kanamycin, streptomycin, dihydrostreptomycin, paromomycin, amikacin, and neomycin). The reaction was initiated by the addition of enzyme and carried out in a total volume of 200µl (wells of a 96-well plate for UV measurement) for 20 min. at 37°C. Initial velocity data were obtained 3 min. after the start of reaction.
4. PDB accession codes
The coordinates and structure factors for BA2930 apo-enzyme were deposited in PDB with accession code 3E4F, while the coordinates and structure factors for complex of BA2930 with CoA were deposited with accession code 3IJW. For complexes of AcCoA with H183A and H183G mutants coordinates and structure factors were deposited in PDB with accession codes 3N0S and 3KZL, 3N0M, respectively.
Supplementary Material
Acknowledgments
The authors would like to thank Wayne Anderson and Alexander Wlodawer for valuable comments on the manuscript. This research was funded with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272200700058C. The results shown in this report are derived from work performed at Argonne National Laboratory, at the Structural Biology Center of the Advanced Photon Source. Argonne is operated by University of Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). We would like to acknowledge the Consortium for Functional Glycomics Grant number GM62116 for glycan array screening.
Abbreviations
- AAC
N-acetyltransferases
- ANT
O-nucleotidyltransferases
- APH
O-phosphotransferases
- AcCoA
Acetyl Coenzyme A
- CoA
Coenzyme A
- DDM
Dodecyl maltoside
- IPTG
Isopropyl-β-D-thiogalactoside
- ITC
Isothermal titration calorimetry
- MSA
Multiple sequence alignment
- PMSF
Phenylmethylsulphonyl fluoride
- TCEP
(tris(2-carboxyethyl)phosphine)
- TEV
Tobacco Etch Virus (TEV) Protease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Maria M. Klimecka, Email: majka@iwonka.med.virginia.edu.
Maksymilian Chruszcz, Email: mc3h@cms.mail.virginia.edu.
Jose Font, Email: font@iwonka.med.virginia.edu.
Igor Shumilin, Email: ias2n@cms.mail.virginia.edu.
Przemyslaw J. Porebski, Email: przemek@iwonka.med.virginia.edu.
Marcin Cymborowski, Email: marcel@iwonka.med.virginia.edu.
Matthew D. Zimmerman, Email: matt@iwonka.med.virginia.edu.
Ian J. Glomski, Email: ijg2b@Virginia.EDU.
Lukasz Lebioda, Email: lebioda@chem.sc.edu.
Alexei Savchenko, Email: alexei.savchenko@utoronto.ca.
References
- 1.Tenover FC. Development and spread of bacterial resistance to antimicrobial agents: an overview. Clin Infect Dis. 2001;33 Suppl 3:S108–s115. doi: 10.1086/321834. [DOI] [PubMed] [Google Scholar]
- 2.Levy SB. The challenge of antibiotic resistance. Sci Am. 1998;278:46–53. doi: 10.1038/scientificamerican0398-46. [DOI] [PubMed] [Google Scholar]
- 3.Davies J, Wright GD. Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol. 1997;5:234–240. doi: 10.1016/S0966-842X(97)01033-0. [DOI] [PubMed] [Google Scholar]
- 4.Vakulenko SB, Mobashery S. Versatility of aminoglycosides and prospects for their future. Clin Microbiol Rev. 2003;16:430–450. doi: 10.1128/CMR.16.3.430-450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azucena E, Mobashery S. Aminoglycoside-modifying enzymes: mechanisms of catalytic processes and inhibition. Drug Resist Updat. 2001;4:106–117. doi: 10.1054/drup.2001.0197. [DOI] [PubMed] [Google Scholar]
- 6.Giamarellou H. Aminoglycosides plus beta-lactams against gram-negative organisms. Evaluation of in vitro synergy and chemical interactions. Am J Med. 1986;80:126–137. doi: 10.1016/0002-9343(86)90490-0. [DOI] [PubMed] [Google Scholar]
- 7.Burk DL, Ghuman N, Wybenga-Groot LE, Berghuis AM. X-ray structure of the AAC(6')-Ii antibiotic resistance enzyme at 1.8 A resolution; examination of oligomeric arrangements in GNAT superfamily members. Protein Sci. 2003;12:426–437. doi: 10.1110/ps.0233503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Llano-Sotelo B, Azucena EF, Jr, Kotra LP, Mobashery S, Chow CS. Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem Biol. 2002;9:455–463. doi: 10.1016/s1074-5521(02)00125-4. [DOI] [PubMed] [Google Scholar]
- 9.Miller GH, Sabatelli FJ, Hare RS, Glupczynski Y, Mackey P, Shlaes D, Shimizu K, Shaw KJ. The most frequent aminoglycoside resistance mechanisms--changes with time and geographic area: a reflection of aminoglycoside usage patterns? Aminoglycoside Resistance Study Groups. Clin Infect Dis. 1997;24 Suppl 1:S46–S62. doi: 10.1093/clinids/24.supplement_1.s46. [DOI] [PubMed] [Google Scholar]
- 10.Jana S, Deb JK. Molecular understanding of aminoglycoside action and resistance. Appl Microbiol Biotechnol. 2006;70:140–150. doi: 10.1007/s00253-005-0279-0. [DOI] [PubMed] [Google Scholar]
- 11.Qi G, Lee R, Hayward S. A comprehensive and non-redundant database of protein domain movements. Bioinformatics. 2005;21:2832–2838. doi: 10.1093/bioinformatics/bti420. [DOI] [PubMed] [Google Scholar]
- 12.Ponstingl H, Kabir T, Thornton JM. Automatic inference of protein quaternary structure from crystals. Journal of Applied Crystallography. 2003;36:1116–1122. [Google Scholar]
- 13.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 14.Ponstingl H, Henrick K, Thornton JM. Discriminating between homodimeric and monomeric proteins in the crystalline state. Proteins. 2000;41:47–57. doi: 10.1002/1097-0134(20001001)41:1<47::aid-prot80>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 15.Magnet S, Lambert T, Courvalin P, Blanchard JS. Kinetic and mutagenic characterization of the chromosomally encoded Salmonella enterica AAC(6')-Iy aminoglycoside N-acetyltransferase. Biochemistry. 2001;40:3700–3709. doi: 10.1021/bi002736e. [DOI] [PubMed] [Google Scholar]
- 16.Vetting MW, LP SdC, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Zhang RG, Skarina T, Katz JE, Beasley S, Khachatryan A, Vyas S, Arrowsmith CH, Clarke S, Edwards A, Joachimiak A, Savchenko A. Structure of Thermotoga maritima stationary phase survival protein SurE: a novel acid phosphatase. Structure. 2001;9:1095–1106. doi: 10.1016/s0969-2126(01)00675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 19.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 20.Rosenbaum G, Alkire RW, Evans G, Rotella FJ, Lazarski K, Zhang RG, Ginell SL, Duke N, Naday I, Lazarz J, Molitsky MJ, Keefe L, Gonczy J, Rock L, Sanishvili R, Walsh MA, Westbrook E, Joachimiak A. The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J Synchrotron Radiat. 2006;13:30–45. doi: 10.1107/S0909049505036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Otwinowski Z, Minor W. Methods in enzymology: Macromolecular crystallography, part A. Vol. 276. New York: Academic Press; 1997. Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 22.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 23.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 24.Otwinowski Z, editor. Isomorphous replacement and anomalous scattering. W. Wolf, P.R. Evans & A. G. W. Leslie edit. Proceedings of the CCP4 study weekend. Edited by W. Wolf, P. R. E., and A.G.W. Leslie. Warrington, UK: Daresbury Laboratory; 1991. [Google Scholar]
- 25.Cowtan KD, Main P. Improvement of macromolecular electron-density maps by the simultaneous application of real and reciprocal space constraints. Acta Crystallogr D Biol Crystallogr. 1993;49:148–157. doi: 10.1107/S0907444992007698. [DOI] [PubMed] [Google Scholar]
- 26.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 27.Collaborative Computional Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr, Sect D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 28.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terwilliger TC. Automated structure solution, density modification and model building. Acta Crystallogr D Biol Crystallogr. 2002;58:1937–1940. doi: 10.1107/s0907444902016438. [DOI] [PubMed] [Google Scholar]
- 30.Terwilliger T. SOLVE and RESOLVE: automated structure solution, density modification and model building. J Synchrotron Radiat. 2004;11:49–52. doi: 10.1107/s0909049503023938. [DOI] [PubMed] [Google Scholar]
- 31.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 34.Yang H, Guranovic V, Dutta S, Feng Z, Berman HM, Westbrook JD. Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystallographica Section D. 2004;60:1833–1839. doi: 10.1107/S0907444904019419. [DOI] [PubMed] [Google Scholar]
- 35.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. Journal of Applied Crystallography. 1997;30:1022–1025. [Google Scholar]
- 36.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 37.Jorgensen WL, Tirado-Rives J. Chemical Theory and Computation Special Feature: Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. PNAS. 2005;102:6665–6670. doi: 10.1073/pnas.0408037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grassetti DR, Murray JF., Jr Determination of sulfhydryl groups with 2,2'- or 4,4'-dithiodipyridine. Arch Biochem Biophys. 1967;119:41–49. doi: 10.1016/0003-9861(67)90426-2. [DOI] [PubMed] [Google Scholar]
- 39.Serpersu EH, Ozen C, Wright E. Studies of enzymes that cause resistance to aminoglycosides antibiotics. Methods Mol Med. 2008;142:261–271. doi: 10.1007/978-1-59745-246-5_20. [DOI] [PubMed] [Google Scholar]
- 40.Williams JW, Northrop DB. Kinetic mechanisms of gentamicin acetyltransferase I. Antibiotic-dependent shift from rapid to nonrapid equilibrium random mechanisms. J Biol Chem. 1978;253:5902–5907. [PubMed] [Google Scholar]
- 41.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeLano W, S The PyMOL Molecular Graphics System. 2002 [Google Scholar]
- 43.Gront D, Kolinski A. BioShell--a package of tools for structural biology computations. Bioinformatics. 2006;22:621–622. doi: 10.1093/bioinformatics/btk037. [DOI] [PubMed] [Google Scholar]
- 44.Pei J, Sadreyev R, Grishin NV. PCMA: fast and accurate multiple sequence alignment based on profile consistency. Bioinformatics. 2003;19:427–428. doi: 10.1093/bioinformatics/btg008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.