

Abstract
Autophagy is an essential, homeostatic process by which cells break down their own components. Perhaps the most primordial function of this lysosomal degradation pathway is adaptation to nutrient deprivation. However, in complex multicellular organisms, the core molecular machinery of autophagy — the 'autophagy proteins' — orchestrates diverse aspects of cellular and organismal responses to other dangerous stimuli such as infection. Recent developments reveal a crucial role for the autophagy pathway and proteins in immunity and inflammation. They balance the beneficial and detrimental effects of immunity and inflammation, and thereby may protect against infectious, autoimmune and inflammatory diseases.
Subject terms: Signal transduction, Autophagy, Immunological disorders
Main
There is only one known mechanism that eukaryotic cells possess to dispose of intracellular organelles and protein aggregates that are too large to be degraded by the proteasome. It is therefore not surprising that this mechanism — the lysosomal degradation pathway known as autophagy — is also used to degrade microorganisms (such as viruses, bacteria and protozoa) that invade intracellularly1,2. Indeed, the mutation of autophagy genes increases susceptibility to infection by intracellular pathogens in organisms ranging from plants to flies to worms to mice, and possibly to humans. Perhaps less apparent, but teleologically as intuitive, the autophagy pathway or unique functions of autophagy proteins also have a central role in controlling other diverse aspects of immunity in multicellular organisms.
The autophagy machinery is thought to have evolved as a stress response that allows unicellular eukaryotic organisms to survive during harsh conditions, probably by regulating energy homeostasis and/or by protein and organelle quality control. The same machinery might therefore be expected to diversify functionally in complex metazoan organisms, so as to regulate new layers of defences used by multicellular organisms to confront different forms of stress. A plethora of genetic, biochemistry, cell biology, systems biology and genomic studies have recently converged to support this notion. The autophagy machinery interfaces with most cellular stress-response pathways3, including those involved in controlling immune responses and inflammation. This interface is not only at the level of the autophagy pathway, but also entails direct interactions between autophagy proteins and immune signalling molecules4. There is a complex reciprocal relationship between the autophagy pathway/proteins and immunity and inflammation; the autophagy proteins function in both the induction and suppression of immune and inflammatory responses, and immune and inflammatory signals function in both the induction and suppression of autophagy. Moreover, similar to cancer, neurodegenerative diseases and ageing5, defects in autophagy — through autophagy gene mutation and/or microbial antagonism — may underlie the pathogenesis of many infectious diseases and inflammatory syndromes.
In this Review, we describe recent advances in our evolving comprehension of the interface between autophagy, immunity and inflammation. We discuss how emerging concepts about the functions of the autophagy pathway and the autophagy proteins may reshape our understanding of immunity and disease. This set of proteins not only orchestrates the lysosomal degradation of unwanted cargo, but also exerts intricate effects on the control of immunity and inflammation. Thus, the autophagy pathway and autophagy proteins may function as a central fulcrum that balances the beneficial and harmful effects of the host response to infection and other immunological stimuli.
Mechanisms and membrane dynamics of autophagy
Autophagy is a general term for pathways by which cytoplasmic material, including soluble macromolecules and organelles, is delivered to lysosomes for degradation6. There are at least three different types of autophagy, including macroautophagy, chaperone-mediated autophagy and microautophagy. Macroautophagy, usually referred to simply as autophagy, is the subject of this Review (Fig. 1). In this pathway, a portion of cytoplasm (usually 0.5–1 μm in diameter) is engulfed by an isolation membrane, or 'phagophore', resulting in the formation of a double-membrane structure known as the autophagosome. The outer membrane of the autophagosome fuses with the lysosome to become an autolysosome, leading to the degradation of autophagosomal contents by lysosomal enzymes. Autophagosomes can also fuse with endosomes or multivesicular bodies and major histocompatibility complex (MHC)- class-II-loading compartments7. Autolysosomes become larger as more autophagosomes and lysosomes fuse, but at a termination phase lysosomes are tubulated and fragmented for renewal8.
Figure 1. Schematic overview of autophagy and its regulation.
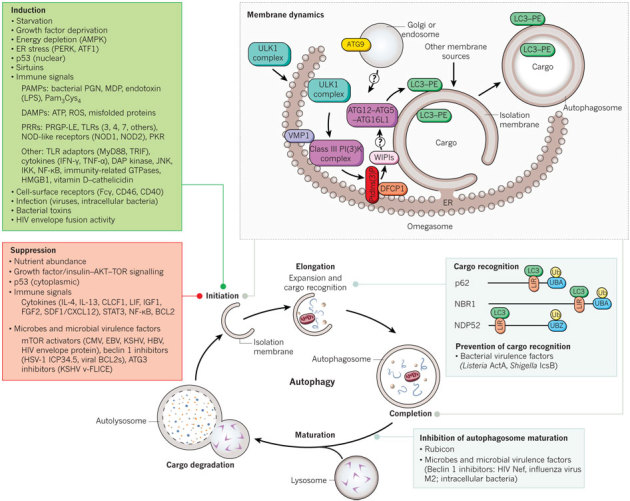
Overview of the autophagy pathway. The top right box shows a model of our current understanding of the molecular events involved in membrane initiation, elongation and completion of the autophagosome. The major membrane source is thought to be the endoplasmic reticulum (ER), although several other membrane sources, such as mitochondria and the plasma or nuclear membrane, may contribute. After induction of autophagy, the ULK1 complex (ULK1–ATG13–FIP200–ATG101) (downstream of the inhibitory mTOR signalling complex) translocates to the ER and transiently associates with VMP1, resulting in activation of the ER-localized autophagy-specific class III phosphatidylinositol-3-OH kinase (PI(3)K) complex, and the phosphatidylinositol-3-phosphate (PtdIns(3)P) formed on the ER membrane recruits DFCP1 and WIPIs. WIPIs and the ATG12–ATG5–ATG16L1 complex are present on the outer membrane, and LC3–PE is present on both the outer and inner membrane of the isolation membrane, which may emerge from subdomains of the ER termed omegasomes. The cellular events that occur during autophagy are depicted in the bottom diagram, including the major known cellular and microbial proteins that regulate autophagy initiation, cargo recognition and autophagosome maturation. Only those cellular proteins known to be adaptors for targeting microbes are shown; other proteins (not shown) also function in cargo recognition of mitochondria and other organelles. CMV, cytomegalovirus; DAMP, danger-associated molecular pattern; DAP, death-associated protein; EBV, Epstein–Barr virus; HBV, hepatitis B virus; HSV-1, herpes simplex virus 1; KSHV, Kaposi's sarcoma-associated herpesvirus; LIR, LC3-interacting region (motif); LPS, lipopolysaccharide; MDP, muramyl dipeptide; Pam3Cys4, a synthetic TLR2 agonist; PAMP, pathogen-associated molecular pattern; PERK, an eIF2α kinase; PGN, peptidoglycan; PRGP-LE, a peptidoglycan-recognition protein; PRR, pathogen-recognition receptor; ROS, reactive oxygen species; Ub, ubiquitin; UBA, ubiquitin-associated domain; UBZ, ubiquitin-binding zinc finger; v-FLICE, viral FLICE.
The membrane dynamics of autophagosome formation involve complex processes, which are not completely understood. Nonetheless, the molecular dissection of autophagy membrane dynamics, stimulated by the discovery of ATG (autophagy-related) genes in yeast9, has shed considerable light on this topic (Table 1). Several recent studies suggest that the endoplasmic reticulum (ER) is crucial for autophagosome formation. The ER cisternae often associate with developing autophagosomes, and electron tomography analysis has demonstrated direct connections between the ER and autophagosomal membranes10,11. Moreover, the function of several key autophagy proteins seems to be at the level of the ER (Fig. 1).
Table 1.
Key proteins involved in mammalian autophagosome formation and their immune functions
| Protein complex | Function of protein complex in autophagy | Specific protein | General properties | Immunological/host defence functions |
|---|---|---|---|---|
| Nucleation step of autophagosome formation | ||||
| ULK complex | This complex is negatively regulated by mTORC1 in a nutrient-dependent manner. After autophagy induction, this complex translocates to early autophagic structures. Although FIP200 and ATG13 are known to be phosphorylated by ULK1, physiologically relevant substrates remain unknown. FIP200 and ATG101 may have functions similar to yeast Atg17, 29 and 31, although they show no sequence similarity with these proteins. |
ULK1/2 ATG13 FIP200 ATG101 |
Protein kinase, phosphorylated by mTORC1 Phosphorylated by mTORC1 Scaffold for ULK1/2 and ATG13 Interacts with ATG13 |
Antibacterial47*,48*; antiviral46* Unknown Maintains numbers of fetal haematopoietic stem cells72 Unknown |
| Class III PI(3)K complex | Beclin 1 is negatively regulated by BCL2 and by BCL-XL through direct binding. This complex produces PtdIns(3)P, probably on the ER. VPS34, VPS15 and beclin 1 are shared with the UVRAG complex, which seems to function in the late endocytic pathway. Rubicon negatively regulates autophagosome–lysosome fusion through interaction with the UVRAG complex. |
VPS34 VPS15 Beclin 1 ATG14 AMBRA1 UVRAG Rubicon |
PI(3) kinase Myristoylated BH3-only protein, interacts with BCL2 and BCL-XL Autophagy-specific subunit Interacts with and activates beclin 1 A VPS38 homologue; interacts with class C VPS (HOPS) complex Interacts with beclin 1 |
Antiviral45*; phagosome maturation33 Unknown Antibacterial45*,48*; antiviral46*,57,58; apoptotic corpse clearance84; decreases inflammation in tumours5; regulates germinal centre induction5; phagosome maturation31,33,49; interacts with TLR signalling adaptors3 Unknown Unknown Unknown Unknown |
| Others | DFCP1 forms an omegasome on the ER, at which other ATG proteins are assembled. ATG9, WIPIs and VMP1 are present on the autophagic membrane. ATG9 also exists in other compartments such as endosomes and the Golgi apparatus. |
ATG2 ATG9 WIPI1–4 DFCP1 VMP1 |
Interacts with Atg18 in yeast Transmembrane protein PtdIns(3)P-binding proteins PtdIns(3)P-binding ER protein Transmembrane protein |
Antiviral46* Antiviral46*; inhibits innate immune signalling4 Unknown Unknown Unknown |
| Elongation step | ||||
| ATG12-conjugation system | The ATG12–ATG5–ATG16L1 dimer is important for LC3–PE conjugation. This complex is present on the outer side of the isolation membrane and is essential for proper elongation of the isolation membrane. |
ATG12 ATG7 ATG10 ATG5 ATG16L1 |
Ubiquitin-like, conjugates to ATG5 E1-like enzyme E2-like enzyme Conjugated by ATG12 Homodimer, interacts with ATG5 |
Antiviral46*; antibacterial34; antigen presentation7,32,74; inhibits type I IFN production78 Antiviral45*,46*; antibacterial48*; antigen presentation32; phagosome maturation31; maintains number of T cells44,72; intestinal immune epithelial cell function90; inhibits type I IFN production78; inhibits pro-inflammatory cytokine production82 Unknown Antiviral40,46*; antibacterial1,30,35,47*,100; antiparasitic35; antigen presentation27,32,73; phagosome maturation31; apoptotic corpse clearance84; maintains number of T cells44,72; maintains number of B1a B cells44; intestinal immune epithelial cell function90; inhibits type I IFN production77,78; increases type I IFN production76 Antibacterial87,88; antigen presentation52; Crohn's disease risk allele85; intestinal immune epithelial cell function81,90; inhibits pro-inflammatory cytokine production82 |
| LC3-conjugation system | The formation of LC3–PE conjugates and their deconjugation by ATG4 is important for isolation membrane elongation and/or complete closure. LC3 is present on both the inner and outer membrane of the autophagosomes, and also serves as an adaptor for selective substrates such as p62, NBR1, NDP52 and the yeast mitophagy protein Atg32. |
LC3 (GATE16, GABARAP) ATG4A–D ATG7 ATG3 |
Ubiquitin-like, conjugates to PE LC3 carboxy-terminal hydrolase, deconjugating enzyme E1-like enzyme E2-like enzyme |
Antiviral46*; antibacterial48*; antigen presentation7; adaptor for substrates of selective autophagy of microbes (xenophagy)38,41,61 Antiviral46* Antiviral45*,46*; antibacterial48*; antigen presentation32; phagosome maturation31; maintains number of T cells44,72; intestinal immune epithelial cell function90; inhibits type I IFN production78; inhibits pro-inflammatory cytokine production82 Antiviral45* |
The components of the autophagy machinery that have been shown to participate in the immune and inflammatory processes depicted in Fig. 3. Owing to space limitations, primary papers are cited only for those citations also included in the main text; otherwise, see references contained within cited review articles.
*Function observed in model organism (for example, Dictyostelium discoideum, Nicotiana benthamiana, Arabidopsis thaliana, Drosophila melanogaster or Caenorhabditis elegans).
Autophagy is induced by nutrient starvation through the inhibition of mammalian target of rapamycin (mTOR), resulting in translocation of the mTOR substrate complex (ULK1/2, ATG13, FIP200 (also known as RB1CC1) and ATG101) from the cytosol to certain domains of the ER or closely attached structures12,13. This leads to the recruitment of the class III phosphatidylinositol-3-OH kinase (PI(3)K) complex, which includes at least VPS34 (also known as PIK3C3), VPS15 (PIK3R4 and p150), beclin 1 and ATG14, to the ER13,14. The PI(3)K complex produces phosphatidylinositol-3-phosphate (PtdIns(3)P), which recruits effectors such as double FYVE-containing protein 1 (DFCP1) and WD-repeat domain phosphoinositide-interacting (WIPI) family proteins. DFCP1 is diffusely present on the ER or the Golgi, but translocates to the autophagosome formation site in a PtdIns(3)P-dependent manner to generate ER-associated Ω-like structures termed omegasomes15. Among the four WIPI isoforms, WIPI2 is the major form in most cell types and functions downstream of DFCP1, and it may promote the development of omegasomes into isolation membranes or autophagosomes16.
An ER-associated multispanning membrane protein, VMP1, is also important for autophagosome formation. Although VMP1 interacts with beclin 1 and is present at the autophagosome formation site at an early stage, it seems to function at a late stage in autophagy13,17,18. This is perhaps consistent with other evidence that beclin 1–class III PI(3)K complexes may function in autophagosomal maturation (in addition to vesicle nucleation), a process that can be regulated by other beclin-1-interacting partners such as rubicon (Table 1). At the final step of autophagosome formation, elongation of the isolation membrane and/or completion of enclosure require two ubiquitin-like conjugates. The first is the ATG12–ATG5 conjugate, which is produced by the ATG7 (E1-like) and ATG10 (E2-like) enzymes, and functions as a dimeric complex together with ATG16L1 (ref. 19) . The second is the phosphatidylethanolamine (PE)-conjugated ATG8 homologues — LC3, GATE16 and GABARAP — which are produced by the ATG7 and ATG3 (E2-like) enzymes9,20. Although the proteins involved in autophagosome membrane formation have been characterized as discrete complexes (Table 1), several potential interconnections between components of the different complexes were identified by a recent proteomics study21. Such interconnections may function in autophagosome membrane formation or other distinct cellular functions. For example, the ATG12–ATG3 conjugate is implicated in mitochondrial homeostasis but not in autophagosome membrane formation22.
In addition to the ER, other membranes may be involved in autophagosome formation (Fig. 1). ATG9, another multispanning membrane protein, is essential for autophagy23 and traffics between the trans-Golgi network, endosomes and autophagosome precursors24. Studies suggest that mitochondria, the plasma membrane and the nuclear membrane could also be membrane sources for autophagosome formation25,26,27. However, the lack of detection of specific protein markers for these structures on the autophagosomal membrane leaves the decades-old question of the membrane source of the autophagosome unanswered. It is possible that cells may use different membrane sources to form the autophagosome in different contexts, thereby permitting specialization of membrane dynamics in a manner that allows divergent autophagy-inducing signals to stimulate the capture of spatially distinct cargo.
Selective autophagy tackles microbes
Autophagy was originally considered to be a non-selective bulk degradation process, but it is now clear that autophagosomes can degrade substrates in a selective manner28. In addition to endogenous substrates, autophagy degrades intracellular pathogens in a selective form of autophagy, termed xenophagy. Similar to bulk autophagy (such as that induced by nutrient deprivation) and other forms of selective autophagy (such as degradation of damaged mitochondria, peroxisomes, aggregate-prone proteins or damaged ER), the precise membrane dynamics and specificity determinants of xenophagy are not fully understood. Nonetheless, considerable advances have been made, and interesting similarities and differences are beginning to emerge between cellular recognition and degradation of self versus foreign microbial components through autophagy-like pathways (Figs 1 and 2).
Figure 2. Possible autophagy-protein-dependent pathways of pathogen degradation.
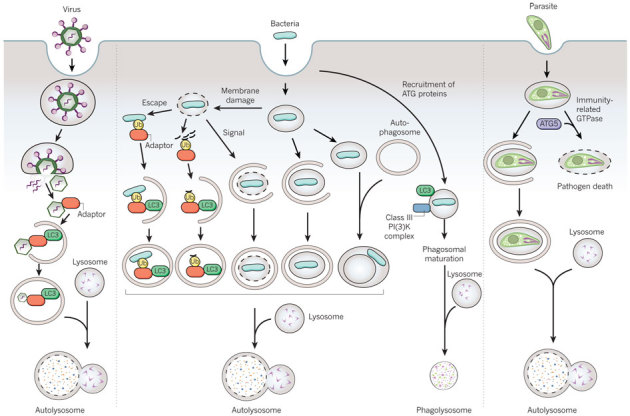
Possible pathways involving the autophagy machinery by which viruses, bacteria (and damaged membranes of bacteria-containing vacuoles) and parasites may be targeted to the lysosome. Adaptor refers to the proteins shown in the cargo-recognition box in Fig. 1; however, as yet undiscovered adaptors may be involved in pathogen recognition, and pathogen targeting may involve ubiquitin-dependent or -independent mechanisms.
The vacuoles used for the engulfment of intracytoplasmic bacteria are similar to autophagosomes, and their formation requires the core autophagy machinery. But one apparent difference is the vacuole size; for example, the diameter of group A Streptococcus-containing autophagosome-like vacuoles (GcAV) can be as big as 10 μm. These large GcAVs are generated by the RAB7-dependent fusion of small isolation membranes29. By contrast, the formation of starvation-induced autophagosomes requires RAB7 later in the autophagy process, at the autophagosome–lysosome fusion step.
A more complex question is how autophagosomes (or components of the autophagy pathway) capture pathogens that are inside vacuolar compartments (Fig. 2). There are at least four general pathways that may be used for autophagy-protein-dependent targeting of bacteria to the lysosome. These include autophagy-protein-facilitated fusion of bacteria-containing phagosomes with lysosomes, the envelopment of bacteria-containing phagosomes or endosomes by autophagosomal membranes, the fusion of bacteria-containing phagosomes or endosomes with autophagosomes, or the xenophagic capture of bacteria that have escaped inside the cytoplasm. In some cases, the route of autophagy-dependent targeting to the lysosome has been well defined, such as for group A Streptococcus that escapes from endosomes30. For several bacteria, however, the precise route is unclear. Many studies define bacterial autophagy as the co-localization of bacteria and LC3, but we now know that LC3 can decorate membranous compartments other than autophagosomes (including phagosomes).
Several lines of evidence suggest that autophagy proteins function more broadly, not only in classical macroautophagy, but also in the process of phagolysosomal maturation during antigen presentation and microbial invasion. Autophagy proteins are required for the fusion of phagosomes that contain Toll-like receptor (TLR)-ligand-enveloped particles with lysosomes in macrophages31, and for the fusion of phagosomes that contain TLR-agonist-associated apoptotic cell antigens with lysosomes in dendritic cells during MHC class II antigen presentation32. The self ligand and cell-surface receptor SLAM functions as a microbial sensor that recruits the beclin 1–class III PI(3)K complex to phagosomes containing Gram-negative bacteria, facilitating phagolysosomal fusion and activation of the antibacterial NADPH oxidase (NOX2) complex33. In addition, the engagement of TLR or Fcγ receptors during phagocytosis recruits LC3 (and ATG12) to the phagosome through NOX2-dependent generation of reactive oxygen species (ROS)34. Thus, in bacterial infections, a paradigm is emerging in which the coordinated regulation of microbial sensing, phagolysosomal maturation and antibacterial activity involves the recruitment of autophagy proteins to the phagosome. As a corollary, an interesting speculation is that impaired recruitment of autophagy proteins to the phagosome may contribute to the pathogenesis of chronic granulomatous disease, a genetic disorder caused by mutations in the NOX2 gene (also known as CYBB) and characterized by recurrent bacterial and fungal infections and inflammatory complications, such as inflammatory bowel disease.
Another autophagosome-independent function of autophagy proteins in pathogen destruction has been described in interferon-γ (IFN-γ)-treated macrophages infected with the parasite Toxoplasma gondii. The parasite-derived membrane, termed the parasitophorous vacuole, undergoes destruction through a mechanism that involves ATG5-dependent recruitment of the immunity-related GTPase proteins to the parasitophorous vacuole35,36, leading to the death of the parasite in the infected cell35,37. Together, these studies suggest that autophagy proteins have diverse roles in membrane dynamics to benefit the host in the removal of invading pathogens (Fig. 2), through xenophagy, phagolysosomal maturation, the recruitment of molecules that damage pathogen-derived membranes, and presumably, many other as yet undiscovered mechanisms.
The mechanisms that cells use to target intracellular bacteria (and probably viruses) to autophagosomal compartments are notably similar to those used for selective autophagy of endogenous cargo. Cellular cargo is commonly targeted to autophagosomes by interactions between a molecular tag (such as polyubiquitin), adaptor proteins such as p62 (also known as SQSTM1 or sequestome 1) or NBR1 (which recognize these tags and contain an LC3-interacting region (LIR) characterized by a WXXL or WXXI motif), and LC3 (ref. 28). These adaptor molecules enable autophagy to target designated cargo selectively to nascent LC3-positive isolation membranes. As reviewed elsewhere38, a similar mechanism involving ubiquitin and p62 seems to be involved in the targeting of intracellular bacteria, such as Salmonella enterica serotype Typhimurium (S. Typhimurium), Shigella flexneri and Listeria monocytogenes, to autophagosomes.
After escape into the cytoplasm or in vacuolar membrane compartments damaged by type III secretion system (T3SS) effectors, bacteria or bacteria-containing compartments, respectively, may become coated with ubiquitin and associate with p62 and nascent LC3-positive isolation membranes. The autophagosomal targeting of Salmonella also requires another cellular factor, NDP52 (nuclear dot protein 52), an autophagy adaptor protein that, like p62, contains an LIR and ubiquitin-binding domains and restricts intracellular bacterial replication. A ubiquitin-independent pathway (that does not involve p62 or NDP52) could also function in targeting damaged Salmonella-containing vacuoles (SCVs) to the autophagosome. In this pathway, a lipid second messenger, diacylglycerol, acts as a signal for the co-localization of SCVs with LC3-positive autophagosomes by a mechanism that involves protein kinase C and its downstream targets, JNK and NADPH oxidase39. The autophagic targeting of a cytoplasmic positive-strand RNA virus, Sindbis virus, also occurs in a ubiquitin-independent manner, but involves the interaction of p62 with the viral capsid protein40. Thus, diverse molecular strategies, including ubiquitin-dependent and -independent mechanisms, may be used to target microbes inside the cytoplasm or vacuolar compartments to the autophagosome.
Beyond targeting intracellular pathogens for degradation, p62 may have further beneficial effects in infected host cells. For example, Shigella vacuolar membrane remnants generated by bacterial T3SS-dependent membrane damage are targeted by polyubiquitination, p62 and LC3 for autophagosomal degradation41 (Fig. 2). These membrane remnants also accumulate numerous molecules involved in sensing and transduction of pathogen-associated molecular pattern (PAMP) and danger-associated molecular pattern (DAMP) signals, and there is an increase in nuclear factor-κB (NF-κB)-dependent cytokine production, ROS production and necrotic cell death in autophagy-deficient cells. Thus, the ubiquitin–p62-dependent autophagic targeting of pathogen-damaged membranes could help to control detrimental downstream inflammatory signalling during bacterial invasion into host cells. Another emerging concept is that selective autophagy of viral proteins, similar to selective autophagy of aggregate-prone toxic cellular proteins, may protect post-mitotic cells such as neurons against cell death. For example, in Sindbis-virus-infected mice with neuron-specific inactivation of Atg5, there is an accumulation of Sindbis virus antigens (without increased levels of infectious virus), increased neuronal cell death and increased animal mortality40. Moreover, p62 is required for starvation and IFN-γ-induced targeting of Fau (and perhaps other ubiquitylated protein complexes) to mycobacteria-containing phagosomes, resulting in the generation of antimycobacterial Fau-derived peptides42. The role of p62 in innate immunity is probably evolutionarily ancient, as the Drosophila p62 orthologue REF(2)P was originally identified in a screen for modifiers of sigma virus replication43.
We speculate that p62, as well as the other known LC3-interacting adaptor proteins (NBR1 and NDP52), may represent the tip of the iceberg in terms of cellular adaptor proteins that bind to ubiquitin (or other molecular tags) and target microbial substrates and cytosolic material to autophagosomes to coordinate innate immune responses. A recent proteomics study showed that the mammalian ATG8 family, which includes LC3, GATE16 and GABARAP, has 67 high-confidence interactions with other cellular proteins21. Some of these new ATG8-family-member-interacting partners may have an as yet undiscovered role in innate immunity. Another open question is whether the known proteins involved in selective autophagy of mitochondria (called mitophagy), such as Nix (also known as BNIP3L) and parkin44, also function in microbial autophagy.
Autophagy and resistance to infection
The autophagy pathway and/or autophagy proteins have a crucial role in resistance to bacterial, viral and protozoan infection in metazoan organisms. The genetic deletion or knockdown of autophagy genes protects plants from viral, fungal and bacterial infection by preventing the uncontrolled spread of programmed cell death during the plant innate immune or hypersensitive response45. In other organisms, autophagy proteins function in a cell-autonomous manner to control infection by intracellular pathogens. In Drosophila, autophagy gene mutation increases susceptibility to viral (vesicular stomatitis virus)46 and bacterial (L. monocytogenes)47 infection. In Dictyostelium and Caenorhabditis elegans, autophagy gene mutation increases susceptibility to lethal S. Typhimurium infection48. In mice, knockout of Atg5 in macrophages and neutrophils increases susceptibility to infection with L. monocytogenes and the protozoan T. gondii35, and neuron-specific Atg5 knockout increases susceptibility to central nervous system Sindbis virus infection40. As noted in the next section, the autophagy pathway and proteins may also have 'proviral' or 'probacterial' effects in in vitro studies; however, in vivo evidence for such effects is so far lacking. The mechanisms by which autophagy genes mediate in vivo resistance to infection are not fully understood, but are likely to involve a combination of xenophagy, other autophagy-protein-dependent effects on microbial replication or survival, activation of innate and adaptive immune responses, and/or alterations in pathogen-induced cell death (Fig. 3).
Figure 3. Functions of the autophagy pathway and/or proteins in immunity.

A summary of the known functions of the autophagy pathway and/or proteins in adaptive and innate immunity, and as effectors during infection.
An important question is whether this function of autophagy in broad resistance to infection with intracellular pathogens extends to humans. Recent human genetic studies provide some clues. The immunity-related GTPase human IRGM, which regulates autophagy-dependent clearance of mycobacteria in vitro49, was identified as a genetic risk locus for tuberculosis in a West African population50. Numerous studies have shown a crucial role for autophagy in defence against mycobacterial infection in human cells1, and a genome-wide analysis of host genes that regulate Mycobacterium tuberculosis replication demonstrated that a predominance of factors were autophagy regulators51. Thus, it is possible that autophagy has a central role in resistance to one of the most important global infectious diseases — tuberculosis. Mutations in NOD2, which encodes an intracellular pathogen-recognition receptor (nucleotide-binding oligomerization-domain-containing protein 2) that functions in bacterial autophagy52,53, are also associated with susceptibility to infection with another mycobacterial agent, Mycobacterium leprae, the aetiological agent of leprosy54. An exciting future venture will be to confirm whether IRGM, NOD2 and other autophagy-related genes are involved in resistance to infection with mycobacteria and other infections in further human populations and, if so, whether this resistance is mediated by autophagy.
Microbes fight back
Microbes undergo strong selective pressure to develop strategies to block host defence mechanisms; the number of such strategies is a surrogate measure of the importance of the host defence mechanism in immunity. As reviewed elsewhere1,55, viruses and intracellular bacteria have evolved several ways to adapt to host autophagy. They can antagonize autophagy initiation or autophagosomal maturation, evade autophagic recognition, or use components of the autophagy pathway to facilitate their own replication or intracellular survival. An emerging theme is that microbial antagonism of autophagy not only blocks the xenophagic degradation of intracellular pathogens, but also blocks the functions of autophagy in innate and adaptive immunity. A relatively unexplored yet crucially important frontier is how microbial antagonism may contribute more broadly to the role of microbes in diseases characterized by defective autophagy, such as cancer, neurodegenerative diseases, ageing and, potentially, autoimmune and inflammatory diseases.
Viral strategies to shut off autophagy include the blockade of positive upstream regulators of autophagy (such as the IFN-inducible RNA-activated eIF2α protein kinase (PKR) signalling pathway), the activation of negative upstream regulators of autophagy (such as the nutrient-sensing TOR kinase signalling pathway) or direct antagonism of the autophagy machinery55. The overlapping functions of the eIF2α kinase signalling pathway in stress-induced general translational arrest, transcriptional activation of stress-response genes and stress-induced autophagy enable viruses to disarm several facets of the cellular stress response to infection by one mechanism — that is, antagonism of eIF2α kinase signalling. The mTOR signalling pathway has a central role in the control of cell growth and metabolism, and interestingly, many of the viruses that activate mTOR are oncogenic (for example, Epstein–Barr virus, Kaposi's sarcoma-associated herpesvirus, hepatitis B virus and retroviruses). This suggests another type of pluripotent viral weapon — one that can promote oncogenesis by simultaneously inactivating autophagy and promoting cell growth through TOR activation. HIV envelope protein-dependent activation of mTOR signalling is also proposed to be a mechanism for HIV evasion of innate and adaptive immune responses in dendritic cells, including the degradation of incoming virions by lysosomes, blockade of HIV transfer to CD4+ T cells, stimulation of TLR4 and TLR8 ligand responses, and presentation of HIV Gag antigen to CD4+ T cells56. It will be important to determine whether these effects of HIV-mediated mTOR activation and autophagy inhibition contribute to impaired dendritic cell function during HIV infection in vivo.
Several viral proteins target the core autophagy protein beclin 1. Autophagosome initiation is blocked by interactions between beclin 1 and the herpes simplex virus 1 (HSV-1) neurovirulence factor ICP34.5 or the oncogenic γ-herpesvirus-encoded viral BCL2-like proteins, whereas autophagosome maturation is blocked by interactions between beclin 1 and the HIV accessory protein Nef or the influenza virus matrix protein 2 (ref. 55). The interactions between beclin 1, HSV-1 ICP34.5 and viral BCL2 are probably physiologically important in vivo; a mutant HSV-1 virus lacking the beclin-1-binding domain of ICP34.5 is attenuated in mouse models of encephalitis (presumably through its failure to control xenophagy and innate immunity)57 and of corneal disease (through its failure to control adaptive immunity)58. Moreover, a mouse γ-herpesvirus that encodes a mutant viral BCL2 unable to bind to beclin 1 demonstrates impaired ability to maintain chronic infection59. Thus, viral antagonism of host autophagy can manipulate distinct aspects of viral pathogenesis and immunity in vivo.
It is not yet clear whether compared with other autophagy proteins, beclin 1 is preferentially targeted by viral virulence proteins because of its central role in autophagosome formation, or more likely, whether we are just beginning to identify pairs of viral proteins and their autophagy pathway targets. In support of the latter, viral FLICE-like inhibitors encoded by Kaposi's sarcoma-associated herpesvirus and molluscum contagiosum virus suppress autophagy by interacting with the ATG3 E2-like enzyme, thereby preventing it from binding and processing LC3 (ref. 60).
Bacteria possess diverse strategies to avoid degradation by autophagolysosomal pathways. As reviewed elsewhere1,38, many bacteria that reside in phagosomes or other vacuolar compartments have methods to inhibit lysosomal fusion or maturation, which, in the case of mycobacteria, can be partially overcome by treatments that stimulate autophagy. Another possible mechanism for bacterial evasion of autophagy has emerged from a genome-wide screen to identify host factors that regulate the intracellular survival of M. tuberculosis51. According to bioinformatics analyses, M. tuberculosis infection of human macrophage-like cells activates cellular pathways that inhibit autophagy. Intracellular bacteria that escape into the cytoplasm, such as S. flexneri and L. monocytogenes, use strategies to camouflage themselves to avoid autophagic recognition. The Shigella T3SS effector IcsB competitively binds to VirG, thereby preventing the interaction between ATG5 and VirG, a bacterial surface protein required for actin-based motility and Shigella targeting to autophagosomes38. The Listeria protein ActA interacts with cytosolic actin polymerization machinery (ARP2/3, VASP and actin), which blocks bacterial association with ubiquitin, p62 recruitment and autophagic targeting61. The precise mechanism of this block is unknown, but it has been proposed that the ActA-dependent recruitment of host cell cytoskeletal proteins may enable the bacterium to disguise itself as a host cell organelle61. This concept sheds light on the autophagy pathway in a fundamental aspect of immunology — the basis for discrimination between self and non-self.
Microbes have evolved not only to antagonize autophagy (as a cellular defence mechanism that threatens their survival), but also to exploit its components and functions in membrane trafficking for their own self-serving purposes1,55. An early concept in the field is that autophagosomes may serve as a protected niche for intracellular bacteria (provided fusion with acidic compartments is blocked) and/or serve as a source of nutrients for intracellular pathogens (which would require intact autophagolysosomal fusion)1. Trafficking of the intracellular bacteria Yersinia pseudotuberculosis to acidic compartments was recently shown to be enhanced by genetic inhibition of autophagy62. This seemingly contradicts other evidence that the autophagy proteins promote phagosomal maturation, but is consistent with the concept that autophagosomes function as a protected intracellular niche for bacteria. The role of the autophagy machinery in promoting and/or inhibiting vacuolar acidification — and the counter effects of microbes that reside in vacuolar compartments — needs to be explored further.
The function of autophagy proteins in membrane formation and/or trafficking is exploited by numerous viruses, including poliovirus, rotavirus, HIV, coronaviruses, Dengue virus, and the hepatitis B and C viruses55. Autophagosomes (defined as LC3-positive membranes, see caveat below) may act as a scaffold for intracellular membrane-associated replication of certain cytoplasmic RNA viruses55. Autophagy may assist in HIV biogenesis, because the processing of the HIV envelope precursor protein Gag and extracellular viral release are enhanced by the autophagy machinery63. Similarly, autophagy proteins are required for maximal levels of poliovirus egress55. Another newly defined proviral function of autophagy is its role in productive hepatitis C virus replication; several different autophagy proteins (such as beclin 1, ATG4B, ATG5, ATG7 and ATG12) assist in the translation of incoming, but not progeny, viral RNA64. ATG7 and class III PI(3)K activity also enhance hepatitis B virus DNA replication65.
The mechanisms by which autophagy proteins facilitate the replication and/or egress of certain viruses are not yet understood. Some observations may relate to the role of autophagy proteins in remodelling the ER (vis-à-vis viral replication) or the role of autophagosomes in fusing with multivesicular bodies (vis-à-vis viral egress). It is possible that autophagy proteins function to provide membrane for viral replication complexes or translation machinery. This may be true for viruses such as hepatitis C virus, for which genetic knockdown of several different autophagy genes decreases productive replication64. However, for other viruses such as coronaviruses, the biogenesis of double-membrane, ER-derived vesicles used for replication proceeds through a pathway that involves the non-lipidated form of LC3 (LC3-I) but not the general autophagy machinery66. Thus, caution must be exercised in interpreting the significance of the co-localization (or biochemical interaction) of viral proteins and LC3, as LC3 may have autophagy-independent roles in membrane dynamics.
Autophagy regulation by immune signalling molecules
The central importance of autophagy in immunity is further underscored by the multitude of immune-related signalling molecules that regulate autophagy. As reviewed in detail elsewhere2,3,4,38, autophagy is induced by different families of pathogen-recognition receptors (such as TLRs, NOD-like receptors and the double-stranded RNA-binding protein PKR), DAMPs (such as ATP, ROS and misfolded proteins), pathogen receptors (such as CD46), IFN-γ and downstream immunity-related GTPases, and DAP kinase, JNK, CD40, tumour necrosis factor-α (TNF-α), inhibitor of NF-κB (IKK) and NF-κB (Fig. 1). High mobility group box (HMGB) proteins have also been shown to function as both universal sensors of nucleic acids in innate immune signalling67 and inducers of autophagy68. Autophagy is inhibited by BCL2, NF-κB, T helper 2 (TH2) cytokines and the canonical nutrient-sensing insulin–AKT–TOR pathway. Inactivation of this nutrient-sensing pathway may contribute to vesicular stomatitis virus stimulation of autophagy in Drosophila46, and autophagy activation in C. elegans with loss-of-function mutations in this pathway may mediate pathogen resistance in long-lived mutant nematodes48. Thus, both 'immune-specific' and more general nutrient-response signals control autophagy in response to infection.
Studies with vitamin D3 have uncovered a possible link between nutrition, innate immunity and the control of autophagy during mycobacterial infection. Low vitamin D3 levels are associated with increased susceptibility to tuberculosis. Vitamin D3 generates an antimycobacterial peptide, cathelicidin, and induces autophagy and mycobacterial killing in human monocytes through cathelicidin-dependent effects69. Although cathelicidin is required for vitamin-D3-dependent transcriptional upregulation of autophagy genes such as BECN1 and ATG5, and vitamin D3 enhances the recruitment of cathelicidin to autophagosomes, it is not yet clear how cathelicidin promotes autophagy. Nonetheless, these observations may begin to provide some insight into the century-old Nobel prize award (Niels Ryberg Finsen, 1903) for the use of ultraviolet-light therapy (which generates active vitamin D3) in the treatment of diseases such as tuberculosis.
In most instances, the mechanisms of autophagy control by immune-related signalling molecules are not understood. However, there are some examples of specific interactions between immune signals and autophagy proteins that may be relevant to these mechanisms. For example, the interaction between beclin 1 and BCL2 (which inhibits its activity) is thought to be disrupted by the TLR adaptors MyD88 and TRIF, as well as by HMGB1, which bind to beclin 1 and displace BCL2 (refs 3, 68). Two intracellular sensors responsible for inducing autophagy in response to bacterial infection, NOD1 and NOD2, interact with ATG16L1 and recruit it to the plasma membrane, resulting in enhanced association of invasive bacteria (S. flexneri) with LC3 (ref. 53). Interestingly, a NOD2 mutation associated with Crohn's disease impairs ATG16L1 plasma membrane recruitment and bacterial co-localization with LC3 (ref. 53).
The identification of other possible protein–protein interactions between core autophagy proteins and immune signals by a large proteomics screen21 has the potential to foster further advances in understanding how different immune signals regulate the autophagy machinery. For example, tectonin proteins with multivalent β-propeller folds are known to function in pathogen recognition and innate immunity in invertebrates70. Thus, the interactions between previously uncharacterized human proteins of this tectonin family, TECPR1 and TECPR2, with the ATG5–ATG12–ATG16L1 complex and ATG8 family members, respectively21, may contribute to pathogen-induced autophagy stimulation or selective autophagic targeting of pathogens in mammals.
Further links between immune signalling molecules and autophagy regulation were suggested by a genome-wide short interfering RNA screen71. The analysis identified 219 genes that suppressed basal autophagy, largely in a mammalian TOR complex 1 (mTORC1)-independent fashion. These included several cytokines such as CLCF1, LIF, IGF1, FGF2 and the chemokine SDF1 (also known as CXCL12), as well as cellular signalling molecules regulated by cytokines such as STAT3. These findings raise the possibility that cytokines may have a broader role in the control of autophagy than previously thought. Moreover, because these cytokine signalling pathways are important in immune cells, another central question is to what extent cytokine-mediated regulation of autophagy governs immune cell function. Given the general function of autophagy in cellular homeostasis5, and the more specific functions in regulating immune and inflammatory signalling (discussed in 'Regulation of immune signalling by autophagy proteins'), cytokine-mediated changes in autophagy levels in immune cells may have a central role in immunity and inflammation.
Autophagy and adaptive immunity
Autophagy proteins function in adaptive immunity, including in the development and homeostasis of the immune system and in antigen presentation (Table 1 and Fig. 3). The knockout of different autophagy genes in specific lymphocyte populations in mice has shown a crucial role for autophagy proteins in the maintenance of normal numbers of B1a B cells, CD4+ T cells, CD8+ T cells and fetal haematopoietic stem cells2,44,72. In T cells, in which mitochondrial numbers are developmentally regulated during the transition from thymocyte to mature circulating T cell, the developmental defect in autophagy-deficient cells may be related to the defective clearance of mitochondria44. Another crucial function of autophagy in the development and homeostasis of the immune system is the elimination of autoreactive T cells in the thymus44. High levels of autophagy are present in thymic epithelial cells, in which autophagy participates in the delivery of self-antigens to MHC class II loading compartments. Genetic disruption of Atg5 in thymic epithelial cells leads to the altered selection of certain MHC class II restricted T-cell specificities and autoimmunity73. Beyond these functions in lymphocyte survival and thymic negative selection, autophagy may exert other functions in lymphocyte differentiation, perhaps, in part, indirectly through effects on cytokine expression (see the next section). It is not yet known whether autophagy is involved in the development and/or homeostasis of immune cell populations other than lymphocytes and haematopoietic stem cells.
Autophagy proteins may participate in different facets of antigen presentation, including the delivery of endogenous antigens for MHC class II presentation to CD4+ T cells74,75, the enhancement of antigen donor cell cross-presentation to CD8+ T cells75, dendritic cell cross-presentation of phagocytosed antigens to CD4+ T cells32 and, in one report, MHC class I presentation of intracellular antigens to CD8+ T cells27. The discovery that autophagosomes could deliver endogenous antigens to MHC class II loading compartments sheds light on one of the central mysteries of antigen presentation — how the immune system elicits CD4+ T-cell responses to antigens that originate in all parts of the cell. The autophagic delivery of endogenously synthesized antigens for MHC class II presentation has been demonstrated in vitro for certain viral antigens75, and probably explains the essential role of Atg5 in vivo in negative thymic selection73. However, the relative importance of this pathway in antigen presentation during infection in vivo is not yet known. There is nonetheless interest in exploiting this pathway for optimizing vaccine-elicited CD4+ T-cell responses, by either pre-treating dendritic cells with autophagy-inducing agents in cell-based vaccine strategies or fusing antigens with LC3 to enhance their targeting to autophagosomes1.
Of note, autophagy proteins are required for antigen cross-presentation during infection in vivo32. Dendritic-cell-specific deletion of Atg5 in mice results in defects in priming CD4+ T-cell responses after HSV and Listeria infections, and mice succumb more rapidly to lethal disease after intravaginal HSV infection. Atg5-deficient dendritic cells have normal migration, innate responses, endocytic and phagocytic activity and cross-presentation of peptides on MHC class I molecules. However, they exhibit defects in phagosome-to-lysosome fusion and in cross-presentation by MHC class II molecules of phagocytosed antigens containing TLR ligand. Thus, the interior of the phagosome, like that of the autophagosome, is a cellular compartment that autophagy-protein-dependent antigen presentation accesses to generate peptides for presentation to CD4+ T cells. A potential evolutionary advantage of this autophagy-protein-dependent cross-presentation is that, by delegating antigen presentation duties to uninfected dendritic cells, the host can bypass the blockade of antigen presentation that may result from microbial antagonism of autophagy in infected cells.
Regulation of immune signalling by autophagy proteins
In response to infection, the host must activate those arms of the innate and adaptive immune system (including autophagy-dependent functions; Fig. 3) that help to control infection while, in parallel, triggering specific responses that limit detrimental, uncontrolled immune activation and inflammation. An exciting new frontier in autophagy research is the growing recognition of the function of autophagy proteins in achieving this balance (Fig. 4).
Figure 4. Autophagy/autophagy proteins act to achieve a balance between activation and inactivation of innate immune signalling.
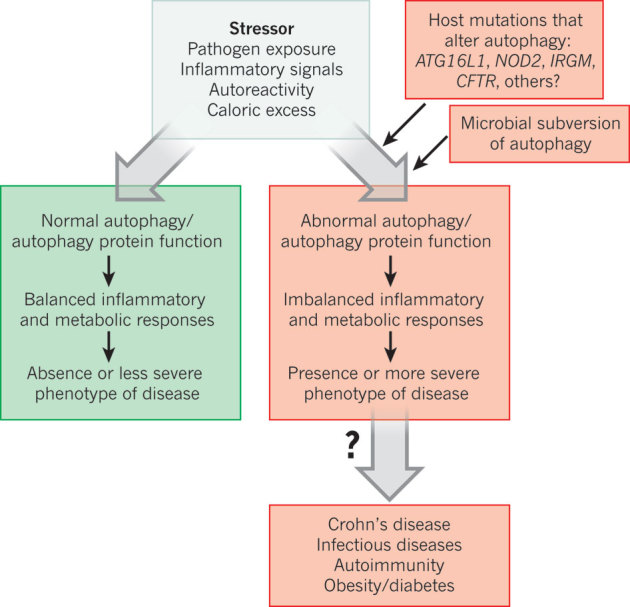
A general model in which the levels of autophagy and autophagy proteins control disease in response to stressors. Normal autophagy protein function (green) contributes to balanced inflammatory and metabolic responses, resulting in protection against disease. Altered autophagy protein function (red) results in maladaptive inflammatory and metabolic responses, increased inflammation and more severe disease.
Autophagy proteins function in both the activation and inactivation of innate immune signalling4. The autophagy pathway activates type I IFN production in plasmacytoid dendritic cells by delivering viral nucleic acids to endosomal TLRs76. By contrast, autophagy proteins negatively regulate RIG-I-like receptor (RLR)-mediated induction of type I IFN production through the autophagic elimination of damaged mitochondria (and reduction of ROS)77, and by the binding of ATG5–ATG12 to caspase recruitment domains of RLR signalling molecules78. Moreover, the autophagy protein ATG9A, but not ATG7, negatively regulates the activation of STING, a transmembrane protein that is required for efficient activation of type I IFN and pro-inflammatory cytokine production in response to stimulatory DNA23. Thus, it seems that autophagy proteins can negatively regulate IFN production by both autophagy-dependent and -independent mechanisms. With respect to the latter, different autophagy proteins may be specialized to target different innate immune signalling molecules.
The autophagy pathway and/or proteins also have a crucial role in the control of inflammatory signalling. A major effect is on the regulation of inflammatory transcriptional responses. Increased levels of the adaptor protein p62, which accumulates in autophagy-deficient cells, activate the pro-inflammatory transcription factor NF-κB through a mechanism involving TRAF6 oligomerization79. The accumulation of p62 in Atg7-deficient hepatocytes results in enhanced activity of the stress-responsive transcription factor NRF2 and NRF2-dependent liver injury80. In addition, Paneth cells (intestinal immune epithelial cells) from mice hypomorphic for Atg16l1 (Atg16l1HM) show enhanced transcription of pro-inflammatory cytokines and adipokines81.
A second important effect of autophagy proteins on inflammatory signalling is at the level of the inflammasome. This complex contains NOD-like receptor cryopyrin proteins, the adaptor protein ASC and caspase 1, and is activated by cellular infection or other stress to promote the maturation of pro-inflammatory cytokines such as interleukin-1β (IL-1β) and IL-18 (ref. 4). Atg16l1- or Atg7-deficient mouse macrophages produce increased levels of mature IL-1β and IL-18 after TLR4 stimulation by endotoxin82. In addition, mouse chimaeras engrafted with Atg16l1−/− fetal liver haematopoietic progenitors have increased serum concentrations of IL-1β and IL-18 after treatment with dextran sodium sulphate (DSS), which contributes to increased DSS-induced colitis82.
The mechanism(s) by which autophagy proteins negatively regulate inflammasome activation are not yet understood. Mutually non-exclusive possibilities include direct interactions between autophagy proteins and inflammasome components, indirect inhibition of inflammasome activity through autophagic suppression of ROS accumulation, or autophagic degradation of danger signals that activate the inflammasome. In line with the latter model, the autophagic degradation of amyotrophic-lateral-sclerosis-linked mutant superoxide dismutase has been proposed to limit caspase 1 activation and IL-1β production83.
In addition to regulating inflammatory signalling, the autophagy pathway may prevent tissue inflammation through its role in apoptotic corpse clearance. The efficient clearance of apoptotic corpses during development and tissue homeostasis prevents secondary necrosis, which releases danger signals (DAMPs) that trigger inflammation. Autophagy genes are essential for the heterophagic clearance of dying apoptotic cells during developmental programmed cell death (by the generation of ATP-dependent engulfment signals)84, and the retinas and lungs of embryonic mice lacking Atg5 have a defect in apoptotic corpse engulfment that is associated with infiltration of inflammatory cells84. On the basis of growing evidence that autophagy proteins function in TLR-mediated phagolysosomal pathways, it is possible that autophagy also functions in phagocytes to facilitate apoptotic corpse clearance. Thus, in tissues such as the intestine, in which physiological regeneration involves continuous shedding or apoptosis of epithelial cells, autophagy-dependent functions in dying cells and/or phagocytic cells may promote efficient corpse clearance, thereby limiting inflammation.
Autophagy and inflammatory disease
Perturbations in autophagy-protein-dependent functions in immunity may contribute not only to increased susceptibility to infection, but also to chronic inflammatory diseases and autoimmune diseases. The only well-characterized link thus far is between mutations in autophagy regulators and Crohn's disease, a chronic inflammatory disorder of the small intestine, in which a breakdown in clearance or recognition of commensal bacteria, as well as altered mucosal barrier function and cytokine production, is thought to lead to intestinal inflammation (Fig. 5). Other emerging links include the autoimmune disease systemic lupus erythematosus (SLE), inflammation-associated metabolic diseases such as obesity and diabetes, and inflammation associated with cystic fibrosis lung disease (Fig. 4).
Figure 5. The link between mutations in autophagy regulators and the chronic inflammatory disorder Crohn's disease.
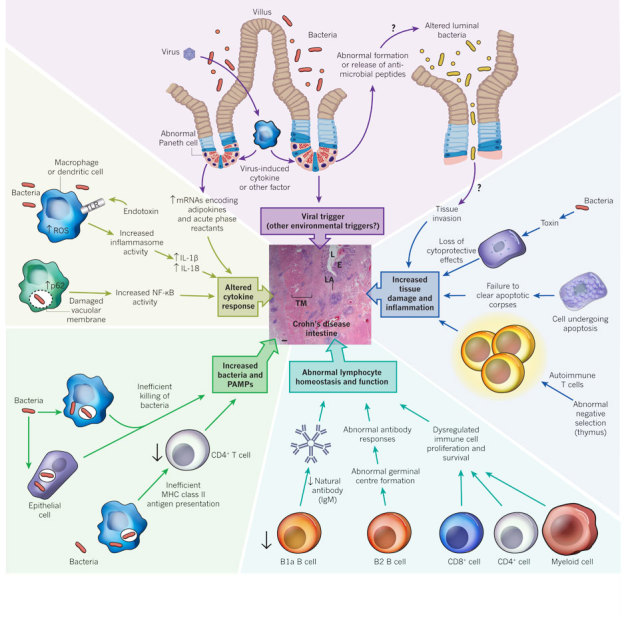
An overview of the many possible mechanisms by which defects in autophagy and autophagy protein function may contribute to the pathogenesis of a type of inflammatory bowel disease, Crohn's disease. A micrograph of a human small intestine from a patient with Crohn's disease is shown (centre), demonstrating the severe transmural inflammation that is characteristic of this disease. The postulated mechanisms by which defects in autophagy protein function might contribute to the development or perpetuation of intestinal inflammation are based on studies in vitro and animal models. There is no direct evidence that autophagy defects contribute to human Crohn's disease, although mutations in three autophagy-related genes, ATG16L1, NOD2 and IRGM, are known to enhance risk of the disease. E, epithelium; IgM, immunoglobulin M; L, lumen; LA, lymphoid aggregates; TM, thickened muscle. Scale bar, 200 μm.
The role of autophagy proteins in Crohn's disease was not suspected until genome-wide association studies identified three Crohn's disease susceptibility genes, IRGM, NOD2 and ATG16L1, that are involved in autophagy85. The IRGM risk allele contains a deletion in the promoter region of the gene that may be associated with changes in IRGM protein expression and may contribute to Crohn's disease, given IRGM's role in autophagy-dependent control of bacterial infection49. However, this hypothesis has not yet been tested. The three major Crohn's-disease-associated NOD2 variants (a frameshift mutant and two missense mutants) may be loss-of-function mutants, with impaired muramyl dipeptide (MDP)-induced inflammatory signalling86. How the loss of function of a pro-inflammatory signal mechanistically contributes to an inflammatory disorder has been unclear, but the recently discovered links between NOD2 and autophagy may solve this conundrum. In primary immature human dendritic cells, NOD2 is required for MDP-induced autophagy, a process that is essential for the MHC class II presentation of bacterial antigens to CD4+ T cells and for bacterial targeting to lysosomes52. Dendritic cells expressing Crohn's disease NOD2 risk variants are defective in both of these functions52. Thus, in patients with Crohn's disease and NOD2 risk variants, aberrant autophagy-dependent bacterial clearance and immune priming could act as a trigger for intestinal inflammation.
A mechanistic link may also exist between ATG16L1 mutation and Crohn's disease pathogenesis. Similar to findings with NOD2 variants, dendritic cells from patients with the Crohn's-disease-associated ATG16L1(T300A) risk variant are defective in presenting bacterial antigen to CD4+ T cells52. However, it is not yet known how the T300A mutation affects the function of the mammalian ATG16L1 protein. This mutation resides in the carboxy-terminal WD-repeat domain that is absent in yeast Atg16 and is dispensable for autophagy. Although some studies have suggested that the ATG16L1(T300A) variant has reduced autophagic clearance of enteric pathogens such as adherent-invasive Escherichia coli87 or S. Typhimurium88, it remains controversial whether the risk versus protective alleles of ATG16L1 have differences in stability or antibacterial autophagic activity89.
Despite the uncertain nature of the effects of the T300A mutation on ATG16L1 function, Atg16l1 mutation (null or hypomorphic alleles) in mice results in abnormalities that are relevant to Crohn's disease pathogenesis. As noted earlier, loss of Atg16l1 function in mice results in enhanced TLR-agonist-induced pro-inflammatory cytokine production by macrophages82, enhanced DSS-induced colitis82,90 and altered inflammatory gene transcriptional profiles in Paneth cells81,90. In addition, the Paneth cells of mice expressing low Atg16l1 levels (Atg16l1HM) show defects in the packaging and extrusion of antimicrobial granules into the gut lumen; Paneth cells from patients with Crohn's disease and the ATG16L1(T300A) risk variant show similar defects81. This suggests that, in addition to the overlapping functions of NOD2 and ATG16L1 in a common bacterial-sensing pathway that promotes bacterial antigen presentation, ATG16L1 may have unique protective functions, including Paneth cell antimicrobial peptide release and the negative regulation of pro-inflammatory cytokine production. To connect the striking phenotypes in Atg16l1-mutant mice and the pathogenesis of Crohn's disease in humans with the ATG16L1(T300A) risk allele, the precise effects of the T300A mutation on ATG16L1 protein function need to be uncovered.
A new dimension in understanding the multifactorial basis of chronic inflammatory diseases such as Crohn's disease has emerged from the discovery that a virus trigger is required to observe intestinal abnormalities in Atg16l1HM mice90. In mice raised in a pathogen-free facility, only Atg16l1HM mice (and not wild-type mice) infected with a virus found in routine conventional animal facilities, a murine norovirus, showed abnormal Paneth cell granule secretion, Paneth cell pro-inflammatory gene-expression profiles, and intestinal inflammation in response to DSS treatment90. This mucosal inflammation depended on the presence of the microbiome and pro-inflammatory cytokines, as it was reversed by antibiotic treatment or by TNF-α or IFN-γ inhibition. Thus, variations in a host autophagy gene, exposure to a specific virus and the microbiome can act together to trigger intestinal inflammation in mice that is similar to that in patients with Crohn's disease. Although environmental factors, including the gut microbiome, have long been suspected to contribute to Crohn's disease in genetically susceptible individuals, formal proof of this concept was lacking, and viruses were a previously unsuspected trigger. Another implication of this work is the concept that autophagy proteins, through their diverse roles in immunity and the control of inflammation, may serve as a central rheostat that prevents inflammatory diseases triggered by environmental stress (Fig. 4).
An important unanswered question is whether perturbations in autophagy may also result in inflammatory autoimmune disease. Genome-wide association studies have linked several single nucleotide polymorphisms (SNPs) in ATG5 to SLE susceptibility91,92,93. SLE is a multifactorial, heterogeneous disease characterized by autoimmune responses against self-antigens generated from dying cells. Although the effects of these SNPs on ATG5 expression and function are not known, the lack of Atg5-dependent negative thymic selection generates autoimmunity and multi-organ inflammation in mice73. Loss of other ATG5-dependent effects, including regulation of IFN and pro-inflammatory cytokine secretion77,78, clearance of dying cells84 and dendritic cell antigen presentation32, might also contribute to the autoimmunity and inflammation associated with SLE. Thus, a link between ATG5 mutation (or mutation of other autophagy genes) and SLE pathogenesis is biologically plausible, although not yet proven.
Defects in autophagy may contribute to inflammation-associated metabolic diseases such as diabetes and obesity, which are both linked to insulin resistance. The metabolic inflammasome — a complex composed of signalling molecules such as PKR, eIF2α, JNK, IRS and IKK— may act as a link between ER stress and more global stress responses, including inflammation and metabolic dysfunction (as observed in insulin resistance and obesity)94. Although most components of the metabolic inflammasome promote autophagy, the induction of autophagy by this signalling complex would be expected to serve as a negative-feedback mechanism that limits ER stress and disease progression. Consistent with this postulated protective effect of autophagy, hepatic suppression of the autophagy gene Atg7 in mice results in increased ER stress and insulin resistance95, and mice deficient in the autophagy adaptor protein p62 develop mature-onset obesity and insulin resistance96. Furthermore, obesity is associated with the accumulation and activation of macrophages and subsets of T cells in adipose tissue and the production of cytokines such as TNF-α and IL-6 (ref. 97). Thus, the failure of autophagy-dependent control of ER stress, immune cell homeostasis, immune cell activation and/or pro-inflammatory cytokine secretion may contribute to inflammation-associated responses that underlie the pathogenesis of metabolic diseases.
Another potential link between autophagy deficiency and chronic inflammation is in cystic fibrosis98, a life-threatening genetic disorder caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR). Mutations in CFTR lead to autophagy inhibition in lung epithelial cells through a mechanism that may involve ROS-mediated sequestration of the beclin 1–class III PI(3)K complex in perinuclear aggregates (redirecting it from its site of autophagy action at the ER). Restoration of beclin 1 and autophagy in cystic fibrosis epithelial cells rescues the disease phenotype, and antioxidants reverse the airway inflammation in a cystic fibrosis mouse model by a mechanism postulated to involve autophagy.
Future directions
The first series of studies demonstrating that the autophagy machinery is used to attack invading intracellular bacteria was published in 2004 (refs 30, 99, 100). Although autophagy had been observed at the ultrastructural level in cells infected with intracellular bacteria and viruses decades earlier, these studies were a seminal advance. For the first time, pharmacological and genetic manipulation of autophagy, which built on the discoveries of the yeast screens that identified the autophagy machinery, challenged the very notion of autophagy as an 'auto'- (self), 'phagy' (eating) pathway. Indeed, we learned that the same genes that are used to orchestrate the degradation of self-constituents, either for nutritional/energy homeostasis or cellular damage control, are also used to orchestrate the degradation of foreign invaders, termed xenophagy.
In the past few years, research in the field has uncovered new layers of complexity and functional diversity in terms of how this set of genes — originally characterized in the context of macroautophagy — may function to protect multicellular organisms against not only the threats of infection but also the threats of the host's own response to infection. The autophagy machinery does much more than form autophagosomes to engulf microbes — it somehow allows microbes in phagosomes and vacuoles to be targeted to the lysosome; it enables crucial cells in the immune system to develop properly and perform some of their 'normal' functions (such as produce IFN, secrete antimicrobial peptides or present antigens to stimulate adaptive immunity); and it ensures that these responses do not become out of control by functioning in central immunological tolerance and the negative regulation of innate and inflammatory signalling. Thus, recent advances may not only modify our understanding of immunity (in terms of understanding new roles of the autophagy machinery in immune regulation) but also reshape our understanding of the pathogenesis of inflammatory diseases (in terms of understanding how perturbations in autophagy protein function may contribute to such diseases).
Clearly, our understanding of the molecular mechanisms of the plethora of functions of autophagy proteins in immune-related processes is still quite primitive. We speculate that, similar to the way in which the initial genetic screens in yeast transformed autophagy research, current proteomic and genomic screens have the potential to transform research on autophagy and immunity. Such a transformation would include facilitating a much deeper understanding of the molecular mechanisms of the existing known immunological functions of autophagy through the use of the tools of modern systems biology to understand autophagy protein–protein interaction and signalling regulatory networks on a broad scale. Perhaps more exciting is the possibility that such a transformation will uncover new ways in which this ancient self-defence machinery can function in immunity.
Acknowledgements
The work in the authors' laboratories was supported by National Institutes of Health (NIH) grants RO1 CA109618 and U54 AI057156 (B.L.); by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and by the Takeda Science Foundation (N.M.); and by NIH grants RO1 AI054483, U54 AI057160, RO1 AI084887 and RO1 CA096511 and the Broad Medical Foundation (H.W.V.). We thank T. Stappenbeck for discussions, and A. Diehl and M. Harstein for scientific illustration. We apologize to those authors whose work could not be cited owing to space limitations.
Competing interests
The authors declare no competing financial interests.
Footnotes
Reprints and permissions information is available at http://www.nature.com/reprints.
References
- 1.Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe. 2009;5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Virgin HW, Levine B. Autophagy genes in immunity. Nature Immunol. 2009;10:461–470. doi: 10.1038/ni.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol. Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J. Cell Biol. 2010;189:925–935. doi: 10.1083/jcb.201002021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nature Rev. Mol. Cell Biol. 2009;10:458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi-Nishino M, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nature Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 11.Yla-Antilla P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5:1180–1185. doi: 10.4161/auto.5.8.10274. [DOI] [PubMed] [Google Scholar]
- 12.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–776. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsunaga K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nature Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 15.Axe EL, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polson HE, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6:506–522. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 17.Ropolo A, et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J. Biol. Chem. 2007;282:124–133. doi: 10.1074/jbc.M706956200. [DOI] [PubMed] [Google Scholar]
- 18.Tian Y, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010;141:1042–1055. doi: 10.1016/j.cell.2010.04.034. [DOI] [PubMed] [Google Scholar]
- 19.Fujita N, et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell. 2008;19:2092–2100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weidberg H, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29:1792–1802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Radoshevich L, et al. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell. 2010;142:590–600. doi: 10.1016/j.cell.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saitoh T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl Acad. Sci. USA. 2009;106:20842–20846. doi: 10.1073/pnas.0911267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Webber JL, Tooze SA. New insights into the function of Atg9. FEBS Lett. 2010;584:1319–1326. doi: 10.1016/j.febslet.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 25.Hailey DW, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nature Cell Biol. 2010;12:747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.English L, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nature Immunol. 2009;10:480–487. doi: 10.1038/ni.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nature Cell Biol. 2010;12:836–841. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 29.Yamaguchi H, et al. An initial step of GAS-containing autophagosome-like vacuoles formation requires Rab7. PLoS Pathogens. 2009;5:e1000670. doi: 10.1371/journal.ppat.1000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakagawa I, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 31.Sanjuan MA, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 32.Lee HK, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity. 2010;32:227–239. doi: 10.1016/j.immuni.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berger SB, et al. SLAM is a microbial sensor that regulates bacterial phagosome functions in macrophages. Nature Immunol. 2010;11:920–927. doi: 10.1038/ni.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang J, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl Acad. Sci. USA. 2009;106:6226–6231. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Z, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khaminets A, et al. Coordinated loading of IRG resistance GTPases on to the Toxoplasma gondii parasitophorous vacuole. Cell. Microbiol. 2010;12:939–961. doi: 10.1111/j.1462-5822.2010.01443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao YO, Khaminets A, Hunn JP, Howard JC. Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNγ-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathogens. 2009;5:1–17. doi: 10.1371/journal.ppat.1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sumpter R, Jr, Levine B. Autophagy and innate immunity: triggering, targeting and tuning. Semin. Cell Dev. Biol. 2010;21:699–711. doi: 10.1016/j.semcdb.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shahnazari S, et al. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe. 2010;8:137–146. doi: 10.1016/j.chom.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orvedahl A, et al. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dupont N, et al. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009;6:137–149. doi: 10.1016/j.chom.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 42.Ponpuak M, et al. Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity. 2010;32:329–341. doi: 10.1016/j.immuni.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guillemain, A. & Plus, N. Contrôle génique de la multiplication du virus de la sensibilité héréditaire au CO2 chez Drosophila melanogaster . Caryologia (suppl.) 1211–1213 (1954).
- 44.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nature Cell Biol. 2010;12:823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, et al. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005;121:567–577. doi: 10.1016/j.cell.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 46.Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity. 2009;30:588–598. doi: 10.1016/j.immuni.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yano T, et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nature Immunol. 2008;9:908–916. doi: 10.1038/ni.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jia K, et al. Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proc. Natl Acad. Sci. USA. 2009;106:14564–14569. doi: 10.1073/pnas.0813319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 50.Intemann CD, et al. Autophagy gene variant IRGM –261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathogens. 2009;5:e1000577. doi: 10.1371/journal.ppat.1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar D, et al. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell. 2010;140:731–743. doi: 10.1016/j.cell.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 52.Cooney R, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nature Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 53.Travassos LH, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nature Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 54.Zhang FR, et al. Genomewide association study of leprosy. N. Engl. J. Med. 2009;361:2609–2618. doi: 10.1056/NEJMoa0903753. [DOI] [PubMed] [Google Scholar]
- 55.Dreux M, Chisari FV. Viruses and the autophagy machinery. Cell Cycle. 2010;9:1295–1307. doi: 10.4161/cc.9.7.11109. [DOI] [PubMed] [Google Scholar]
- 56.Blanchet FP, et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity. 2010;32:654–669. doi: 10.1016/j.immuni.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orvedahl A, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 58.Leib DA, Alexander DE, Cox D, Yin J, Ferguson TA. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T cell responses. J. Virol. 2009;83:12164–12171. doi: 10.1128/JVI.01676-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.EX, et al. Viral Bcl-2-mediated evasion of autophagy aids chronic infection of γherpesvirus 68. PLoS Pathogens. 2009;5:e1000609. doi: 10.1371/journal.ppat.1000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee JS, et al. FLIP-mediated autophagy regulation in cell death control. Nature Cell Biol. 2009;11:1355–1362. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshikawa Y, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nature Cell Biol. 2009;11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 62.Moreau K, et al. Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell. Microbiol. 2010;12:1108–1123. doi: 10.1111/j.1462-5822.2010.01456.x. [DOI] [PubMed] [Google Scholar]
- 63.Kyei GB, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009;186:255–268. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl Acad. Sci. USA. 2009;106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sir D, et al. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl Acad. Sci. USA. 2010;107:4383–4388. doi: 10.1073/pnas.0911373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reggiori F, et al. Coronaviruses hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host Microbe. 2010;7:500–508. doi: 10.1016/j.chom.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yanai H, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462:99–103. doi: 10.1038/nature08512. [DOI] [PubMed] [Google Scholar]
- 68.Tang D, et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yuk JM, et al. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe. 2009;6:231–243. doi: 10.1016/j.chom.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 70.Low DH, et al. A novel human tectonin protein with multivalent β-propeller folds interacts with ficolin and binds bacterial LPS. PLoS ONE. 2009;4:e6260. doi: 10.1371/journal.pone.0006260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lipinski MM, et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev. Cell. 2010;18:1041–1052. doi: 10.1016/j.devcel.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu F, et al. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood. 2010;116:4806–4814. doi: 10.1182/blood-2010-06-288589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. 2008;455:396–400. doi: 10.1038/nature07208. [DOI] [PubMed] [Google Scholar]
- 74.Paludan C, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–596. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 75.Munz C. Antigen processing via autophagy—not only for MHC class II presentation anymore? Curr. Opin. Immunol. 2010;22:89–93. doi: 10.1016/j.coi.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 77.Tal MC, et al. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl Acad. Sci. USA. 2009;106:2770–2775. doi: 10.1073/pnas.0807694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jounai N, et al. The Atg5–Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl Acad. Sci. USA. 2007;104:14050–14055. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Komatsu M, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nature Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 81.Cadwell K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 83.Meissner F, Molawi K, Zychlinsky A. Mutant superoxide dismutase 1-induced IL-1β accelerates ALS pathogenesis. Proc. Natl Acad. Sci. USA. 2010;107:13046–13050. doi: 10.1073/pnas.1002396107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Qu X, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 85.Barrett JC, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nature Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brain O, Allan P, Simmons A. NOD2-mediated autophagy and Crohn disease. Autophagy. 2010;6:412–414. doi: 10.4161/auto.6.3.11389. [DOI] [PubMed] [Google Scholar]
- 87.Lapaquette P, Glasser AL, Huett A, Xavier RJ, Darfeuille-Michaud A. Crohn's disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell. Microbiol. 2010;12:99–113. doi: 10.1111/j.1462-5822.2009.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. Impaired autophagy of an intracellular pathogen induced by a Crohn's disease associated ATG16L1 variant. PLoS ONE. 2008;3:e3391. doi: 10.1371/journal.pone.0003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fujita N, et al. Differential involvement of Atg16L1 in Crohn disease and canonical autophagy: analysis of the organization of the Atg16L1 complex in fibroblasts. J. Biol. Chem. 2009;284:32602–32609. doi: 10.1074/jbc.M109.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cadwell K, et al. Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harley JB, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nature Genet. 2008;40:204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gateva V, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nature Genet. 2009;41:1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Han JW, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nature Genet. 2009;41:1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 94.Nakamura T, et al. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell. 2010;140:338–348. doi: 10.1016/j.cell.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rodriguez A, et al. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006;3:211–222. doi: 10.1016/j.cmet.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 97.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 98.Luciani A, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nature Cell Biol. 2010;12:863–875. doi: 10.1038/ncb2090. [DOI] [PubMed] [Google Scholar]
- 99.Gutierrez MG, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 100.Ogawa M, et al. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]