

Abstract
Autotaxin (ATX) is a secreted phosphodiesterase that hydrolyzes the abundant phospholipid lysophosphatidylcholine (LPC) to produce lysophosphatidic acid (LPA). The ATX-LPA signaling axis has been implicated in inflammation, fibrosis, and tumor progression, rendering ATX an attractive drug target. We recently described a boronic acid-based inhibitor of ATX, named HA155 (1). Here, we report the design of new inhibitors based on the crystal structure of ATX in complex with inhibitor 1. Furthermore, we describe the syntheses and activities of these new inhibitors, whose potencies can be explained by structural data. To understand the difference in activity between two different isomers with nanomolar potencies, we performed molecular docking experiments. Intriguingly, molecular docking suggested a remarkable binding pose for one of the isomers, which differs from the original binding pose of inhibitor 1 for ATX, opening further options for inhibitor design.
Introduction
The secreted glycoprotein autotaxin (ATX) is a phosphodiesterase responsible for the hydrolysis of lysophosphatidylcholine (LPC) into lysophosphatidic acid (LPA) and choline, as depicted in Scheme 1.1,2 The bioactive lipid LPA stimulates migration, proliferation and survival of cells by activating specific G protein-coupled receptors.(3) The ATX-LPA signaling axis is involved in cancer, inflammation and fibrotic disease.4−6 Potent and selective ATX inhibitors are needed to elucidate the contribution of ATX action to signaling cascades that may result in disease in case of malfunction.
Scheme 1. Autotaxin (ATX) is Responsible for Hydrolyzing the Lipid Lysophosphatidylcholine (LPC) into Lysophosphatidic Acid (LPA) and Choline.

ATX, also known as eNPP2, is a unique member of the ecto-nucleotide pyrophosphatase/phosphodiesterase (eNPP) family of proteins. It is the only family member capable of producing LPA by hydrolysis of LPC.(7) Recently reported crystal structures of mouse(8) and rat(9) ATX confirmed that a threonine residue and two zinc ions are necessary for activity of ATX.(10) From these structures, it could be concluded that ATX hydrolyzes its substrates through a typical alkaline phosphatase/phosphodiesterase mechanism.11,12 Furthermore, these structures showed that ATX specifically binds its lipid substrates in a hydrophobic pocket extending from the active site of ATX. This pocket accommodates the alkyl chain of the lipids in different poses as was also shown in various crystal structures.(8)
Recently, we described the discovery of a boronic acid-based ATX inhibitors that helped to reveal the short half-life (∼5 min) of LPA in vivo.13,14 We introduced a boronic acid moiety in the inhibitor structure to rationally target the threonine oxygen nucleophile of ATX with a hard matching Lewis acid. The crystal structure of ATX in complex with HA155 (1)(9) confirmed our hypothesis that this inhibitor targets the threonine oxygen nucleophile in the ATX active site via the boronic acid moiety, while the hydrophobic 4-fluorobenzyl moiety of inhibitor 1 targets the hydrophobic pocket responsible for lipid binding (Figure 1).
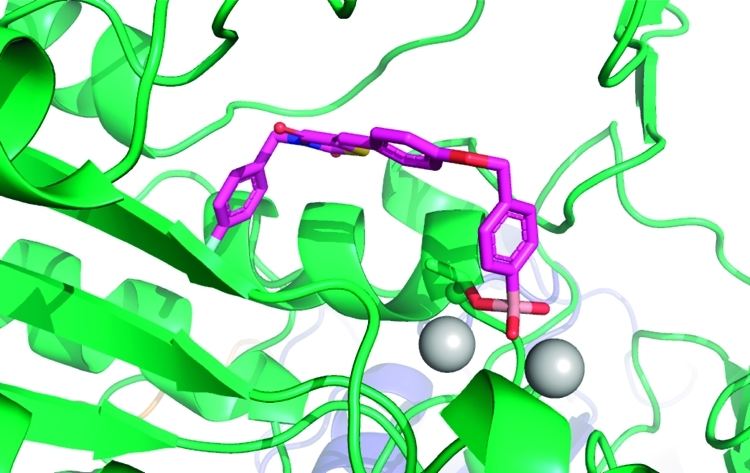
Figure 1.

ATX structure liganded with inhibitor 1 (PDB ID 2XRG). (A) Surface representation of ATX with inhibitor 1 (magenta). (B) Binding of inhibitor 1 to the threonine oxygen nucleophile and two zinc ions. (C) Visualizing the ether linker of inhibitor 1 bound to ATX. (D) Visualizing the degree of freedom for the thiazolidine-2,4-dione core of inhibitor 1 in the ATX binding site.
Here, we report a number of synthetic routes, systematically substituting linkers and the thiazolidine-2,4-dione core in 1, while keeping the boronic acid moiety untouched. The observed structure–activity relations could well be explained from the ATX structure in complex with inhibitor 1. A remarkable binding pose of a novel inhibitor, as predicted from molecular docking experiments, suggests additional avenues for further inhibitor design.
Results and Discussion
Design of Inhibitors
The structure of inhibitor 1 bound to the ATX active site (Figure 1) showed that its 4-fluorobenzyl moiety binds into the hydrophobic lipid binding pocket of ATX (Figure 1C,D).(9) This pocket also accommodates the lipid tail of LPA, the hydrolysis product of LPC.(8) The thiazolidine-2,4-dione core of 1 and the conjugated aromatic ring are located between the hydrophobic pocket and the catalytic site (Figure 1D). The ether linker, bridging the two aromatic rings in 1, and especially a methylene and arylboronic acid moiety are well accessible to solvent (Figure 1C). Binding of inhibitor 1 to the ATX active site is predominately driven by hydrophobic interactions (the interaction interface is approximately 500 Å2) and by the boronic acid binding to the threonine oxygen nucleophile of ATX.(9) The boron–oxygen distance observed is ∼1.6 Å, which is consistent with a covalent bond. As expected, this binding is reversible evidenced by the fact that ATX activity can be fully restored upon washing out the inhibitor.(13) In addition, one of the boronic acid hydroxyl moieties is tethered by the two zinc ions in the ATX active site. Thus, the boronic acid moiety targets not only the threonine oxygen nucleophile, but also the two zinc ions that are essential for catalytic activity of ATX (Figure 1B). Remarkably, there are no hydrogen bonds or salt bridges that participate in binding of inhibitor 1 to ATX. Inhibitor 1 is locked in a pose with reduced molecular flexibility, forming an ideal starting point for a structure-based approach to further modifications.
Previously, we determined that the 4-fluorobenzyl moiety is preferred from over 40 benzylic substituents tested.(13) For this reason, we left the 4-fluorobenzyl moiety untouched in this study. We investigated new design options, starting by modifying the ether linker in inhibitor 1. We decided to replace the ether linkage (OCH2) with various amides, an amine, and an E-configured double bond (CONH (17), CONCH2 (18), NHCO (19), NHCH2 (36), and (E)-CH=CH (20); see Table 1). The thiazolidine-2,4-dione core was investigated next. We substituted the sulfur atom present with (substituted) amino and methylene moieties (NH (26), NCH3 (28), or CH2 (32)) (Table 2). The carbon double bond conjugated to the thiazolidine-2,4-dione carbonyl moiety forms a possible Michael acceptor. Although this Michael acceptor is resistant to nucleophilic additions in vitro (Supporting Information Figure S1) it may be a biologically active Michael acceptor in vivo, and therefore, we investigated its removal.
Table 1. IC50 Values of the Inhibitors Resulting from the Linker Modification.

IC50 values have been determined in the choline release assay using 40 μM LPC and 10 nM ATX. The dose–response curve of inhibitor 17 shows biphasic curve (see Supporting Information Figure S2).
Table 2. IC50 Values of the Inhibitors Resulting from the Core Modification.

IC50 values have been determined in the choline release assay using 40 μM LPC and 10 nM ATX. E-28 contains 20% of the Z-isomer.
Chemical Synthesis of Modified Inhibitors
We first explored synthetic routes to replace the ether linkage in 1. The synthesis of target molecules 17–20 (Scheme 2) starts with palladium catalyzed borylation of appropriate aldehydes via a Suzuki-Miyaura reaction(15) as depicted in Scheme 2 (for syntheses of aldehydes 2–6 see Supporting Information). This reaction smoothly provided intermediates 7–11. Next, the pinacol protecting group was hydrolyzed under acidic conditions and oxidatively destroyed by NaIO4 giving boronic acid aldehydes 12–16. In the final step 3-(4-fluorobenzyl)thiazolidine-2,4-dione is reacted with the boronic acid aldehyde by Knoevenagel condensation to selectively give the Z-isomer of the final products (1 and 17–20).
Scheme 2. Synthetic Route Towards Linker Modified Inhibitors.

In order to remove the potential Michael acceptor present, we reduced the double bond in inhibitor 1 using hydrogen and palladium on carbon (Scheme 3A), to give compound 21. To reduce the carbonyl moiety also in the thiazolidine-2,4-dione core of 1, we used NaBH4 resulting in hemiaminal 22. After addition of sulfuric acid to the reaction mixture to eliminate the hydroxyl moiety in 22, unsaturated inhibitor 23 was obtained.
Scheme 3. Synthetic Routes Towards Core Modified Inhibitors.

For the syntheses of imidazolidine-2,4-dione-based inhibitors 26 and 28, imidazolidine-2,4-dione (24) was mono N-alkylated with 4-fluorobenzyl bromide to give intermediate 25 (Scheme 3B). Reaction of 25 by a Knoevenagel condensation with aldehyde 12 selectively resulted in the formation of the Z-isomer of 26. In parallel, intermediate 25 was methylated to give compound 27. Finally, 27 was condensed with aldehyde 12 resulting in target molecule 28. Both the Z- and E-isomers were formed in a 1:4 (Z:E) ratio. To obtain and isolate solely the Z-isomer 28, we N-methylated compound 26.
Core-hopping from thiazolidine-2,4-dione to pyrrolidine-2,5-dione is depicted in Scheme 3C. The route starts with the formation of a Wittig reagent starting from 2,5-pyrroledione (29), which is reacted with triphenylphosphine to form ylide 30.(16) The Wittig reaction of the carbonyl stabilized ylide 30 with aldehyde 12 selectively leads to the E-isomer of intermediate 31 as expected. Finally, compound 31 is N-alkylated with 4-fluorobenzyl bromide resulting in the pyrrolidine-2,5-dione product 32.
For the synthesis of a final tetrahydroisoquinoline-based core with a more rigid structure, the secondary amine in R- or S-tetrahydroisoquinoline 33 is reacted with 4-fluorobenzyl isocyanate in the presence of sodium hydroxide to form a urea intermediate. The imidazolidine ring is then formed upon acidification with hydrochloric acid, resulting in compound R- or S-34. In the final step, intermediate 34 is O-alkylated with 4-(bromomethyl)phenylboronic acid resulting in R- or S-35.
Structure–Activity Relations of Inhibitors and Autotaxin
Activity of the new molecules resulting from the linker and core modifications were determined in an LPC hydrolysis assay described previously,13,17 in which ATX-mediated release of choline from LPC is detected by a two-step enzymatic colorimetric reaction. The IC50 values observed for inhibitors with modified linkers and cores are listed in Tables 1 and 2.
When we replaced the ether moiety (OCH2, inhibitor 1, IC50 = 5.7 nM) for an amide linker (CONH, 17), we observed a significant loss of activity (IC50 = 147 nM). The dose–response curve of inhibitor 17 shows a biphasic curve(18) that could suggest several binding sites of this inhibitor for ATX (Supporting Information Figure S2). Expanding the CONH linker (17) to a more flexible CONHCH2 linker (18) improved the IC50 value (71 nM) by 2-fold compared to inhibitor 17. Reversing the amide linker in 17 to yield compound 19, results in high potency (IC50 = 10 nM), similar to inhibitor 1. However, inhibitor 19 is not able to achieve full inhibition, and 10% residual ATX activity is observed (Supporting Information Figure S3). Apparently, a more rigid amide linker results in suboptimal binding of the inhibitor. Therefore, we synthesized the more flexible amine analogue 36 (for the synthesis of inhibitor 36 see Supporting Information). This resulted indeed in a potent inhibitor (IC50 = 8.3 nM), similar to 1, and with no residual ATX activity (Supporting Information Figure S3). Introduction of an (E)-CH=CH linker resulted in compound 20, which was inactive in the nanomolar range (IC50 > 5 μM). This observation can be explained by the fact that the two aromatic rings linked with a flexible OCH2 linker in inhibitor 1 are positioned in an angle of roughly 90° in the ATX structure (see Figure 1B) which cannot be achieved by the (E)-CH=CH linker due to its rigid planar conformation.
Next, we explored the activity of compounds with a modified core. Reducing the carbon double bond in 1 resulted in little loss of activity in 21 with an IC50 value of 25 nM. Thus, although rigidity is preferred, the Michael acceptor can easily be removed without significant loss in activity. Reducing both the carbon double bond and the neighboring carbonyl in inhibitor 1 to hemiaminal 22, led to a significant loss in potency (IC50 = 1.6 μM). Inhibitor 23, where the hydroxyl moiety in compound 22 is removed is not very potent either (IC50 = 683 nM), indicates that the carbonyl moiety is important for binding. It appears from the crystal structure of inhibitor 1 bound to ATX that π-stacking between the phenyl ring of phenylalanine residue 274 (F274) and the carbonyl moiety in 1 is very likely seen their distance (4.1 Å, Supporting Information Figure S4).(19) By removing the carbonyl moiety in 1 or by changing it into a hydroxyl moiety, π-stacking will be lost resulting in lower potencies as observed for inhibitor 22 and 23.
The sulfur heteroatom in the thiazolidine-2,4-dione core was replaced with other atoms and moieties. We started by replacing the sulfur atom in 1 with a methylene moiety gives compound 32 with an IC50 value of 7.3 nM, comparable to inhibitor 1. Replacement of the sulfur atom for an amino group (26, IC50 = 26 nM) resulted in little loss in activity compared to inhibitor 1. When the amine in compound 26 is methylated, potency for the resulting inhibitor Z-28 (IC50 = 6.7 nM) is slightly increased. Interestingly, the E-isomer of 28 (IC50 = 5.3 nM) is marginally more potent than Z-28 or inhibitor 1, a finding that we did not anticipate.
Intrigued by the characteristics of inhibitors Z-28 and E-28, we decided to calculate likely binding poses. For this purpose, we used the Glide docking software.20−22 To validate our docking approach where we constrain the boronic acid moiety, we first docked inhibitor 1 back into the ATX active site, resulting in a pose (Figure 2B) very similar to the original crystal structure (Figure 2A) with a rmsd value of 1.1 Å (for superimposed image see Supporting Information Figure S5). Next, we docked the Z- and E-isomers of inhibitor 28 (the three best docking poses are depicted in Figure 2C and D). The docking poses of Z-28 correspond to the pose of inhibitor 1 in the ATX structure. However, two of the best three docking poses for E-28 suggest that the 4-fluorobenzyl moiety likely binds to a different area in the hydrophobic pocket (Figure 2D). The imidazolidine-2,4-dione core of E-28 is flipped around the double bond axis in the ATX binding site compared to its Z-isomer (compare Figure 2C with D). This observation is in agreement with the conformations of both isomers. In addition, the binding poses of Z-28 and E-28 resulting from our docking study suggest that the current 4-fluorobenzyl moiety could be expanded from the methylene moiety in the 4-fluorobenzyl substituent with other substituents in future ATX inhibitors.
Figure 2.

(A) Focus from inside the protein on the thiazolidine-2,4-dione core of inhibitor 1 bound to ATX. (B) Inhibitor 1 docked into the active site of ATX to validate our docking approach. (C) The three best docking poses for the Z-isomer of 28. (D) The three best docking poses for the E-isomer of 28. Docking poses were generated using the docking program Glide.
Finally, we evaluated inhibitors in which we introduced a rigidified three-ring system (Table 2) incorporating a tetrahydroisoquinoline motif that we deemed likely to bind. This modification avoids the presence of a Michael acceptor, while introducing rigidity and a new core structure. This modification resulted in the chiral inhibitor 35, which is still very potent although some activity is lost compared to 1. No significant difference is observed in potency for either the S or R enantiomers of inhibitor 35, with IC50 values of 55 and 59 nM, respectively.
In summary, we explored structure–activity relations building on boronic acid-based ATX inhibitor 1, which resulted in a number of potent inhibitors. We used the crystal structure of ATX liganded with inhibitor 1(9) to explain the structure–activity relationships observed for a rational inhibitor modification approach. Our results suggest that this approach allows rapid structure guided modification. Finally, molecular docking efforts proved useful to explain unexpected high potency of E-isomer 28 and suggested that the lipophilic pocket near the ATX active site may be better exploited in the future for the design of new inhibitors.
Experimental Section
Chemistry
The R and S enantiomers of building block 33 were purchased from CSPS Pharmaceuticals, San Diego, USA. All other chemicals were obtained from Sigma-Aldrich and used without further purification unless otherwise noted. Analytical thin layer chromatography was performed on aluminum sheets precoated with silica gel 60 F254. Column chromatography was carried out on silica gel (0.035–0.070, 90 Å, Acros).
For isolation by centrifugation a Heraeus Multifuge 3 S-R centrifuge was used. Products were spun at 4400g, at 298 K for 5 min. Nuclear magnetic resonance spectra (1H and 13C NMR) were determined in deuterated dimethyl sulfoxide (d6-DMSO) using a Bruker Avance 300 (1H, 300 MHz; 13C, 75 MHz) at 298 K, unless indicated otherwise. Peak shapes are indicated with the symbols ‘d’ (doublet), ‘dd’ (double doublet), ‘s’ (singlet), ‘bs’ (broad singlet), and ‘m’ (multiplet). Chemical shifts (δ) are given in ppm and coupling constants J in Hz. Dimethyl sulfoxide (δH = 2.50 ppm; δC = 39.51 ppm) was used as internal reference.
The purity of all tested compounds was determined by high-performance liquid chromatography coupled to mass spectrometry (HPLC-MS) and was greater than 95%. HPLC-MS measurements were performed on a system equipped with a Waters 2795 Separation Module (Alliance HT), Waters 2996 Photodiode Array Detector (190–750 nm), Atlantis T3 C18 column (2.1 × 100 mm, 3 μm), and an LCT Orthogonal Acceleration Time of Flight Mass Spectrometer. Samples were run at a flow rate of 0.40 mL min–1 at 313 K, using gradient elution (water/acetonitrile/formic acid) from 950/50/1 (v/v/v) to 50/950/1 (v/v/v).
The preparative HPLC system was equipped with a Waters 1525 Binary HPLC Pump, a Waters 2487 Dual λ Absorbance Detector, and an Atlantis C18 column (19 × 250 mm, 10 μm). Samples were run at a flow rate of 18 mL min–1 using gradient elution (water/acetonitrile) from 60/40 (v/v) to 10/90 (v/v).
General Procedure for Borylation of Aldehydes and Pinacol Deprotection (12–16)
In a dry flask, bis(pinacolato)diboron (1.34 g, 5.28 mmol), the appropriate aldehyde (1.80 mmol) and potassium acetate (0.542 g, 5.52 mmol) were added to a solution of Pd(dppf)Cl2 (46.6 mg, 0.0637 mmol) in dimethylformamide (15 mL). The reaction mixture was stirred under an atmosphere of argon for 18 h at 353 K. The reaction mixture was filtered over Hyflo Super Cel medium and diluted with ethyl acetate (100 mL). The solution was washed with brine (50 and 25 mL), dried over magnesium sulfate, and concentrated.
The crude product was dissolved in tetrahydrofuran (11 mL) and sodium periodate (2.22 g, 10.4 mmol) and water (2.8 mL) were added. After stirring for 30 min, 1 M hydrochloric acid (1.1 mL) was added, and after 2 h, additional sodium periodate (1.17 g, 5.47 mmol) was added and the solution was stirred for another 2 h. The reaction mixture was diluted with ethyl acetate (20 mL) and washed with water (10 mL). The water layer was extracted with ethyl acetate (15 mL). The combined organic layers were washed with brine (15 mL), dried over magnesium sulfate, and the solution was concentrated under vacuum resulting in a light yellow solid. The resulting product was used without further purification.
(4-((4-Formylphenoxy)methyl)phenyl)boronic Acid (12)
Yield: 70%. 1H NMR: δ = 9.86 (s, 1H), 8.09 (s, 1H), 7.87 (d, J = 8.8, 1H), 7.82 (d, J = 8.1, 1H), 7.42 (d, J = 8.1, 1H), 7.20 (d, J = 8.7, 1H), 5.24 (s, 1H). 13C NMR: δ = 191.75, 163.73, 138.49, 134.74, 132.26, 130.22, 127.15, 115.76, 70.09, 39.95 (C–B(OH)2 not visible). MS: m/z [M+H]+ calc. 257.10, obs. 257.10.
For experimental details of compounds 13–16, see Supporting Information.
General Method for Knoevenagel Condensation (1, 17–20, 26, and E-28)
To a solution of 3-(4-fluorobenzyl)thiazolidine-2,4-dione (0.293 mmol) in ethanol (2.5 mL), piperidine (20 μL, 0.207 mmol), and the appropriate aldehyde (0.352 mmol) were added and the solution was refluxed for 22 h.
(Z)-4-[(4-{[3-(4-Fluorobenzyl)-2,4-dioxo-1,3-thiazolan-5-yliden]methyl}phenoxy) methyl]benzene Boronic Acid (1)
Upon cooling the reaction mixture to room temperature, the product precipitated out of solution. Dissolving the product in dimethyl sulfoxide and precipitating it with 0.5 M hydrochloric acid resulted in pure compound. Yield: 81%. 1H NMR: δ = 8.03 (s, 2H), 7.92 (s, 1H), 7.80 (d, J = 8.1, 2H), 7.60 (d, J = 8.9, 2H), 7.41 (d, J = 8.0, 2H), 7.39–7.31 (m, J = 5.5, 8.8, 2H), 7.26–7.09 (m, J = 4.5, 8.9, 4H), 5.21 (s, 2H), 4.82 (s, 2H). 13C NMR: δ = 167.38, 165.59, 161.66 (d, 1JCF = 244), 160.33, 138.19, 134.28, 133.48, 132.33, 131.81 (d, 4JCF = 3), 129.95 (d, 3JCF = 8), 126.67, 125.55, 117.89, 115.81, 115.47 (d, 2JCF = 21), 69.52, 43.90 (C–B(OH)2 not visible). MS: m/z [M+H]+ calc. 464.11, obs. 464.19.
For experimental details of compounds 17–20, 26, and E-28 see Supporting Information.
(4-((4-((3-(4-Fluorobenzyl)-2,4-dioxothiazolidin-5-yl)methyl)phenoxy)methyl) phenyl)boronic Acid (21)
A mixture of compound 1 (50.1 mg, 0.108 mmol) and 10 wt % Pd/C (24.0 mg) in degassed methanol (3 mL) was stirred under a hydrogen atmosphere for 2 h. Extra 10 wt % Pd/C was added (12.0 mg) and the reaction was allowed to continue for one night. The mixture was filtrated and concentrated to dryness. Preparative HPLC afforded the title compound. Yield: 18.3 mg, 77%. 1H NMR: δ = 8.03 (s, 2H), 7.80 (d, J = 8.1, 2H), 7.39 (d, J = 8.1, 2H), 7.28–6.99 (m, 6H), 6.88 (d, J = 8.7, 2H), 5.06 (s, 2H), 5.00 (dd, J = 4.4, 8.0, 1H), 4.60 (dd, J = 15.0, 21.5, 2H), 3.14 (dd, J = 8.0, 14.2, 1H). 13C NMR: δ = 173.71, 171.00, 161.50 (d, 1JCF = 244), 157.44, 138.74, 134.16, 131.58 (d, 4JCF = 3), 130.56, 129.66 (d, 3JCF = 8), 127.98, 126.52, 115.21 (d, 2JCF = 21), 114.57, 69.12, 50.94, 43.56, 35.79 (C–B(OH)2 not visible). MS: m/z [M+H]+ calc. 466.13, obs. 466.25, [M-H2O+H]+ calc. 448.12, obs. 448.23.
(4-((4-((3-(4-Fluorobenzyl)-4-hydroxy-2-oxothiazolidin-5-yl)methyl)phenoxy) methyl)phenyl)boronic Acid (22)
To a solution of compound 1 (50.0 mg, 0.108 mmol) in dimethyl sulfoxide (0.5 mL), sodium borohydride (16.3 mg, 0.430 mmol) was slowly added. After 9 h of stirring, the reaction mixture was diluted with ethyl acetate (4 mL) and was washed with water (2 × 2 mL). The organic layer was dried over calcium chloride and concentrated in vacuo, resulting in the title compound. Yield: 30.2 mg, 60%. 1H NMR: δ = 8.06 (s, 2H), 7.80 (d, J = 8.1, 2H), 7.39 (d, J = 8.0, 2H), 7.36 – 7.11 (m, 5H), 6.94 (d, J = 8.7, 2H), 6.87 (d, J = 8.7, 2H), 6.71 (d, J = 6.2, 1H), 5.06 (s, 2H), 4.72 (dd, J = 7.7, 15.1, 1H), 4.10 (d, J = 15.1, 1H), 3.67 (t, J = 7.9, 1H), 3.35 (s, 1H), 2.81 (dd, J = 7.3, 13.9, 1H), 2.70 (dd, J = 8.4, 13.9, 1H). 13C NMR: δ = 169.70, 161.55 (d, 1JCF = 244), 157.13, 138.85, 134.15, 133.14 (d, 4JCF = 3.0), 130.01, 130.00 (d, 3JCF = 8), 129.73, 126.54, 115.38 (d, 2JCF = 21), 114.64, 84.02, 69.11, 51.42, 44.02, 39.65. (C–B(OH)2 not visible). MS: m/z [M+H]+ calc. 468.15, obs. 468.23.
(4-((4-((3-(4-Fluorobenzyl)-2-oxo-2,3-dihydrothiazol-5-yl)methyl)phenoxy) methyl)phenyl)boronic Acid (23)
To a solution of compound 1 (45.7 mg, 0.0989 mmol) in dimethyl sulfoxide (0.75 mL), sodium borohydride (29.1 mg, 0.769 mmol) was slowly added. After 7 h of stirring the reaction mixture, concentrated sulfuric acid (2 × 50 μL) was added over a 15 min interval. The reaction mixture was stirred for an additional 5 h. Ethyl acetate (25 mL) was added and the mixture was washed with water (4 × 10 mL). The organic layer was dried over calcium chloride and concentrated in vacuo, affording pure compound 23. Yield: 31.8 mg, 72%. 1H NMR: δ = 8.05 (s, 2H), 7.79 (d, J = 8.1, 2H), 7.38 (d, J = 8.1, 2H), 7.36–7.16 (m, 4H), 7.13 (d, J = 8.7, 2H), 6.95 (d, J = 8.7, 2H), 6.87 (s, 1H), 5.07 (s, 2H), 4.80 (s, 2H), 3.73 (s, 2H). 13C NMR: δ = 170.43, 161.61 (d, 1JCF = 244), 157.13, 138.85, 134.17, 133.10 (d, 4JCF = 3), 130.63, 130.63, 129.76 (d, 3JCF = 8), 129.42, 126.48, 121.56, 117.78, 115.50 (d, 2JCF = 21), 114.86, 69.16, 46.78, 32.91 (C–B(OH)2 not visible). MS: m/z [M+H]+ calc. 450.13, obs. 450.22.
3-(4-Fluorobenzyl)imidazolidine-2,4-dione (25)
To a cooled solution (273 K) of hydantoin (8.01 g, 80.1 mmol) in dimethylformamide (140 mL) sodium hydride (60% in oil, 1.80 g, 45.0 mmol) was added. A solution of 1-(bromomethyl)-4-fluorobenzene (5.0 mL, 41 mmol) in dimethylformamide (5 mL) was added to the reaction mixture. The mixture was allowed to warm up to room temperature and was stirred for 6 h. Then, the mixture was poured into water (200 mL) and hexane (200 mL) was added. After a night at 277 K, the precipitate was filtered and dried to give a white solid. Yield: 4.7 g, 56%. 1H NMR: δ = 8.14 (s, 1H), 7.45–7.21 (m, 1H), 7.26–7.09 (m, 1H), 4.51 (s, 2H), 3.97 (s, 2H). 13C NMR: δ = 171.91, 161.45 (d, 1JCF = 244), 157.29, 133.04 (d, 4JCF = 3), 129.66 (d, 3JCF = 8), 115.21 (d, 2JCF = 21), 46.00, 40.29. MS: m/z [M+H]+ calc. 209.07, obs. 208.93.
3-(4-Fluorobenzyl)-1-methylimidazolidine-2,4-dione (27)
To a cooled solution (273 K) of 3-(4-fluorobenzyl)imidazolidine-2,4-dione (98.1 mg, 0.471 mmol) in dimethylformamide (0.5 mL), sodium hydride (60% in oil, 21.1 mg, 0.530 mmol) was added. Subsequently, iodomethane (33 μL, 0.53 mmol) was added to the reaction mixture. The mixture was allowed to warm up to room temperature and was stirred for 3 h. Then, the mixture was poured into ice water (2.5 mL) and hexane (2.5 mL) was added. After a night at 277 K, the precipitate was filtered and dried to give a white solid. Yield: 75 mg, 72%. 1H NMR: δ = 7.34–7.12 (m, 4H), 4.52 (s, 2H), 4.01 (s, 2H), 2.86 (s, 3H). 13C NMR: δ = 170.20, 161.46 (d, 1JCF = 244), 156.24, 132.92 (d, 4JCF = 3), 129.68 (d, 3JCF = 8), 115.21 (d, 2JCF = 21), 51.36, 40.74, 29.21. MS: m/z [M+H]+ calc. 223.09, obs. 223.06.
(Z)-(4-((4-((1-(4-Fluorobenzyl)-3-methyl-2,5-dioxoimidazolidin-4-ylidene)methyl) phenoxy)methyl)phenyl)boronic Acid (Z-28)
To a cooled solution (273 K) of hydantoine 26 (10.1 mg, 0.0224 mmol) and sodium hydride (60% in oil, 1.46 mg, 0.0365 mmol) in DMF (0.15 mL), iodomethane (2.25 μL, 0.0361 mmol) was added. The mixture was allowed to warm up to room temperature and was stirred for 4 h. Final product was isolated by using preparative HPLC. Z-configuration confirmed by the chemical shift of the vinyl and methyl proton reported in literature.(24)
Yield: 6.97 mg, 68%. 1H NMR: δ = 8.11 (s, 1H), 7.80 (d, J = 8.1, 2H), 7.50–7.30 (m, 6H), 7.26–7.10 (m, 2H), 7.05 (d, J = 8.8, 2H), 6.76 (s, 1H), 5.14 (s, 2H), 4.67 (s, 2H), 2.92 (s, 3H). 13C NMR: δ = 163.01, 159.92, 156.78 (d, 1JCF = 249), 138.52, 134.19, 132.55 (d, 4JCF = 3), 131.28, 129.75 (d, 3JCF = 8), 128.50, 126.58, 124.66, 115.31 (d, 2JCF = 21), 114.55, 111.56, 69.30, 41.12, 30.35 (C–B(OH)2 not visible). MS: m/z [M+H]+ calc. 461.17, obs. 461.12.
(E)-(4-((4-((2,5-Dioxopyrrolidin-3-ylidene)methyl)phenoxy)methyl)phenyl)boronic Acid (31)
To a heated (343 K) solution of compound 30 (159 mg, 0.442 mmol)(16) in methanol (5 mL) aldehyde 12 (106 mg, 0.414 mmol) was added. After 1 h of heating, the reaction mixture was cooled using an ice bath resulting in precipitation of the title compound. The precipitate was filtered and washed with ice-cold methanol resulting in compound 31. Yield: 88 mg, 63%. 1H NMR: δ = 11.34 (s, 1H), 8.04 (s, 2H), 7.80 (d, J = 8.0, 2H), 7.57 (d, J = 8.9, 2H), 7.41 (d, J = 8.0, 2H), 7.33 (t, 4J = 2.1, 1H), 7.10 (d, J = 8.8, 2H), 5.19 (s, 2H), 3.60 (d, 4J = 2.2, 2H). 13C NMR: δ = 175.80, 172.09, 159.47, 138.42, 134.22, 132.00, 131.31, 126.96, 126.54, 124.16, 115.31, 69.31, 34.71 (C–B(OH)2 not visible). MS: m/z [M+H]+ calc. 338.12, obs. 338.11.
(E)-(4-((4-((1-(4-Fluorobenzyl)-2,5-dioxopyrrolidin-3-ylidene)methyl)phenoxy) methyl)phenyl)boronic Acid (32)
To a solution of compound 31 (30 mg, 0.0890 mmol) in dimethylformamide (0.3 mL) sodium hydride (60% in oil, 3.65 mg, 0.0913 mmol) was added. After addition of 4-fluorobenzyl bromide (24 μL, 0.19 mmol), the reaction mixture was stirred for 4 h. In addition, potassium carbonate (2.04 mg, 0.0148 mmol) was added and the reaction mixture was stirred overnight. Then, the mixture was poured into ice water (0.9 mL) and hexane (0.3 mL) was added. After a night at 277 K, the precipitate was filtered, dried, and purified using preparative HPLC to give a white solid. E-Configuration confirmed by the chemical shift of the vinyl proton reported in literature.(16) Yield: 13 mg, 32%. 1H NMR: δ = 8.04 (s, 2H), 7.80 (d, J = 8.1, 2H), 7.61 (d, J = 8.9, 2H), 7.45 (t, 4J = 2.1, 1H), 7.41 (d, J = 8.0, 2H), 7.38–7.29 (m, 2H), 7.23–7.04 (m, 4H), 5.19 (s, 2H), 4.66 (s, 2H), 3.74 (d, 4J = 2.1, 2H). 13C NMR: δ = 174.19, 170.59, 161.44 (d, 1JCF = 244), 159.65, 138.39, 134.22, 132.59 (d, 4JCF = 3), 132.31, 132.17, 129.73 (d, 3JCF = 8), 126.85, 126.54, 122.27, 115.36, 115.22 (d, 2JCF = 21), 69.33, 40.71, 33.74 (C–B(OH)2 not visible). MS: m/z [M+H]+ calc. 446.16, obs. 446.12.
(S)-2-(4-Fluorobenzyl)-7-hydroxy-10,10a-dihydroimidazo[1,5-b]isoquinoline-1,3(2H,5H)-dione (S-34)
Compound S-33 (100 mg, 0.518 mmol) was dissolved in a mixture of dioxane and water (3:1, 4 mL) and 30 wt % of sodium hydroxide solution was used to adjust the pH to 14. The reaction mixture was heated to 313 K and 4-fluorobenzyl isocyanate (100 μL, 0.785 mmol) was added. After 2 h of stirring, the mixture was cooled to room temperature and the resulting solid was removed using centrifugation. From the resulting solution, dioxane was evaporated and concentrated hydrochloric acid was used to adjust the pH to 1. After refluxing the reaction mixture for 2 h 30 min, it was cooled to 278 K affording a white precipitate. Washing the precipitate with ice-cold water (3 × 1 mL) afforded pure compound S-34. Yield: 30 mg, 18%. 1H NMR: δ = 9.37 (s, 1H), 7.35–7.30 (m, 2H), 7.18–7.12 (m, 2H), 7.06–7.03 (m, 1H), 6.65–6.63 (m, 2H), 4.77 (d, J = 16.8, 1H), 4.58 (s, 2H), 4.32–4.27 (m, 2H), 3.05 (m, 1H), 2.82–2.64 (m, 1H). 13C NMR: δ = 172.76, 161.44 (d, 1JCF = 244), 156.07, 154.60, 132.82 (d, 4JCF = 3), 132.35, 130.13, 129.51 (d, 3JCF = 8), 121.45, 115.26 (d, 2JCF = 21), 114.30, 112.67, 54.54, 41.23, 40.56, 29.02. MS: m/z [M+H]+ calc. 327.11, obs. 327.09.
(R)-2-(4-Fluorobenzyl)-7-hydroxy-10,10a-dihydroimidazo[1,5-b]isoquinoline-1,3(2H,5H)-dione (R-34)
For reaction details, see compound S-34. Yield: 29%. 1H NMR, 13C NMR, and MS, see Supporting Information.
(S)-(4-(((2-(4-Fluorobenzyl)-1,3-dioxo-1,2,3,5,10,10a-hexahydroimidazo[1,5-b]isoquinolin-7-yl)oxy)methyl)phenyl)boronic Acid (S-35)
To a heated solution (323 K) of compound S-34 (15.3 mg, 0.0469 mmol) in acetone (0.3 mL), potassium carbonate (10.2 mg, 0.0738 mmol) and 4-(bromomethyl)phenylboronic acid (12.4 mg, 0.0577 mmol) were added. After 4 h of stirring and heating, additional potassium carbonate (10.1 mg, 0.0730 mmol) was added and the suspension was stirred overnight. Finally, the reaction mixture was diluted with ethyl acetate and the organic layer was washed with 1 M hydrochloric acid (2 × 0.5 mL) and brine (0.5 mL), dried over sodium sulfate, and concentrated in vacuo. The resulting solid was further purified using preparative HPLC affording pure compound S-35. Yield: 11 mg, 52%. 1H NMR: δ = 8.06 (s, 2H), 7.78 (d, J = 8.1, 2H), 7.50–7.27 (m, 4H), 7.27–7.05 (m, 3H), 7.02–6.76 (m, 2H), 5.09 (s, 2H), 4.82 (d, J = 17.0, 1H), 4.58 (s, 2H), 4.42–4.20 (m, 2H), 3.19–3.02 (m, 1H), 2.80–2.71 (m, 1H). 13C NMR: δ = 172.72, 161.46 (d, 1JCF = 244), 157.09, 154.60, 138.74, 134.17, 132.81 (d, 4JCF = 3), 132.60, 130.23, 129.55 (d, 3JCF = 8), 126.49, 123.60, 115.28 (d, 2JCF = 21), 113.87, 112.47, 69.20, 54.40, 41.30, 40.60, 29.00 (C–B(OH)2 not visible). MS: m/z [M+H]+ calc. 461.17, obs. 461.14.
(R)-(4-(((2-(4-Fluorobenzyl)-1,3-dioxo-1,2,3,5,10,10a-hexahydroimidazo[1,5-b]isoquinolin-7-yl)oxy)methyl)phenyl)boronic acid (R-35)
For reaction details, see compound S-35. Yield: 56%. 1H NMR, 13C NMR, and MS, see Supporting Information.
Choline Release Assay.(13)
Measuring ATX activity using LPC (18:1) as substrate was determined as follows. In an opaque flat-bottom 96-wells plate (Greiner) was added 0.5 μL dimethyl sulfoxide containing inhibitor, to 25 μL recombinant ATX (∼20 nM) in Tris-HCl buffer (0.01% Triton X-100 and 50 mM Tris-HCl, pH 7.4). Finally, 25 μL of 80 μM LPC (18:1) in Tris-HCl buffer (10 mM MgCl2, 10 mM CaCl2, 0.01% Triton X-100 and 50 mM Tris-HCl, pH 7.4) was added to each well and the plate was incubated at 310 K. The above-described mixture with dimethyl sulfoxide alone was used as a control. LPC without ATX was taken as control for autohydrolysis of LPC. For each inhibitor, ten concentrations were measured covering a range of 0.01 to 30 μM to determine IC50 values. After 4 h of incubation, 50 μL ABTS (2 mM) and horseradish peroxidase (5 U mL–1) was added to the reaction mixture and absorbance was measured and used to correct for absorbance of the molecules. Finally, 50 μL choline oxidase (5 U mL–1) in Tris-HCl (0.01% Triton X-100 and 50 mM Tris-HCl, pH 7.4) was added for colorimetric reaction. Absorbance was measured in a Perkin-Elmer Envision plate reader (λ = 405 nm). Data were analyzed using Graphpad Prism software.
In addition, the effect of the inhibitors on the enzymatic coloring reaction was investigated using 40 μM choline at 30 μM inhibitor using the above-described coloring reagents. The enzymatic coloring reaction was not effected by the inhibitors.
Docking Experiments, Protein, and Ligand Preparation
The X-ray structure of ATX in complex with inhibitor 1 (PDB ID: 2XRG) was used for the docking studies. The protein structure was prepared using the Schrödinger Suite 2010 Protein Preparation Wizard (with Epik 2.1,(25) Impact 5.6, and Prime 2.2). The initial 3D structures of the ligands were generated using LigPrep 2.4 and the ligand partial charges were ascribed using the OPLS2005 force-field as performed by Glide 5.6.20−22 We defined the binding region by a 20 Å × 20 Å × 20 Å box centered on the central position of inhibitor 1 in the crystal ATX complex. We used positional constraints for the two oxygen atoms of the boronic acid and the arylic carbon direct next to the boron atom in inhibitor 1. The Glide Emodel score was used to rank the docking poses. Pictures were made using PyMOL 1.3.
Acknowledgments
In memory of Loes J. D. Hendrickx 06-01-1986/28-05-2010. We want to thank Irene Farre Gutierrez for helpful discussions. This work was supported by grants from The Netherlands Organization for Scientific Research (NWO), the Dutch Cancer Society (KWF), and The Netherlands Proteomics Centre supported by The Netherlands Genomics Initiative.
Glossary
Non-Standard Abbreviations
- ABTS
2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid)
- ATX
autotaxin
- HPLC-MS
high-performance liquid chromatography mass spectrometry
- LPA
lysophosphatidic acid
- LPC
lysophosphatidylcholine
- eNPP
ectonucleotide pyrophosphatase and phosphodiesterase
Supporting Information Available
Contains Michael acceptor study of inhibitor 1, dose–response curves of inhibitors 17, 19, and 36, image visualizing π-stacking of carbonyl inhibitor 1 with ATX residue F274, image of docked and X-ray pose of inhibitor 1 superimposed, experimental details of compounds 13–20, 26, and E-28, details on the syntheses of aldehydes 2–6 and amine linker-based inhibitor 36, and spectral data of all intermediates and target molecules. This material is available free of charge via the Internet at http://pubs.acs.org.
Author-Status
† Deceased.
Supplementary Material
References
- Tokumura A.; Majima E.; Kariya Y.; Tominaga K.; Kogure K.; Yasuda K.; Fukuzawa K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [DOI] [PubMed] [Google Scholar]
- Umezu-Goto M.; Kishi Y.; Taira A.; Hama K.; Dohmae N.; Takio K.; Yamori T.; Mills G. B.; Inoue K.; Aoki J.; Arai H. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moolenaar W. H.; van Meeteren L. A.; Giepmans B. N. The ins and outs of lysophosphatidic acid signaling. Bioessays 2004, 26, 870–881. [DOI] [PubMed] [Google Scholar]
- Kanda H.; Newton R.; Klein R.; Morita Y.; Gunn M.; Rosen S. Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat. Immunol. 2008, 9, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meeteren L.; Moolenaar W. Regulation and biological activities of the autotaxin-LPA axis. Prog. Lipid Res. 2007, 46, 145–160. [DOI] [PubMed] [Google Scholar]
- Tager A.; LaCamera P.; Shea B.; Campanella G.; Selman M.; Zhao Z.; Polosukhin V.; Wain J.; Karimi-Shah B.; Kim N.; Hart W.; Pardo A.; Blackwell T.; Xu Y.; Chun J.; Luster A. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [DOI] [PubMed] [Google Scholar]
- Stefan C.; Jansen S.; Bollen M. Modulation of purinergic signaling by NPP-type ectophosphodiesterases. Purinergic. Signal 2006, 2, 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H.; Okudaira S.; Hama K.; Mihara E.; Dohmae N.; Inoue A.; Ishitani R.; Takagi J.; Aoki J.; Nureki O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 2011, 18, 205–212. [DOI] [PubMed] [Google Scholar]
- Hausmann J.; Kamtekar S.; Christodoulou E.; Day J.; Wu T.; Fulkerson Z.; Albers H.; van Meeteren L.; Houben A.; van Zeijl L.; Jansen S.; Andries M.; Hall T.; Pegg L.; Benson T.; Kasiem M.; Harlos K.; Kooi C.; Smyth S.; Ovaa H.; Bollen M.; Morris A.; Moolenaar W.; Perrakis A. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijsbers R.; Aoki J.; Arai H.; Bollen M. The hydrolysis of lysophospholipids and nucleotides by autotaxin (NPP2) involves a single catalytic site. FEBS Lett. 2003, 538, 60–64. [DOI] [PubMed] [Google Scholar]
- Gijsbers R.; Ceulemans H.; Stalmans W.; Bollen M. Structural and catalytic similarities between nucleotide pyrophosphatases/phosphodiesterases and alkaline phosphatases. J. Biol. Chem. 2001, 276, 1361–1368. [DOI] [PubMed] [Google Scholar]
- Zalatan J.; Fenn T.; Brunger A.; Herschlag D. Structural and functional comparisons of nucleotide pyrophosphatase/phosphodiesterase and alkaline phosphatase: implications for mechanism and evolution. Biochemistry 2006, 45, 9788–9803. [DOI] [PubMed] [Google Scholar]
- Albers H.; van Meeteren L.; Egan D.; van Tilburg E.; Moolenaar W.; Ovaa H. Discovery and optimization of boronic acid based inhibitors of autotaxin. J. Med. Chem. 2010, 53, 4958–4967. [DOI] [PubMed] [Google Scholar]
- Albers H. M.; Dong A.; van Meeteren L. A.; Egan D. A.; Sunkara M.; van Tilburg E. W.; Schuurman K.; van Tellingen O.; Morris A. J.; Smyth S. S.; Moolenaar W. H.; Ovaa H. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 7257–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaura N.; Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar]
- Mizufune H.; Nakamura M.; Mitsudera H. Process research on arylnaphthalene lignan aza-analogues: a new palladium-catalyzed benzannulation of [alpha],[beta]-bisbenzylidenesuccinic acid derivatives. Tetrahedron 2006, 62, 8539–8549. [Google Scholar]
- Cui P.; Tomsig J.; McCalmont W.; Lee S.; Becker C.; Lynch K.; Macdonald T. Synthesis and biological evaluation of phosphonate derivatives as autotaxin (ATX) inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 1634–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer G.; Mutel V.; Trube G.; Malherbe P.; Kew J. N. C.; Mohacsi E.; Heitz M. P.; Kemp J. A. Ro 25–6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. characterization in vitro. J. Pharmacol. Exp. Therapeutics 1997, 283, 1285–1292. [PubMed] [Google Scholar]
- Jain A.; Purohit C.; Verma S.; Sankararamakrishnan R. Close contacts between carbonyl oxygen atoms and aromatic centers in protein structures: π-π or lone-pair-π interactions?. J. Phys. Chem. B 2007, 111, 8680–8683. [DOI] [PubMed] [Google Scholar]
- Friesner R.; Banks J.; Murphy R.; Halgren T.; Klicic J.; Mainz D.; Repasky M.; Knoll E.; Shelley M.; Perry J.; Shaw D.; Francis P.; Shenkin P. Glide: a new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Friesner R.; Murphy R.; Repasky M.; Frye L.; Greenwood J.; Halgren T.; Sanschagrin P.; Mainz D. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- Halgren T.; Murphy R.; Friesner R.; Beard H.; Frye L.; Pollard W. T.; Banks J. Glide: a new approach for rapid, accurate docking and scoring. 2. enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- Tan S.; Ang K.; Fong Y. (Z)- and (E)-5-Arylmethylenehydantoins: spectroscopic properties and configuration assignment. J. Chem. Soc., Perkin Trans. 2 1986, 1941–1944. [Google Scholar]
- Shelley J.; Cholleti A.; Frye L.; Greenwood J.; Timlin M.; Uchimaya M. Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.