

Abstract
Lipopolysaccharide (LPS) is known to damage hepatocytes by cytokines released from activated Kupffer cells, but the ancillary role of LPS as a direct hepatotoxin is less well characterized. The aim of this study was to determine the direct effect of LPS on hepatocyte viability and the underlying signaling mechanism. Rat hepatocyte cultures treated overnight with LPS (500 ng/mL) induced apoptosis as monitored morphologically (Hoechst 33258) and biochemically (cleavage of caspase 3 and 9 and the appearance of cytochrome C in the cytoplasm). LPS-induced apoptosis was additive to that induced by glycochenodeoxycholate or Fas ligand, was associated with activation of c-Jun N-terminal kinase B (JNK) and p38 mitogen-activated protein kinases (MAPK), and inhibition of protein kinase (AKT). Inhibition of JNK by SP600125, but not of p38 MAPK by SB203580 attenuated LPS-induced apoptosis, indicating JNK dependency. CPT-2-Me-cAMP, an activator of cAMP-GEF, decreased apoptosis due to LPS alone or in combination with glycochenodeoxycholate or Fas ligand. CPT-2-Me-cAMP also prevented LPS-induced activation of JNK and inhibition of AKT Taken together, these results suggest that LPS can induce hepatocyte apoptosis directly in vitro in a JNK-dependent manner and activation of cAMP-GEF protects against the LPS-induced apoptosis most likely by reversing the effect of LPS on JNK and AKT
Keywords: apoptosis, cAMP-GEF, AKT, exchange protein activated by cAMP (EPAC), lipopolysaccharide, JNK
Introduction
Bacterial cell wall products absorbed from the gastrointestinal tract and delivered to the liver by the portal circulation both induce and aggravate liver injury.1–4 Absorption of lipopolysaccharide (LPS), a glycolipid found on the outer membrane of bacteria, from the gastrointestinal tract, occurs with alterations in intestinal permeability, and contributes to the pathology in many liver diseases including; alcoholic and nonalcoholic steatohepatitis, viral and immune hepatitis, and cirrhosis.2–9 In laboratory rodents, in vivo administration of LPS results in hepatic injury and exacerbates damage from known hepatotoxins.1,2,10–12 In addition, virally infected cholestatic, and cirrhotic livers have heightened sensitivity to LPS mediated damage.2,13–15 Although the hepatotoxic effect of LPS is considered to be secondary to the release of inflammatory cytokines from LPS activated Kupffer and stellate cells,16 a direct effect of LPS on hepatocytes has not been ruled out.
Hepatocytes express toll like receptors 4 (TLR4), the plasma membrane receptor for LPS,4,17 but the extent to which they respond to LPS by the stimulation of downstream signaling pathways has not been well characterized. When LPS binds to TLR 4 in monocytes/macrophages, pro-inflammatory signaling is promoted through activation of the transcription factor, nuclear factor-κB (NK-κB), and the stress activated kinases, C-jun-N-terminal kinase (JNK), and p3 8 mitogen activated kinase (p38 MAPK).3,4 Kupffer and stellate cells are triggered by LPS/TLR to activate these signal transduction pathways that promote the synthesis and release of damaging cytokines and inflammatory molecules. Previous studies looking at the direct effect of LPS on hepatocyte function have primarily examined production of acute phase and inflammatory proteins and not hepatocyte signaling mechanisms. 18–21 A previous study examining the effect of LPS on hepatocyte signaling demonstrated that LPS activates NF-κB and p42/p44 MAPK in primary mouse hepatocytes.17 However, the functional consequence of this activation was not studied. A more recent study has shown that hepatocytes are desensitized by LPS in a TLR4 dependent manner.22 Considering the increasing recognition of the importance of LPS/TLR4 signaling in hepatic diseases, the possibility of a direct effect of LPS on hepatocyte toxicity warrants further investigation.
Increasing intracellular cAMP protects hepatocytes from bile acid Fas ligand and cytokine tumor necrosis factor (TNF-a) mediated cell death.23–27 Cyclic AMP transduces intracellular signals through three pathways: a) the serine threonine protein kinase, protein kinase A, b) cyclic nucleotide gated channels, and c) the cAMP binding guanine nucleotide exchange factors (cAMP-GEFs, also known as exchange proteins regulated by cAMP, [EPAC]).28 Cyclic AMP-GEFs catalyze the formation of the active guanosine triphosphate (GTP) bound form of the small GTPase, Rap. Cyclic AMP/cAMP-GEF/Rap signaling pathways exist in hepatocytes and studies show that cAMP-GEF activation is responsible for cAMP’s pro-survival effect in hepatocytes.23,24,27 Cyclic AMP is known to modulate the liver’s response to LPS. In vivo, oral administration of phosphodiesterase inhibitors,29,31 that increase hepatic cAMP levels, parenteral administration of glucagon32 or cell permeable cAMP analogues33 protect against LPS-induced liver damage. Although there is some evidence that cAMP’s protective effect is at the level of the Kupffer cell,34,35 the effect of cAMP on the potential toxic effects of LPS in hepatocytes is unknown, as is information on which arm of cAMP signaling is important in LPS cytoprotection.
In a previous study Yao and colleagues reported that LPS can induce apoptosis in cultured rat hepatocytes.36 However, this study used high doses of LPS (5 to 10 μg/mL) and apoptosis was not confirmed using morphologic criteria. The aim of the present study was to confirm the direct apoptotic effect of LPS in hepatocytes using both morphologic and biochemical criteria. Additional aims were to characterize the signaling pathway involved in LPS-induced hepatocyte apoptosis and to determine if activation of cAMP-GEF’s would protect against LPS mediated damage.
Materials and methods
Cell culture
Hepatocytes were isolated from rat livers by collagenase perfusion as previously described.23,24 This isolation procedure has been shown to yield pure hepatocyte cultures free of Kupffer and stellate cell contamination.37 In addition, purity of the cultures was assessed at >99% by light microscopy. These studies were conducted in accordance with Tufts University institutional animal care and use committee and the National Institute of Health guidelines. Cells were plated on to Type I collagen in minimal essential media supplemented with 10% fetal calf serum, 100 ng/mL of insulin, 100 ng/mL of penicillin, and 100 ng/mL of streptomycin. After 1 hour, the media was changed to non-supplemented minimal essential media for 3 hours. LPS (500 ng/mL) from E. coli 0127: B8 (Sigma-Aldrich Chemical, St Louis, MO) was added at this time and allowed to incubate overnight. The next day cells were treated with either 100 μM glycochenodoxycholate (GCDC, Sigma-Aldrich Chemical, St Louis, MO) or 50 ng/mL Fas ligand (Axxora, LLC, San Diego, CA). Some cultures were pretreated for 30 minutes with the p38 MAPK inhibitor, SB 203580 (5 μM), the JNK inhibitor, SP600125 (15 μM) (EMD Chemicals, Gibbstown, NJ) or with 20 μM of the cAMP-GEF activator, 4-(4-chlorophenylthio)-2’-O-methyladenosine-3’5’cyclic monophosphate, (CPT-2-Me-cAMP, Axxora, LLC, San Diego, CA) prior to the addition of LPS.
Immunoblotting
Whole cell lysates were prepared in cell lysis buffer (20 mM Tris, 150 mM NaCl, 1% Triton, 1 mM phenylmethlysulfonyl fluoride, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM ß-glycerophosphate, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 500 nM okadaic acid and 1 mM orthovanadate, pH 7.5). Proteins were separated by SDS-PAGE and transferred to a polyvinylidene fluoride membrane (PVDF, Millipore, Billerica, MA). Immunoblotting was performed to detect levels of Bim, Bad Bax (Santa Cruz Biotechnology, Santa Cruz, CA), Bcl-xL, cleaved caspase 3 or 9, phospho Jun N-terminal Kinase (JNK) (Thr 183 and Tyr 185), phospho protein kinase (AKT) (Ser 473), and phospho p38 MAPK (Thr 180 and Tyr 182) (Cell Signaling Technology, Danvers MA). Equal protein loading was verified by stripping and immunoblotting with the antibodies to the non-phosphorylated forms of the kinase or with an antibody to actin (EMD Chemicals, Gibbstown, NJ). The blots were developed by chemiluminescence, digitized with Adobe Photoshop®, and quantified using Sigma Gel® software, as we have done previously23,24
Monitoring of apoptosis
Rat hepatocytes were exposed to LPS overnight or to GCDC for 2 hours or Fas ligand for 4 hours and then monitored morphologically for apoptosis using Hoechst staining.23,24 Apoptotic cells were identified by brightly staining condensed chromatin or nuclear fragmentation. A total of 500 cells were counted from random fields on each slide. Apoptosis was expressed as a percentage of the total number of cells counted. Apoptosis was also confirmed biochemically by immunoblotting cell lysates for the 17 kD or 37 kD proteolytic processing fragment associated with activation of caspase 3 or caspase 9, respectively (Cell Signaling Technology, Danvers, MA), Cytochrome C release from mitochondria was determined in cytosolic extracts prepared by a selective dignitonin permeabilization technique as previously described.38 Cytosolic proteins were separated by SDS-PAGE transferred to PVDF membrane and immunoblotted for cytochrome C (Santa Cruz Biotechnology). Equal protein loading and purity were assessed with by immunoblotting for actin and anti-cytochrome C oxidase (Molecular Probes), respectively. All immunoblots containing cytosolic protein were negative for the presence of cytochrome C oxidase immunoreactivity confirming lack of mitochondrial contamination.
Statistics
The results represent the mean and standard deviation of at least 3 separate experiments. Comparisons were done with Student’s t-test or one way ANOVA and significance was considered at a level of P < 0.05.
Results
LPs induces apoptosis in hepatocytes and enhances fas ligand-and bile acid-induced apoptosis
Treatment of hepatocytes with LPS (500 ng/mL) overnight resulted in the morphologic appearance of apoptosis in 6.5 ± 0.56 % of the hepatocytes (Figures 1A and 1B) representing an almost 3 fold increase in the small amount of basal apoptosis seen in untreated hepatocytes. Biochemically, LPS treatment was associated with an increase in the generation of the 17 kD and 37 kD cleavage fragment of caspase 3 and caspase 9, respectively (Figures 1C and 1D). Analysis of cytosolic extracts revealed the appearance of cytochrome C in LPS treated hepatocytes (Figure 1E), indicating activation of the mitochondrial pathway. These results indicate that LPS, in a clinically relevant dose, induces hepatocyte apoptosis directly in the absence of signals from Kupffer or stellate cells, and that this apoptosis involves a mitochondrial mediated pathway.
Figure 1.
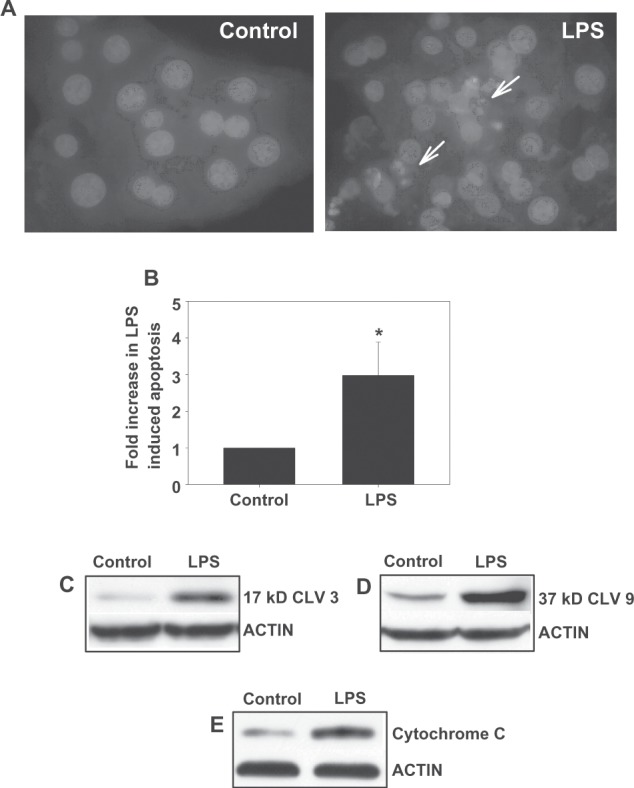
LPS induces apoptosis in hepatocytes. Hepatocytes were treated overnight with 500 ng/mL LPs and cells were evaluated morphologically by Hoechst staining for the presence of apoptosis. Representative photomicrographs (100×) are shown; the arrows depict apoptotic hepatocytes (A). The amount of apoptosis was quantified by counting the number of hepatocytes with apoptotic morphology and is expressed as a percentage of the amount of apoptosis seen in the control and represents mean ± sD [n = 3–5] (B). Biochemical evidence of apoptosis was also obtained by immunoblotting for the 17 kD or 37 kD cleavage product of caspase 3 or caspase 9, respectively (C and D). Cytosolic preparations were immunoblotted for the appearance of mitochondrial cytochrome C (E). *Significantly different from control, P < 0.05.
Abbreviations: LPS, lipopolysaccharide; SD, standard deviation.
Hepatocytes exposed to GCDC (2 hours at 100 μM) or Fas ligand (4 hours at 50 ng/mL) undergo apoptosis with an average of 2.5 and 5.2 fold increase in cell death, respectively, compared to untreated cultures (Figures 2A and 2B). When LPS sensitized hepatocytes were exposed to GCDC or Fas ligand, apoptosis increased approximately 60% over that seen in hepatocytes without pre-treatment with LPS. The increase in apoptosis seen in the presence of GCDC or Fas ligand appeared to be additive to that seen in the presence of LPS alone.
Figure 2.

LPS-mediated apoptosis is additive to that induced by bile acids or Fas ligand. Hepatocytes were treated with 500 ng/mL LPs overnight followed by treatment with 100 μM GCDC for 2 hours (A) or 50 ng/mL Fas L for 4 hours (B). Cells were evaluated morphologically by Hoechst staining for the presence of apoptosis. Results are expressed as a percentage of the amount of apoptosis seen in the control and represent mean ± SD (n = 3–5). *Significantly different from control, #Significantly different from LPs alone, P < 0.05.
Abbreviations: LPS, lipopolysaccharide; GCDC, glycochenodoxycholate; SD, standard deviation; FAS, Fas ligand.
LPS activates stress-activated kinases in hepatocytes
The stress-activated protein kinases, JNK39–43 and p38 MAPK44,45 have been implicated in facilitating hepatocyte apoptosis, while AKT has cytoprotective properties.23,24,46–48 Thus we investigated the temporal effect of LPS on the phosphorylation state (and hence activation) of these 3 kinases. LPS induced a 1.5 to 2 fold increase in JNK phosphorylation at 2 hours that persisted for up to 24 hours (Figures 3A and 3D). At the same time LPS induced an early (by 30 minutes) and sustained (up to 24 hours) increase (1.4 to 2 fold) in p38 phosphorylation (Figures 3B and 3D). LPS decreased AKT phosphorylation, but only after overnight incubation (Figures 3C and 3D). In support of a proapoptotic role for decreased AKT phosphorylation, overnight treatment with 20 μM LY 294002, a PI3K/AKT inhibitor, resulted in 12.5% ± 1.2% apoptosis. LPS had no effect on the expression of total p38, JNK or AKT proteins for up to 24 hours (data not shown). It may be noted that LPS did not affect extracellular receptor kinase ERK phosphorylation (data not shown).
Figure 3.

LPS activates stress activated kinases in hepatocytes. Cultured rat hepatocytes were treated with 500 ng/mL LPS for the time points indicated and cell lysates were prepared. The lysates were immunoblotted for phospho JNK (A and D), phospho p38 MAPK (B and D), and phospho AKT (C and D). The blots were developed by chemiluminescence, digitized and quantified. Results of quantification of at least 3 separate experiments are shown in A B and C and representative gels in D. Hepatocytes were pretreated for 30 min with either a JNK (SP 600125, 15 μM) or p38 MAPK inhibitor (SB 203580, 5 μM) and then overnight with 500 ng/mL of LPS (E). Cells were evaluated morphologically by Hoechst staining for the presence of apoptosis. Results are expressed as the percentage increase in apoptosis compared to control and represent mean ± SD (n = 3). *Significantly different from control, #Significantly different from LPS, P < 0.05.
Abbreviations: LPS, lipopolysaccharide; JNK, c-Jun N-terminal kinase; pJNK, phospho JNK; MAPK, p38 mitogen-activated protein kinases; AKT, protein kinase; SD, standard deviation.
In order to determine whether LPS induced the activation of JNK or p38 MAPK was necessary for LPS induced apoptosis, hepatocytes were pre-treated with a specific inhibitor of JNK (SP600125) or p38 MAPK (SB203580) for 30 minutes prior to the application of LPS. The results of these experiments show that the inhibition of JNK, but not p38 MAPK attenuated LPS induced apoptosis (Figure 3E). Treatments with the kinase inhibitors alone had no effect on apoptosis (Figure 3E). Previous studies by others and us demonstrated inhibition of JNK and p38 MAPK by these inhibitors under similar experimental conditions.49,50 These results suggest that LPS-induced apoptosis is JNK-dependent.
Activation of cAMP-GEF attentuates LPS-induced apoptosis and opposes LPS-induced modulation of JNK and AKT phosphorylation
Increases in cAMP protect hepatocytes from a variety of apoptotic stimuli.23,24,27 The cAMP cytoprotective pathway in rat hepatocytes involves protein kinase A independent activation of a cAMP-GEF/phosphoinositol-3-kinase/AKT pathway.23,24 We used a cAMP analogue (CPT-2-Me-cAMP) that selectively activates cAMP-GEF’s, but not protein kinase A in hepatocytes23,27 to investigate the role of this pathway in LPS induced apoptosis. Pretreatment with 20 μM CPT-2-Me-cAMP inhibited morphologic evidence of LPS-induced apoptosis by 60% (Figure 4A) and prevented caspase 3 cleavage (Figure 4B). Since we have already shown that CPT-2-Me-cAMP protects against GCDC and Fas ligand mediated apoptosis in hepatocytes,23 we investigated whether this cytoprotective effect persisted in the presence of LPS. cAMP-GEF activation was capable of inhibiting Fas ligand and GCDC induced apoptosis in the presence of LPS (Figures 4C and 4D).
Figure 4.

Activation of cAMP-GEFs attenuates LPs induced hepatocyte apoptosis. Hepatocytes were pretreated with 20 μM cPT2-Me-cAMP or vehicle for 30 minutes followed by treatment with vehicle or 500 ng/mL LPS overnight. Cells were evaluated morphologically by Hoechst staining for the presence of apoptosis (A). Results are expressed as a percentage of the amount of apoptosis seen in the control and represent mean ± SD (n = 4–6). Whole cell lysates were immunoblotted for the 17 kD cleaved fragment of caspase 3 and equal protein loading was verified by immunoblotting for actin (B). Hepatocytes were pretreated with 20 uM cPT-2-Me-cAMP for 30 minutes and then treated overnight with 500 ng/mL LPS ± pretreatment before exposing to 100 μM GCDC for 2 hours (C) or 50 ng/mL of Fas L for 4 hours (D). *Significantly different from control. #Significantly different from LPS, P < 0.05.
Abbreviations: LPS, lipopolysaccharide; cAMP, cyclic adenosine monophosphate; CPT-2-Me-cAMP, 4-(4-chlorophenylthio)-2’-O-methyladenosine-3’5’cyclic monophosphate; GCDC, glycochenodoxycholate; SD, standard deviation.
In order to determine if the cytoprotective effect of CPT-2-Me-cAMP was associated with modulation of LPS induced changes in JNK or AKT phosphorylation, we treated hepatocytes sequentially with CPT-2-Me-cAMP and LPS and then determined the effect on JNK phosphorylation at 4 and 24 hours and AKT phosphorylation at 24 hours. CPT-2-Me-cAMP opposed the LPS induced increase in JNK and decrease in AKT phosphorylation (Figure 5). CPT-2-Me-cAMP alone had no effect on JNK phosphorylation, and as we have previously reported, stimulated AKT phosphorylation.23,24 These results suggest that cAMP-GEF activation may prevent LPS-induced apoptosis by modulation of AKT and/or JNK.
Figure 5.

Activation of cAMP-GEF inhibits LPs induced phosphorylation of JNK. Hepatocytes were treated with 500 ng/mL of LPs +/− pretreatment with 20 μM cPT-2-Me-cAMP for 4 or 24 hrs. Whole cell lysates were prepared. The lysates were immunoblotted for phospho JNK (A and C) and phospho AKT (B and C) and developed by chemiluminescence, digitized and quantified. Representative gels are shown in C and the results of quantification of at least 3 separate experiments are shown in A and B. *Significantly different from control, P < 0.05. #Significantly different from LPS, P < 0.05.
Abbreviations: LPS, lipopolysaccharide; cAMP cyclic adenosine monophosphate; CPT-2-Me-cAMP,4-(4-chlorophenylthio)-2’-O-methyladenosine-3’5’cyclic monophosphate; JNK, c-Jun N-terminal kinase; AKT, protein kinase; SD, standard deviation.
LPS has no effect on the expression of Bcl-2 proteins
Hepatocytes are Type II cells that rely on the mitochondrial pathway to execute apoptosis. The B-cell lymphoma-2 (Bcl-2) family proteins are important modulators of the mitochondrial response to toxic injury. Previous studies of LPS-induced injury in nonhepatic cells have suggested that LPS can modulate the expression of Bcl-2 proteins.51,52 Thus, we investigated the effect of overnight treatment with LPS on the expression of the proapoptotic proteins, Bcl-2-associated X protein (Bax), Bcl-2 antagonist of cell death protein (Bad), and BH3-only protein (Bim), and antiapoptotic protein, Bcl-xL. LPS had no significant effect on the total expression of these Bcl-2 members (data not shown).
Discussion
The present study demonstrates that treatment of rat hepatocytes with LPS induces hepatocyte apoptosis. LPS treatment of hepatocytes is accompanied by sustained phosphorylation of JNK and p38 MAPK and decreases in AKT phosphorylation. Inhibition of LPS-induced phosphorylation of JNK with chemical inhibitors or by activation of cAMP-GEF’s attenuates LPS-induced apoptosis.
Our study shows that LPS-induced rat hepatocyte apoptosis can result from a direct effect of LPS on hepatocytes. Our findings with a more clinically relevant dose of LPS and morphological and biochemical confirmation of apoptosis establish that LPS can cause hepatocyte apoptosis independent of its ability to activate Kupffer or stellate cells. Thus, it would appear that although LPS-induced hepatocytes apoptosis in vivo results primarily from the release of cytokines from activated Kupffer and stellate cells, LPS can also have a direct toxic effect on hepatocytes.
The present study provides further insight into the signaling mechanism involved in LPS-induced hepatocyte apoptosis. The finding of sustained activation of p38 MAPK and JNK by LPS raises the possibility that these kinases may be involved in LPS-induced apoptosis since both JNK and p38 MAPK have been implicated in hepatocyte apoptosis.39–45 Studies with inhibitors of p38 MAPK and JNK however showed that LPS-induced apoptosis may be mediated via JNK and not p38 MAPK. Sustained JNK activation is associated with several models of hepatocyte apoptosis/cell death including bile acid, fatty acid and TNF-α mediated apoptosis, ischemia-reperfusion injury and acetaminophen toxicity,39–44,53 although a recent in vivo study suggested that it was likely nonparenchymal cell JNK activation that was most important in TNF-α mediated apoptosis.54 The mechanisms whereby JNK controls hepatocyte cell death has not been fully elucidated, but has been associated with modulation of Bim and TNF related apoptosis inducing ligand (TRAIL) death receptors, trafficking of Fas death receptors to plasma membrane and accelerated turnover of the antiapoptotic protein, cFLIP.39,42,44,52,55
The mechanism by which LPS activates JNK and p38 MAPK in hepatocytes is unclear at this time. Studies in non-hepatic cells suggest that LPS binding to toll-like receptor 4 protein (TLR4) results in the activation of the transcription factor, NF-κB and the stress activated kinases, JNK and p38 MAPK.3,4 Since hepatocytes express TLRs, it is likely that the activation of JNK and p38 MAPK observed after LPS exposure in the present study is mediated by TLR activation. However, a recent study with mouse hepatocytes showed that LPS failed to activate JNK and p38 MAPK, although LPS-induced TLR4-dependent phosphorylation of p42/p44 mitogen-activated kinase and activation of NF-κB17 This discrepancy may be due to the time frames examined (not mentioned in the mouse study) or represent differences due to culture conditions or variation within species. It is, however, also possible that LPS-induced activation of JNK and p38 MAPK may not be mediated via TLR4 in hepatocytes. Experiments to determine if LPS activates these stress activated kinases in hepatocytes isolated from TLR 4−/− mice would help to answer this question.
Our studies show that LPS mediated apoptosis in hepatocyte proceeds through a mitochondrial dependent pathway as evidenced by cytochrome C release and caspase 9 cleavage. Activation of JNK can mediate mitochondrial dysfunction. In acetaminophen and ischemia-reperfusion injury in hepatocytes, JNK translocates to the mitochondria and promotes cell death by induction of the mitochondrial membrane permeability transition (MPT).40,43 Since our studies concur with previous studies which support a role for mitochondrial dysfunction in LPS-mediated toxicity in the liver56,57 the role of JNK in LPS-mediated mitochondrial damage deserves further clarification. Another mechanism by which JNK modulates mitochondrial integrity is by controlling the balance of pro- and antiapoptotic Bcl-2 proteins. Free fatty acid-induced JNK activation is associated with an increase in expression of the pro-apoptotic proteins Bax and Bim expression.41,58 We were unable to show any changes in the total expression of Bax or Bim with LPS treatment. However, since these proteins are also controlled by post-translational events such as phosphorylation and translocation to the mitochondria we cannot rule out a role for LPS in the modulation of their action. Indeed, acetaminophen induced JNK activation results in mitochondrial translocation of Bax without changes in total Bax expression.40
We and others have shown that increases in intracellular cAMP can protect hepatocytes from a variety of toxic injuries including bile acid, Fas ligand- and TNF-α-mediated apoptosis.23–27 We extend these observations to show that cAMP acting through cAMP-GEF’s, also protects hepatocytes from LPS-mediated damage. This cytoprotective effect of cAMP-GEF against a wide variety of apoptotic stimuli in hepatocytes may suggest that cAMP-GEF exerts its cytoprotective effect at the level of mitochondria. In Type II cells, such as hepatocytes, death receptor-mediated apoptosis is believed to require a mitochondrial amplification loop and cAMP-GEF has been shown to produce a cytoprotective effect by inhibiting the mitochondrial pathway.50
The cytoprotective effect is accompanied by attenuation of LPS-mediated activation of JNK- and LPS-induced dephosphorylation of AKT, both of which would oppose hepatocyte apoptosis.23,24,27,38,39,41 In addition, cAMP-GEF activates the antiapoptotic AKT signaling pathway. Thus, cAMP appears to reverse LPS-induced hepatocyte apoptosis by inhibiting LPS-induced pro-apoptotic stimuli as well as activating antiapoptotic signals. Hepatocytes express several receptors, including those for glucagon and epinephrine, which increase cAMP synthesis. It is tempting to speculate that increases in intracellular cAMP which occur in response to these stress hormones may serve as a protective surveillance mechanism against hepatocyte damage from intestinally derived LPS. A recent study showing that lack of the glucagon receptor in mice predisposes them to toxic injury is consistent with this hypothesis.27
Our study also showed that LPS-induced apoptosis is additive to that induced by GCDC or Fas ligand in hepatocytes. This observation is consistent with in vivo studies in which LPS enhances hepatic injury from a variety of insults including viral, cholestatic and toxic events.2,10,13–15 In some cases, this enhanced sensitivity to injury is associated with upregulation of TLR4 in the liver.59–61 Our results raise the possibility that this heightened sensitivity may, in part, be due to the direct effect of LPS on hepatocytes could be even more profound, if the LPS effect is mediated via TLR, in the damaged liver with increased TLR4 expression.
Conclusion
This is the first study to show that LPS induces JNK-dependent hepatocyte apoptosis in vitro in the virtual absence of Kupffer and stellate cells, and that LPS’s effect is additive to the apoptosis caused by bile acids or Fas ligand. Our study in isolated hepatocytes demonstrates that LPS not only causes liver injury independently, but may also augment injury when other pro-apoptotic stimuli are present. Since these results are similar to what has been observed in vivo, it is likely that the direct apoptotic effect of LPS on hepatocytes plays a role in liver disease. Selective strategies to prevent LPS-mediated toxic injury, such as augmenting intracellular cAMP levels, may have therapeutic application in the myriad of hepatic diseases in which LPS has been implicated to play a pathologic role.
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health: NIH R01 DK065975 to Cynthia R Leveille-Webster and NIH R01 DK033436 to M. Sawkat Anwer.
Footnotes
Disclosures
The authors report no conflicts of interest in this work.
References
- 1.Han D. Intestinal endotoxemia as a pathogenetic mechanism in liver failure. W J Gastro. 2002;8:961–965. doi: 10.3748/wjg.v8.i6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jirillo E, Caccavo D, Magrone T, et al. The role of the liver in the response to LPS; experimental and clinical findings. J Endotoxin Res. 2002;8:319–327. doi: 10.1179/096805102125000641. [DOI] [PubMed] [Google Scholar]
- 3.Schwabe RF, Seki E, Brenner DA. Toll-like receptor signaling in the liver. Gastroenterology. 2006;130:1886–1900. doi: 10.1053/j.gastro.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 4.Szabo G, Dolganiuc A, Mandrekar P. Pattern recognition receptors: A contemporary view of liver disease. Hepatology. 2006;44:287–298. doi: 10.1002/hep.21308. [DOI] [PubMed] [Google Scholar]
- 5.Hritz I, Mandrekar P, Velayudham A, et al. The critical role of toll-like receptor 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology. 2008;48:1224–1231. doi: 10.1002/hep.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keshavarzian A, Farhadi A, Forsyth CB, et al. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to the development of alcoholic steatohepatitis in rats. J Hepatol. 2009;50:538–547. doi: 10.1016/j.jhep.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riordan SM, Skinner NA, Kurtovic J, et al. Toll like receptor expression in chronic hepatitis C: correlation with pro-inflammatory cytokine levels and liver injury. Inflamm Res. 2006;55:279–285. doi: 10.1007/s00011-006-0082-0. [DOI] [PubMed] [Google Scholar]
- 8.Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor 4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatoheaptitis. J Hepatol. 2007;47:571–579. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brun P, Castagliuolo I, Di Leo V, et al. Increased intestinal permeability in obese mice: new evidence in the pathogenesis of non-alcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G518–G525. doi: 10.1152/ajpgi.00024.2006. [DOI] [PubMed] [Google Scholar]
- 10.Barton CC, Hill DA, Yee SB, Barton EX, Ganey PE, Roth RA. Bacterial lipopolysaccharide exposure augments aflatoxin B1-induced liver injury. Toxicol Sci. 2000;55:444–452. doi: 10.1093/toxsci/55.2.444. [DOI] [PubMed] [Google Scholar]
- 11.Kirsch R, Clarkson V, Verdonk RC, et al. Rodent nutritional model of steatohepatitis: effect of endotoxin (lipopolysaccharide) and tumor necrosis factor alpha deficiency. Gastroenterol Hepatol. 2006;21:174–182. doi: 10.1111/j.1440-1746.2005.04220.x. [DOI] [PubMed] [Google Scholar]
- 12.Hassan F, Morikawa A, Islam S, et al. Lipopolysaccharide augments the in vivo lethal action of doxorubicin against mice via hepatic damage. Clin Exp Immunol. 2008;151:334–350. doi: 10.1111/j.1365-2249.2007.03568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moazzan F, Brems JJ, Yong SL, et al. Endotoxin potentiates hepatocyte apoptosis in cholestasis. J Am Coll Surg. 2002;194:731–739. doi: 10.1016/s1072-7515(02)01173-0. [DOI] [PubMed] [Google Scholar]
- 14.Tazi KA, Bieche I, Paradis V, et al. In vivo altered protein response and apoptosis in livers from LPS challenged cirrhotic rats. J Hepatol. 2007;46:1075–1088. doi: 10.1016/j.jhep.2007.01.034. [DOI] [PubMed] [Google Scholar]
- 15.Iida A, Yoshidome H, Shida T, et al. Does prolonged biliary obstructive jaundice sensitize the liver to endotoxemia. Shock. 2009;31:397–403. doi: 10.1097/SHK.0b013e31818349ea. [DOI] [PubMed] [Google Scholar]
- 16.Su GL. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol Gastrointest Liver Physiol. 2002;283:G256–G265. doi: 10.1152/ajpgi.00550.2001. [DOI] [PubMed] [Google Scholar]
- 17.Liu S, Gallo DJ, Green AM, et al. Role of toll-like receptors in changes in gene expression and NF-κB activation in mouse hepatocytes stimulated with lipopolysaccaharide. Infect Immun. 2002;70:3433–3442. doi: 10.1128/IAI.70.7.3433-3442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panesar N, Tolman K, Mazuski JE. Endotoxin stimulates hepatocytes interleukin-6 production. J Surg Res. 1999;85:251–225. doi: 10.1006/jsre.1999.5648. [DOI] [PubMed] [Google Scholar]
- 19.Portoles MT, Ainage MJ, Pagani R. The induction of lipid peroxidation by E. coli lipopolysaccharide on rat hepatocytes as an important factor in the etiology of endotoxic liver damage. Biochim Biophys Acta. 1993;1158:287–292. doi: 10.1016/0304-4165(93)90027-6. [DOI] [PubMed] [Google Scholar]
- 20.Satoh S, Nussler AK, Liu ZZ, Thomson AW. Proinflammatory cytokines and endotoxin stimulate ICAM-1 gene expression and secretion by normal human hepatocytes. Immunology. 1994;82:571–576. [PMC free article] [PubMed] [Google Scholar]
- 21.Saad B, Frei K, Scholl FA, Fontana A, Maier P. Hepatocyte-derived interleukin-6 and tumor necrosis factor alpha mediate the lipopolysaccharide induced acute phase response and nitric oxide release by cultured rat hepatocytes. Eur J Biochem. 1995;229:349–355. doi: 10.1111/j.1432-1033.1995.0349k.x. [DOI] [PubMed] [Google Scholar]
- 22.Scott MJ, Lui S, Shapiro RA, Vodovotz Billiar TR. Endotoxin uptake in mouse liver is blocked by endotoxin pretreatment through a suppressor of cytokine signaling-1-dependent mechanism. Hepatology. 2009;49:1695–1708. doi: 10.1002/hep.22839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cullen K, McCool J, Anwer MS, Webster CRL. Activation of cAMP-guanine exchange factor (cAMP-GEF) confers protein kinase A independent protection from hepatocyte apoptosis. Am J Physiol Gastrointest Liver Physiol. 2004;287:G334–G343. doi: 10.1152/ajpgi.00517.2003. [DOI] [PubMed] [Google Scholar]
- 24.Gates A, Hohenester S, Anwer S, Webster CRL. cAMP-GEF cytoprotection by Src tyrosine kinase activation of phosphoinositide-3-kinase p110 β/α in rat hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2009;296:G764–774. doi: 10.1152/ajpgi.90622.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Yang S, Billiar TR. Cyclic nucleotides suppress tumor necrosis factor alpha mediated apoptosis by inhibiting caspase activation and cytochrome c release in primary hepatocytes via a mechanism independent of AKT activation. J Biol Chem. 2000;275:13026–13034. doi: 10.1074/jbc.275.17.13026. [DOI] [PubMed] [Google Scholar]
- 26.Reinehr R, Häussinger D. Inhibition of bile salt-induced apoptosis by cyclic AMP involves serine/threonine phosphorylation of CD95. Gastroenterology. 2004;126:249–262. doi: 10.1053/j.gastro.2003.09.044. [DOI] [PubMed] [Google Scholar]
- 27.Sinclair EM, Yusta B, Streutker C, et al. Glucagon receptor signaling is essential for control of murine hepatocyte survival. Gastroenterology. 2008;135:2096–2106. doi: 10.1053/j.gastro.2008.07.075. [DOI] [PubMed] [Google Scholar]
- 28.Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG. Cell physiology of the cAMP sensor EPAC. J Physiol. 2006;577:5–15. doi: 10.1113/jphysiol.2006.119644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coimbra R, Porcides RD, Melbostad H, et al. Nonspecific phoshodiesterase inhibition attenuates liver injury in acute endotoxemia. Surg Infect (Larchmt) 2005;6:73–85. doi: 10.1089/sur.2005.6.73. [DOI] [PubMed] [Google Scholar]
- 30.Fischer W, Schudt C, Wendel A. Protection by phosphodiesterase inhibitors against endotoxin induced liver injury in galactosamine-sensitized mice. Biochem Pharmacol. 1993;45:2399–2404. doi: 10.1016/0006-2952(93)90219-m. [DOI] [PubMed] [Google Scholar]
- 31.Taguchi I, Oka K, Kitamura K, Sugiura M, Oku A, Matsumoto M. Protection by cyclic AMP-specific phosphodiesterase inhibitor, rolipram and dibutyryl cyclic AMP against propionbacterium acnes and lipopolysaccharide induced mouse hepatitis. Inflamm Res. 1999;48:380–385. doi: 10.1007/s000110050475. [DOI] [PubMed] [Google Scholar]
- 32.Harbrecht BG, Perpetua M, Fulmer M, Zhang B. Glucagon regulates hepatic inducible nitric oxide synthesis in vivo. Shock. 2004;22:157–162. doi: 10.1097/01.shk.0000131579.22409.33. [DOI] [PubMed] [Google Scholar]
- 33.Arai T, Hiromatsu K, Kobayashi N, et al. IL-10 is involved in the protective effect of dibutyryl cyclic adenosine monophosphate on endotoxin induced inflammatory liver injury. J Immunol. 1995;155:5743–579. [PubMed] [Google Scholar]
- 34.Risoe PK, Wang Y, Stuestol JF, Aasen AO, Wang JE, Dahle MK. Lipopolysaccharide attenuates mRNA levels of several adenylyl cyclase isoforms in vivo. Biochim Biophys Acta. 2007;1771:32–39. doi: 10.1016/j.bbadis.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 35.Gobejishvili L, Barve S, Joshi-Barve S, McClain C. Enhanced PDE4B expression augments LPS-inducible TNF expression in ethanol primed monocytes: relevance to alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2008;295:G718–G724. doi: 10.1152/ajpgi.90232.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao Y, Zhang D, Luo Y, et al. Fas ligand expression and apoptosis in primary rat hepatocytes induced by lipopolysaccharide. Zhonghua Gan Zang Bing Za Zhi. 2000;8:285–287. [PubMed] [Google Scholar]
- 37.Smedsrød B, Pertoft H. Preparation of pure hepatocytes and reticuloendothelial cells in high yield from a single rat liver by means of Percoll centrifugation and selective adherence. J Leukoc Biol. 1985;38:213–230. doi: 10.1002/jlb.38.2.213. [DOI] [PubMed] [Google Scholar]
- 38.Higuchi H, Yoon JH, Grambihler A, Werneburg N, Bronk SF, Gores GJ. Bile acids stimulate cFLIP phosphorylation enhancing TRAIL-mediated apoptosis. J Biol Chem. 2003;278:454–461. doi: 10.1074/jbc.M209387200. [DOI] [PubMed] [Google Scholar]
- 39.Corazza N, Jakob S, Schaer C, et al. TRAIL receptor mediated JNK activation and BIM phosphorylation critically regulate Fas-mediated liver damage and lethality. J Clin Invest. 2006;116:2493–2499. doi: 10.1172/JCI27726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondrial bioenergetics in acetaminophen induced liver injury. J Biol Chem. 2008;283:13565–13577. doi: 10.1074/jbc.M708916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 42.Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK 2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49:87–96. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Theruvath TP, Snoddy MC, Zhong Z, Lemasters JJ. Mitochondrial permeability transition in liver ischemia and reperfusion: role of c-jun-N terminal kinase 2. Transplantation. 2008;85:1500–1504. doi: 10.1097/TP.0b013e31816fefb5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higuchi H, Grambihler A, Canbay A, Bronk SF, Gores GJ. Bile acids up-regulate death receptor5/TRAIL-receptor 2 expression via a c-Jun-N terminal kinase dependent pathway involving Sp1. J Biol Chem. 2004;279:51–60. doi: 10.1074/jbc.M309476200. [DOI] [PubMed] [Google Scholar]
- 45.Grambihler A, Higuchi H, Bronk SF, Gores GJ. cFLIP-L inhibits p38 MAPK activation: an additional anti-apoptotic mechanism in bile acid mediated apoptosis. J Biol Chem. 2003;278:26831–26837. doi: 10.1074/jbc.M303229200. [DOI] [PubMed] [Google Scholar]
- 46.Moumen A, Ieraci A, Patané S, et al. Met signals hepatocyte survival by preventing Fas-triggered FLIP degradation in a PI3K-AKT dependent manner. Hepatology. 2007;45:1210–1217. doi: 10.1002/hep.21604. [DOI] [PubMed] [Google Scholar]
- 47.Nitta T, Kim JS, Mohuczy D, Behrns KE. Murine cirrhosis induces hepatocyte epithelial mesenchymal transition and alterations in survival signaling pathways. Hepatology. 2008;48:909–919. doi: 10.1002/hep.22397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osawa Y, Hannun YA, Proia RL, Brenner DA. Roles of AKT and spingosine kinase in the antiapoptotic effect of bile duct ligation in mouse liver. Hepatology. 2005;42:1320–1328. doi: 10.1002/hep.20967. [DOI] [PubMed] [Google Scholar]
- 49.Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. Differential requirement for c-Jun NH2 terminal kinase in TNF alpha and Fas mediated apoptosis in hepatocytes. FASEB J. 2004;18:720–722. doi: 10.1096/fj.03-0771fje. [DOI] [PubMed] [Google Scholar]
- 50.Webster CR, Usechak P, Anwer MS. cAMP inhibits bile acid induced apoptosis by blocking caspase activation and cytochrome C release. Am J Physiol Gastrointest Liver Physiol. 2002;283:G727–G738. doi: 10.1152/ajpgi.00410.2001. [DOI] [PubMed] [Google Scholar]
- 51.Kim MJ, Lee JH, Park SY, et al. Protection from apoptotic cell death by cilostazol, phosphodiesterase type III inhibitor, via cAMP dependent protein kinase activation. Pharmacol Res. 2006;54:261–267. doi: 10.1016/j.phrs.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 52.Das M, Sabio G, Jiang F, Rincón M, Flavell RA, Davis RJ. Induction of hepatitis by JNK-mediated expression of TNF-alpha. Cell. 2009;136:249–260. doi: 10.1016/j.cell.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bourdi M, Korrapati MC, Chakraborty M, Yee SB, Pohl LR. Protective role of c-jun NH2-terminal kinase 2 in acetaminophen induced liver injury. Biochem Biophys Res Commun. 2008;374:6–10. doi: 10.1016/j.bbrc.2008.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gugasyan R, Gerondakis S, Cretney E, et al. Fatal hepatitis mediated by tumor necrosis factor TNF alpha requires caspase 8 and involved BH3-only proteins Bid and Bim. Immunity. 2009;30:56–66. doi: 10.1016/j.immuni.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang L, Kamata H, Solinas G, et al. The E3 ubiquitin ligase itch couples JNK activation to TNF alpha induced cell death by inducing c-FLIP (L) turnover. Cell. 2006;124:601–613. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 56.Kuwabara T, Imajoh-Ohmi S. LPS-induced apoptosis is dependent upon mitochondrial dysfunction. Apoptosis. 2004;9:467–474. doi: 10.1023/B:APPT.0000031453.90821.6a. [DOI] [PubMed] [Google Scholar]
- 57.Crouser ED, Julian MW, Huff JE, et al. Abnormal permeability of inner and outer mitochondrial membranes contributes independently to mitochondrial dysfunction in the liver during acute endotoxemia. Crit Care Med. 2004;32:607–609. doi: 10.1097/01.CCM.0000109449.99160.81. [DOI] [PubMed] [Google Scholar]
- 58.Wang Y, Ausman LM, Russell RM, Greenberg AS, Wang XD. Increased apoptosis in high fat diet induced nonalcoholic steatohepatitis in rats is associated with c-Jun-NH2 terminal kinase activation and elevated proapoptotic Bax. J Nutr. 2008;138:1866–1871. doi: 10.1093/jn/138.10.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hua J, Qui DK, Li JQ, Li EL, Chen XY, Peng YS. Expression of TLR4 in rat liver during the course of carbon tetrachloride induced liver injury. J Gastro Hepatology. 2007;22:862–869. doi: 10.1111/j.1440-1746.2007.04896.x. [DOI] [PubMed] [Google Scholar]
- 60.Mozer-Lisewska I, Sluzewski W, Kaczmarek M, et al. Tissue localization of Toll like receptors in biopsy specimens of liver from children infected with hepatitis C virus. Scand J Immunol. 2005;62:407–412. doi: 10.1111/j.1365-3083.2005.01670.x. [DOI] [PubMed] [Google Scholar]
- 61.Wang AP, Migita K, Ito M, et al. Hepatic expression of toll-like 4 in primary biliary cirrhosis. J Autoimmun. 2005;25:85–91. doi: 10.1016/j.jaut.2005.05.003. [DOI] [PubMed] [Google Scholar]